
Abstract
Herpesviruses are successful pathogens that infect most vertebrates as well as at least one invertebrate species. Six of the eight human herpesviruses are widely distributed in the population. Herpesviral infections persist for the life of the infected host due in large part to the ability of these viruses to enter a non-productive, latent state in which viral gene expression is limited and immune detection and clearance is avoided. Periodically, the virus will reactivate and enter the lytic cycle, producing progeny virus that can spread within or to new hosts. Latency has been classically divided into establishment, maintenance, and reactivation phases. Here we focus on demonstrated and postulated molecular mechanisms leading to the establishment of latency for representative members of each human herpesvirus family. Maintenance and reactivation are also briefly discussed. In particular, the roles that tegument proteins may play during latency are highlighted. Finally, we introduce the term animation to describe the initiation of lytic phase gene expression from a latent herpesvirus genome, and discuss why this step should be separated, both molecularly and theoretically, from reactivation.
Similar content being viewed by others


Review
Herpesvirus Lytic and Latent Infections
Herpesviruses are large double-stranded DNA viruses with a unique virion morphology consisting of a genome-containing capsid, a proteinaceous tegument, and a lipid envelope. Human herpesviruses are divided into three families (alpha, beta, and gamma) based on tissue tropism and sequence similarity [1]. The amplification of virus within an infected cell or host is accomplished by productive, lytic infection where, upon entry into a susceptible cell (Table 1), a specific cascade of viral gene expression is activated, the genome is replicated to high levels, and infectious progeny virions are assembled and released. The lytic cascade of herpesvirus gene expression initiates with the synthesis of the immediate early (IE) genes. Early and late gene expression follows [1]. Provocatively, unlike other large DNA viruses, many herpesvirus IE genes are not controlled by promoters that are efficient and constitutively active within the context of the viral genome. Rather, viral transactivator proteins incorporated into the virion tegument and released into the cell upon infection play critical roles in the activation of viral IE gene expression [2–5]. Such a mechanism permits a far greater level of regulation than a simple constitutive promoter would allow.
The productive, lytic cycle is not the only possible outcome upon viral infection of an individual cell. In certain cell types (Table 1), herpesvirus infections establish the viral genomes in the nucleus but a productive round of replication is not completed in a timely manner. In such cells, a different, significantly smaller subset of viral genes is expressed. Importantly, because these infected cells maintain the potential to undergo productive replication at some later time after receiving the appropriate stimulus, this type of infection is described as latency. The resumption and completion of productive, lytic replication after a period of latency is called a reactivation event [1]. Both the restriction of substantial viral gene expression and the maintenance over time of the viral genome during latency allow herpesviral infections to persist for the life of the host even in the face of intense immune surveillance. Reactivation events allow for dissemination throughout and among hosts. While drugs that suppress lytic replication are available [6, 7], treatments for latently infected cells currently do not exist. As controlling or curing a herpesvirus infection would require modulation or elimination of the reservoir of latently infected cells, understanding the molecular mechanisms that govern latency is of utmost importance.
Latency is Cell Type Specific
Cell types that support latent infection with different herpesviruses are highly exclusive and non-overlapping. The alphaherpesviruses, Herpes Simplex Virus type 1 and 2 (HSV-1 and -2) and Varicella Zoster Virus (VZV) establish latency in neurons [8, 9]. The betaherpesviruses Human Cytomegalovirus (HCMV), and Human Herpesvirus -6 and -7 (HHV-6 and -7) establish latency in different subsets of hematopoietic cells; HCMV in hematopoietic stem cells [10–12], HHV-6 in bone marrow progenitor cells [13, 14] and HHV-7 in T-cells [14, 15]. The gammaherpesviruses Epstein Barr Virus (EBV) and Kaposi's Sarcoma Associated Herpesvirus (KSHV) establish latency in B cells [16–18]. Interestingly, most of these viruses have a broader tropism for lytic replication than latent infections, with many herpesviruses being limited to a single cell type in which latency can be established. The specificity for latent infections seems to indicate that each individual cell type contributes important factors that promote latency. This review presents our interpretations of the molecular mechanisms that control herpesvirus latency. For independent views, readers are directed to several recent literature surveys focusing on HSV-1 [19–23], HCMV [24–28], or EBV [29–33].
The systems used to study alpha-, beta-, and gammaherpesvirus latency vary dramatically. A valuable animal model for HSV-1 latency exists, but in vitro models [34–38] receive less attention. Thus the genetics of HSV-1 latency is established (i.e. which open reading frames are required for latency), but the molecular/cellular biology (i.e. how the products of the open reading frames actually promote latency) is less well understood. An in vitro model for HCMV latency is in its infancy. Despite its technical difficulties, it is amenable to both genetic and molecular analysis. Many cell lines exist to study the maintenance and reactivation of EBV latency, but the difficulty in making high titer virus stocks makes studying the establishment phase challenging. Animal models for HCMV and EBV latency do not exist, although certain aspects can be studied in immunodeficient humanized mice [39, 40]. The general lack of animal models for these viruses makes it difficult to determine the true physiologic relevance of in vitro studies.
One clear component of latency for each virus is the silencing of lytic phase gene expression. Interestingly, in the absence of tegument transactivators, most if not all viral IE promoters are poorly activated even in cell types where lytic infection occurs and that presumably have all the required cellular activating transcriptional factors for high-level lytic gene expression [2, 41]. Thus, the roles of tegument proteins and cellular transcriptional repressors in herpesvirus latency have recently received increased scrutiny. In the sections below, we review the current knowledge of how viral IE gene expression from incoming viral genomes is initially repressed in latently infected cells, and briefly address the maintenance of that repression. Finally, we examine the initial events that may resuscitate latent viral genomes (an event we term animation), and how they relate to full reactivation and infectious progeny virion production.
Establishing Latent HSV-1 Infections
Establishment of herpesviral latency can be operationally defined as the delivery of the viral genome to the nucleus without the initiation of a productive infection. HSV-1 establishes latency upon infection of a subset of neurons found in trigeminal ganglia, but productively replicates in other types of neurons found in these structures [42–48]. Productive replication within the trigeminal ganglia increases the number of latently infected neurons, but is not absolutely required for the establishment of latency [49–53]. Debate remains as to whether or not neurons destined to establish a latent infection may initially express lytic phase genes and then subsequently extinguish them. Inferred activity of an ectopic ICP0 promoter driving expression of the Cre recombinase and the subsequent Cre-mediated activation of a LacZ reporter from the cellular genome has been used to argue that no more than one-third of latently-infected neurons may have experienced ICP0 expression sometime during the infection [44]. However, the correct Cre-mediated recombination events were not confirmed in those experiments, and the proper IE regulation of the ectopic ICP0 promoter was not examined. Nevertheless, the ectopic ICP0 promoter apparently was never activated in the majority of latently infected neurons [44], indicated that in most (if not all) cells, the very first class of lytic genes, the IE genes, are not expressed upon neuronal infection where latency is established. To appreciate how HSV-1 IE genes may be silenced during the establishment of latency, we must understand how they are activated upon lytic infection.
HSV-1 IE gene expression is activated by a complex consisting of the viral tegument protein VP16, which is delivered to the cell upon entry, and two cellular proteins, host cell factor 1 (HCF) [54–56] and the POU homeodomain protein Oct-1 [57–59]. Tegument-delivered VP16 encounters HCF in the cytoplasm, and this association is absolutely required for VP16 translocation to the nucleus. Tegument-delivered VP16 remains in the cytoplasm of infected cells if binding to HCF is disrupted by mutation, or if the nuclear localization sequence (NLS) of HCF is deleted [60]. Under these circumstances, viral IE gene expression is inhibited. Once in the nucleus, the VP16/HCF pair interacts with Oct-1 associated with TAATGARAT motifs (where R is a purine) found in HSV-1 IE promoters [61]. Now tethered to viral genomes, the VP16/HCF/Oct-1 complex activates viral gene expression by recruiting cellular RNA Polymerase II and by modulating both histone occupancy and chromatin structure of the viral genome [62].
VP16 contains a prototypical acidic activation domain [63] that interacts with RNA Polymerase II, as well as several cellular components of the basal transcriptional machinery, including transcription factor IIB (TFIIB) and IIH (TFIIH), TATA-binding protein (TBP) and other transcription associated factors (TAFs) [56, 64–68]. Complex association with the TAATGARAT sequences orients the preinitiation complex facilitating transcriptional initiation. A specific VP16 mutant allele that lacks the acidic activation domain termed RP5 is unable to recruit the basal transcriptional machinery to viral IE promoters [62]. The RP5 virus exhibits a severe growth defect at low multiplicities of infection and is unable to replicate in immunocompetent mice [69].
In addition to recruiting RNA Polymerase II, the VP16/HCF/Oct-1 complex also controls histone occupancy, positioning, and modification at viral IE promoters [62, 70, 71]. Viral genomes packaged into virions and delivered to the nuclei of infected cells are devoid of cellular histone proteins [72, 73]. However, chromatinization occurs rapidly upon nuclear entry and is maintained throughout infection, although histone association with the viral genome is less prevalent and irregularly spaced compared to the cellular genome [21, 22, 62, 72, 74–77]. Histones wrap DNA into nucleosomes. The positioning of nucleosomes, as well as the modification of the resident histones, has profound effects on both transcriptional activation and repression.
The dynamics and importance of histone association with herpesviral genomes are just beginning to be appreciated. However, early work has made it clear that the VP16/HCF/Oct-1 complex modulates cellular histone association with viral IE promoters. The acidic activation domain of VP16 interacts with cellular ATP-dependent chromatin remodeling complexes that include the proteins BRG1 and hBRM, which are the mammalian counterparts of the yeast SWI/SNF complex components [78, 79]. This results in decreased histone occupancy at viral IE promoters. The RP5 mutant virus is unable to recruit these chromatin remodeling complexes and displays increased histone association with viral IE promoters [62]. HCF interaction with the histone chaperone Asf1b may also play a role in regulating viral genome chromatinization [71].
In addition to controlling the occupancy and positioning of cellular histones on viral genomes, the VP16/HCF/Oct-1 complex also regulates their post-translational modifications. VP16 interacts with the cellular histone acetyltransferases (HATs) CBP and p300, and increases the acetylation of histones associated with viral IE promoters [78, 80–83]. Specifically, markers of active euchromatin such as H3K9/K14 acetylation and H3K4 methylation are induced during lytic infection [62, 72, 76, 77]. Interestingly, neither p300 nor CBP activity appear to be required for HSV-1 IE gene expression [84], indicating that either other cellular HATs can compensate, or that histone occupancy is quantitatively more important for controlling viral IE gene expression than is histone modification. In summary, it is becoming increasingly clear that part of the mechanism through which VP16 activates HSV-1 IE gene expression is through reducing the overall level, ensuring the proper positioning, and facilitating the activation-linked modification of histones that become associated with viral promoters upon entry of the genome into the nucleus. Indeed, chromatinization of the viral genome can be a significant barrier to HSV-1 IE gene expression and the initiation of lytic infection that is overcome by the various functions of VP16.
As mentioned above, viral IE genes are likely not expressed at the start of an HSV-1 latent infection. Presumably, this must occur through either a loss of VP16-mediated enhancement, a dominant block to IE gene expression even in the presence of VP16 function, or a combination of both mechanisms. Analyzing the molecular details of the establishment of HSV-1 latency has been monumentally difficult because of the heavy reliance on animal model systems. Mouse and rabbit model systems provide quantitative readouts for various latency parameters, and are invaluable in the analysis of viral genetics in a holistic view of HSV-1 latency. However, in these systems it is either difficult or impossible to perform a detailed molecular analysis, or to divorce lytic replication from latency competence. Animal models require initial lytic infection at epithelial sites (for example, the eye) to seed ganglia with infectious virus that becomes amplified through more lytic replication prior to or concomitant with establishing latency in a subset of sensory neurons. Reactivations scored in animals after various stresses require viruses competent for productive, lytic infection. Alternatively, reactivation can be monitored in explanted neurons. Unfortunately, the explantation process significantly alters neurons, complicating the interpretation of results.
The literature pertaining to HSV-1 latency is enormous and often contradictory, and a full-scale review of it is not our intent here. Below we attempt to address and review a specific aspect of HSV-1 latency, the initial silencing of viral IE gene expression during the establishment of latency. Evolution has provided HSV-1 a mechanism to regulate viral IE gene expression by the use of promoters that are not constitutively active in their native locales, but require a trans-acting, tegument-delivered viral transcription factor (VP16) for their activation. Here we revisit a simple question that, although previously pondered [4, 60, 85, 86] deserves, in our opinion, increased scrutiny: is this potential regulation point used to modulate establishment of latency?
To answer this, one must determine if VP16 is or has the potential to be active in sensory neurons at a time when HSV-1 establishes latency. For tegument-delivered VP16 to activate viral gene expression it must pair with HCF [60, 87] in the cytoplasm and then translocate to the nucleus. HCF is expressed in neurons. However, unlike other cell types where the protein is mainly nuclear, in neurons, HCF is cytoplasmic [86], perhaps sequestered there through its association with proteins called Zhangfei [88, 89] and Luman [90, 91], and is often found in association with the Golgi apparatus [92]. This would seem to indicate that nuclear import of VP16 is most likely impaired in neurons. Cytoplasmic sequestration of VP16 may have a direct (and positive) effect on HSV-1 latency as the IE promoters of latent genomes are highly chromatinized, and the associated histones have heterochromatin-like marks, including the absence of acetylation and the presence of repressive modifications, such as H3K9 tri-methylation [93–95]. Such a configuration would favor the transcriptional repression observed and likely be required for the establishment of latency. In addition to the loss of VP16 function, low levels of Oct-1 [96] or differing ratios of Oct protein family members [97] may also decrease the likelihood of IE gene expression in neurons. Thus, it appears that VP16 function could be compromised during the establishment of latency (Figure 1A). Whether this alone, or other mechanisms in addition to or instead of impaired VP16 function, contribute to the absence of viral IE gene expression observed when HSV-1 establishes latency remains to be demonstrated experimentally.

Establishment of herpesvirus latency. A. Herpes Simplex Virus Type 1 (HSV-1). Infection of a sensory neuron allows for nuclear entry of viral DNA but not the tegument transactivator VP16. Viral immediate early (IE) genes are silenced. B. Human Cytomegalovirus (HCMV). Infection of a CD34+ hematopoietic progenitor cell allows for nuclear entry of viral DNA but not the tegument transactivator pp71. Viral IE genes are silenced by Daxx and an unidentified (?) trans-dominant, HDAC-independent mechanism. C. Epstein-Barr Virus (EBV). Infection of a memory B cell allows for nuclear entry of viral DNA. Tegument transactivators for EBV are uncharacterized. At least one IE gene (Z) and two early genes (BALF1 and BHRF1) are expressed. Z promotes B cell proliferation and BALF1/BHRF1 inhibit apoptosis, both of which appear to be required for the efficient establishment of latency. Z is unable to fully activate lytic phase gene expression because the viral genome is unmethylated.
Maintaining and Reactivating Latent HSV-1 Infections
Maintaining a latent infection requires that the viral genome be perpetuated and the cell kept alive. As neurons are non-dividing, genome replication and faithful partitioning to daughter cells upon division do not represent significant issues for HSV-1 latency. Perhaps to avoid cell death by apoptotic or immune measures, HSV-1 severely restricts viral gene expression during latency. The only major transcript detected is called the LAT for latency-associated transcript [98]. The LAT promoter has binding sites for neuronal-specific transcription factors [99, 100] and, unlike viral lytic phase promoters, is associated with histones exhibiting marks of active euchromatin during latency [101]. Although there are open reading frames encoded by LAT with the potential to encode proteins, such proteins have never been reproducibly detected [102]. Rather, LAT is thought to be processed into microRNAs (miRNAs), at least one of which has the potential to translationally silence mRNAs that encode the viral IE gene ICP0 [103–108]. Like other viral IE proteins, ICP0 can activate the expression of other viral genes and thus promote the lytic replication cycle [109–112]. ICP0 is found in virions but it is unclear if tegument-delivered ICP0 can activate gene expression, whereas de novo expressed ICP0 clearly does. Thus, preventing the synthesis of viral IE proteins appears to be a critical part of maintaining the latent HSV-1 genome.
The presence of LAT can also affect the chromatin structure of viral IE promoters. In the absence of LAT, the normally heterochromatic structure of the IE promoters during latency takes on features more reminiscent of active, euchromatin [94]. It is currently unknown if this is a direct effect of the LAT transcript similar to the way non-coding RNAs can affect chromatic structure [113], or an indirect effect of the ability of LAT to suppress the expression of ICP0, which itself can promote euchromatin structures on viral genomes [114–116]. LAT has also been proposed to have an anti-apoptotic effect [117], but the mechanism through which this might be achieved is unknown. Interestingly, while LAT-null viruses have latency defects, they still establish, maintain, and reactivate latency to a substantial degree [47, 118, 119]. This may indicate the presence of one or more non-LAT measures of latency maintenance. Indeed, a miRNA not encoded by LAT that has the potential to translationally silence the mRNA for the viral IE protein ICP4 has also been detected in latently infected cells [103]. Thus it would appear that a major strategy to maintain HSV-1 latency is simply to curtail inappropriate reactivation events by preventing the translation of any spurious IE messages that might be generated. However, direct repression of viral lytic gene expression and/or inhibition of apoptosis may also occur.
Other control measures in addition to LAT (Figure 2A) also help maintain HSV-1 in a latent state. Recent experiments with an in vitro model system of primary rat superior cervical ganglia neurons indicate that a signaling cascade starting with nerve growth factor and proceeding through PI3K and Akt is essential to prevent lytic reactivation events [34]. The molecular mechanism through which this pathway suppresses reactivation has not been described. However, the in vitro model system utilized appears much more amenable to molecular studies than existing animal models, so the prospects for a more detailed dissection of how this pathway maintains the viral genome in a latent state are appealing.

Maintenance of herpesvirus latency. A. HSV-1. Viral latency associated transcripts (LAT) encode micro RNAs (miRNA) that suppress the expression of viral IE proteins. Nerve growth factor (NGF) induced signaling also helps maintain latency in vitro. Non-dividing neurons do not require a mechanism to replicate or faithfully partition viral genomes. B. HCMV. The contributions of viral mRNAs/transcripts detected during latency (CTLs, LUNA, UL138, US28, vIL10) to the establishment, maintenance, animation, or reactivation from latency have not been fully characterized. Whether or not latently infected progenitor cells divide or self-renew (arrow with question mark) is not known, thus the need for (or presence of) replication or partition functions is also unclear. C. EBV. The viral EBNA1 protein provides replication and partition functions required to maintain latency in dividing B cells. Different types of EBV latency also express other latent genes whose functions appear to be proliferation induction, apoptosis inhibition and immune evasion. EBER and BART transcripts are also expressed during latency. EBERs inhibit protein kinase R (PKR) to maintain translational proficiency, and BARTs are processed into miRNAs. Viral genome methylation prohibits lytic phase gene expression.
Reactivation mechanisms of latent HSV-1 infections have been notoriously controversial. Even the stage of infection at which the reactivation event initiates (IE gene expression or viral DNA replication) has been debated [120–124]. Because the latent genome is without an accompanying tegument, it has always been assumed that reactivation of a lytic infection from latency must be fundamentally different from the de novo initiation of the lytic replication program upon infection of a susceptible cell type. Complex animal experiments with complicated interpretations have been used to explore HSV-1 reactivation. Most or all of these depend on the ability of mutant viruses to reactivate from latency and complete productive replication, often in explant cultures from HSV-1 infected animals.
ICP0 is the protein most often described as an inducer of reactivation. ICP0 mutant viruses clearly fail to reactivate from animal models of HSV-1 latency [125, 126]. However, they also have severe defects during de novo lytic infections [127, 128]. Thus it is unclear if the latency phenotype of ICP0 mutants is due to defects in the initiation of lytic phase gene expression from a latent viral genome, or from the failure to complete the lytic replication cycle after it has been efficiently started (or both). Furthermore, differing readouts of reactivation (explant vs. in vivo) give different results as to the requirement for ICP0 during reactivation [125, 126, 129]. Recently, VP16 has been proposed to be an inducer of reactivation [130], even though earlier work indicated that a VP16 mutant with a non-functional acidic activation domain (in1814) can establish, maintain, and reactivate from latency [131, 132] despite being impaired in its ability to initiate a de novo lytic infection [41]. Readers are directed to numerous recent reviews with a more detailed and complete examination of mechanisms controlling HSV-1 latency maintenance and reactivation [19, 21–23].
Animating Latent HSV-1 Infections
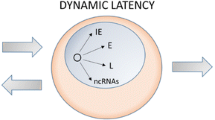
Two significant obstacles hinder our understanding of HSV-1 latency. The first is the underutilization of suitable in vitro systems [34–38] relative to the heavy reliance upon animal models, valuable as they are, for all levels of latency experimentation. The recent resurrection of a tractable cell culture model for HSV-1 latency [34] should catalyze additional molecular studies. The second is the marriage of the initial events that awaken latent genomes with the completion of productive viral replication. This is necessitated by the term reactivation, which requires productive replication as an endpoint, and therefore mixes both inciting and propagating events under the same moniker. Based on the concept that initiating reactivation and completing reactivation represent two separate events [129, 133], we propose the word animation to describe the very first lytic phase event that occurs from latent viral genomes. This theoretical separation has practical application, as it allows one to divorce the completion of productive lytic replication in a previously latent cell from its initiation.
The verb animate means to give life or motion to, thus we define herpesvirus animation as execution of the event that ends latency and initiates the productive replication cycle via reactivation. Unlike designations such as initiation or reactivation that refer to productive infections, animation does not require the eventual generation of infectious virus particles. Applied to latency, animation as a term provides a clear and defined separation between the commencement and completion of a reactivation event. This nomenclature provides concise specific terminology to an important, circumscribed event currently masked by its inclusion with the many subsequent occurrences of reactivation. It allows us to ask what is the animating event in HSV-1 reactivation, and what controls it? Treatment of latently infected mice with acyclovir (an inhibitor of viral DNA replication) concomitant with a reactivation stimulus (heat stress) inhibited late gene expression and reactivation, but did not prevent IE protein production [134]. Thus while viral DNA replication is required for reactivation, it is apparently not required for animation. In similar experiments it was demonstrated that ICP0 mutant viruses could produce IE proteins after heat stress, but did not productively replicate [129]. Thus, while ICP0 is clearly required for reactivation, it apparently is not required for animation.
If the two main protagonists in the reactivation debate (ICP0 and DNA replication) are not responsible for genome animation, than what is? Could VP16, which activates the genome upon de novo lytic infection, also be responsible for animating latent HSV-1 genomes? A caveat to this hypothesis is that a specific VP16 mutant virus (in1814) with a 12 bp insertion in the acidic activation domain can efficiently reactivate from latency in explant assays [131, 132]. However, this same mutant fails to reactivate in heat stressed mice [130]. As the in vivo assay would presumably be more physiologically relevant, it appears that VP16 is required for reactivation, and thus may be required for animation. In a direct test of this model it was observed that viral IE proteins were expressed after heat stress in animals latently infected with an ICP0 mutant virus [129], but not with VP16 mutant viruses [130]. In addition, two different reporter viruses detected VP16 promoter activity after the reactivation stimulus, but prior to IE protein production [130]. While VP16 is expressed as a late gene during de novo lytic infection, it apparently is expressed prior to the classically defined viral IE genes in heat stressed mice. Thus, the de novo expression of the VP16 protein appears to be the animating event that initiates the reactivation of a productive infection from latency (Figure 3A). It is unclear what changes the stressed cell may undergo that facilitate VP16 expression during latency animation. However, at least some of those changes appear to specifically modulate the activity of the VP16 promoter, as the late promoter that drives expression of the gene encoding the major capsid protein (UL19; VP5) could substitute for the VP16 promoter during lytic infection in vitro and during acute ocular infections of mice, but was severely impaired for replication in mouse trigeminal ganglia [130]. Interestingly, this implies that a significant amount of viral replication in the ganglia occurs not by de novo lytic infection but by reactivation of (short duration) latent infections, even during the acute phase. In addition, it is likely that cellular stresses also lead to HCF subcellular relocalization to allow the newly expressed VP16 to enter the nucleus and activate viral IE gene expression. Therefore, animation of the HSV-1 genome may be nearly identical for de novo lytic infections and for latency reactivations, with the only difference being the source of VP16 (tegument delivered or newly synthesized).

Animation and reactivation of herpesvirus latency. A. HSV-1. Stress signals (lightning bolt) induce the de novo expression of VP16 and its recruitment into the nucleus (likely via HCF) to activate viral IE gene expression. A productive reactivation event follows. B. HCMV. Signals (lightning bolt) induce CD34+ cell differentiation into a dendritic cell, inducing animation and subsequent reactivation through unknown molecular mechanisms. C. EBV. Activation by antigen stimulation of the B cell receptor (BCR) induces B cell differentiation into a plasma cell, inducing animation and subsequent reactivation through unknown molecular mechanisms.
Establishing Latent HCMV Infections
Numerous viral genes have been reported to be expressed upon in vitro infection of CD34+ hematopoietic progenitor cells with HCMV [135–139]. This heterogeneous population of cells represents the most widely accepted and utilized model for experimental HCMV latency. It is clear that the viral IE genes are not among the latently expressed transcripts. Thus it appears that silencing viral IE gene expression occurs as HCMV establishes latency. To appreciate how this occurs, we must understand how IE genes are activated during lytic infection.
Expression of the main viral IE proteins, IE1 and IE2, is controlled by the Major Immediate Early Promoter, or MIEP. Often referred to as "the CMV promoter", the MIEP is constitutively active when found in heterologous constructs such as plasmids, but is surprisingly dependent on viral tegument transactivators for activation in the context of the viral genome [2, 140–143]. The major tegument transactivator is pp71 [144]. Upon infection of a cell type permissive for lytic replication, pp71 travels to the nucleus and activates the MIEP by inactivating a cellular intrinsic immune defense that would otherwise silence HCMV gene expression [145]. This defense is mediated by cellular transcriptional co-repressors that localize to promyelocytic leukemia nuclear bodies (PML-NBs) [146–153]. Recombinant viruses with pp71-null or Daxx-binding mutations have severe growth defects and fail to effectively initiate IE gene expression [2, 154]. The hallmark event of this cell defense neutralization is the degradation of Daxx [145]. The mechanism through which pp71 induces Daxx degradation is not understood, although proteasome activity and the ability of pp71 to interact with Daxx is required. Interestingly, all evidence points to an ubiquitin-independent route to the proteasome [155, 156].
The Daxx-mediated defense silences viral IE gene expression by fostering a transcriptionally repressive chromatin structure on the HCMV genome [157]. Virion-packaged DNA lacks histones [158] but is rapidly chromatinized upon entry into the nucleus [159, 160]. Prior to, or in the absence of pp71 function, the histones that become associated with the HCMV genome bear transcriptionally repressive post-translational modifications, such as the absence of acetylation and the presence of H3K9 dimethylation [161–164]. Association with the repressive HP-1 protein is also observed. Knockdown of Daxx by siRNA reduces heterochromatic markings associated with the HCMV genome [157]. When pp71 is present and active, or when Daxx levels are decreased by RNA interference approaches, histones associated with the viral genome are acetylated, a mark of transcriptionally active euchromatin [161–166]. As Daxx associates with histone deacetylases (HDACs), the widely accepted model is that the pp71-mediated degradation of Daxx prevents HDAC association with the HCMV genome and thus facilitates euchromatin formation and transcriptional activity. pp71 also displaces ATRX from Daxx [167], thus further activating IE gene expression. ATRX has homology to the SWI/SNF family of chromatin remodeling proteins [168, 169], but it is currently unclear if this protein alters the occupancy or placement of histones at the MIEP.
As mentioned above, IE genes are not expressed at the start of HCMV latency. Presumably, this must occur through a loss of pp71-mediated de-repression, a dominant block to IE gene expression, or a combination of both mechanisms. In the CD34+ cell populations, it appears both mechanisms are used (Figure 1B). pp71 is prevented from degrading Daxx and de-repressing viral gene expression because it fails to localize to the nucleus in infected CD34+ cells [153]. Knockdown of Daxx in these cells, or treatment with the HDAC inhibitor valproic acid rescues IE gene expression upon infection with the AD169 laboratory-adapted strain of HCMV [153]. Thus, the Daxx-mediated intrinsic defense contributes to the silencing of the MIEP that occurs when latency is established. Essentially identical results [152] were obtained in two other cell culture models for quiescent HCMV infections that appear to faithfully mimic most aspects of latency. Differentiation fails to induce reactivation from these quiescently infected cells, though more recent reports suggest treatment with vasoactive intestinal peptide (VIP), an immunomodulatory neuropeptide [170], or phorbol 12-myristate 13-acetate [171], can induce low levels of reactivation.
As the same intrinsic defense that represses IE1 expression during lytic infection prior to pp71 function also silences expression during latency [145, 153], it is not surprising that indistinguishable chromatin is assembled on this transcriptionally inert viral locus under these two different conditions. Latent genomes either from in vitro infected CD34+ cells or from natural latent infections in vivo display unacetylated and H3K9 dimethylated histones and are associated with the HP-1 transcriptional repressor [162, 163], similar to what is found during lytic infection in the absence of pp71.
Thus, analogous to HSV-1, sequestration of the virion tegument transactivator protein in the cytoplasm appears to be at least one way that viral genes are silenced at the start of latency. While it is likely that the cytoplasmic localization of HCF prevents VP16 nuclear localization in HSV-1 infected neurons, it is unclear what restricts pp71 nuclear entry during HCMV infection of CD34+ cells. Interestingly, de novo expressed pp71 in CD34+ cells localizes to the nucleus [153], so it does not appear that pp71 trafficking is controlled in the same manner as VP16. pp71 cytoplasmic localization is more likely due to a defect in tegument disassembly than to a specific effect on pp71 trafficking because at least one other tegument protein, pp65, is also sequestered in the cytoplasm in undifferentiated NT2 cells quiescently infected with HCMV [172]. pp65 localization upon latent infection of CD34+ cells has not been analyzed. A mechanism for this hypothesized defect in tegument disassembly has not been offered, although it appears that this is not a dominant block imposed by undifferentiated cells, but a recessive trait. Heterologous fusions of undifferentiated and differentiated cells permit tegument-delivered pp71 nuclear localization, leading to the conclusion that differentiated cells possess a dominantly acting factor that drives tegument-delivered pp71 nuclear localization [172]. This fits well with the observed sub-cellular localization of pp71 and IE gene expression competency for matched pairs of undifferentiated and differentiated cells [152].
Interestingly, pp71 cytoplasmic sequestration is not the only mechanism to restrict IE gene expression during establishment of HCMV latency in CD34+ cells. While IE gene expression from the AD169 genome could be rescued by HDAC inhibition, this was not the case during infection with the FIX or TB40/E clinical strains of HCMV [153]. Clinical strains of HCMV have undergone significantly fewer passages in vitro and retain a large section of the viral genome (called the ULb' region) that is absent in laboratory-adapted strains such as AD169 [173–175]. In mixed infections between AD169 and a clinical strain, the clinical-strain imposed restriction of viral IE gene expression in the presence of HDAC inhibition was dominant [153]. Whether this clinical strain encoded, trans-dominant HDAC independent restriction of IE gene expression occurs in a cell autonomous or non-cell autonomous manner, and the viral gene responsible for this restriction, is unknown. Also, whether or not this clinical strain function is newly expressed upon viral infection or is a component of the infecting virion has not been determined. The fact that viral DNA damaged by ultraviolet (UV) light, which is unable to support transcription, fails to be maintained in latently infected cultures over time has been used to conclude that viral gene expression is required for the establishment of HCMV latency [136]. While this may be true, viral DNA clearance under these experimental conditions may (also or instead) be the result of the UV-induced damage itself, and not the lack of gene expression. Thus it is currently unclear if the establishment of HCMV latency requires viral latent gene expression or simply the silencing of viral lytic gene expression. Either way, HCMV uses at least two mechanisms to silence viral IE gene expression during the establishment of latency, pp71 cytoplasmic sequestration and an unidentified HDAC-independent block [153].
Maintaining and Reactivating HCMV Latent Infections
How HCMV maintains latency is poorly understood (Figure 2B). Unlike other betaherpesviruses that have shown the ability to insert their genomes into cellular chromosomes [176–178], the available evidence indicates that latent HCMV genomes are episomal, circular molecules [179]. It is also unclear whether HCMV genomes are replicated during latency. The half-life and dividing potential, in vivo, of latently infected CD34+ cells is not known. If latently infected cells fail to divide but have long half-lives, this would mimic the situation with HSV-1 latent infection of neurons. If cells latently infected with HCMV do divide, it would more resemble the situation with EBV, and would require replication and likely partitioning functions for the viral genome during latency. Alternatively, the latent reservoir may be a short-lived non-dividing cell, which would require that HCMV continually re-seed the latent reservoir to achieve life-long persistence. Clearly, these are important questions that to date have not been sufficiently addressed.
While over 80 genes are reported to be expressed in in vitro latently infected cells [136, 139], only a few are reportedly expressed during natural latent infections in vivo. These are the CLTs, US28, vIL10, LUNA, and UL138 loci. Contributions that these loci may (or may not) make to HCMV latency has been recently reviewed [26], so they will only be briefly mentioned here. The CTLs (CMV latency transcripts) represent sense and antisense RNAs from the MIE locus [180, 181]. While there is potential for CTLs to act in an antisense or interfering way with viral IE gene expression, no such activity has been demonstrated. Antibodies to proteins hypothetically encoded by the CTLs have been detected in HCMV seropositive patients [182], implying that they might be translated. However, deleting the prominent open reading frame of these transcripts (UL94) did not impair in vitro latency [183]. US28 is a chemokine receptor [184, 185] whose role during latency has not been studied. vIL10 is a cytokine that may protect latently infected cells against host immune surveillance [137, 186, 187]. A transcript antisense to the UL81-82 region of the genome encodes an open reading frame termed LUNA for latency unidentified nuclear antigen [138, 188]. While LUNA may indeed be a functional protein, this transcript has the potential to modulate de novo pp71 expression in an antisense or interfering manner (pp71 is the product of the UL82 gene). Analyses examining the requirement or role for LUNA during latency have not been reported. UL138 encodes a protein that localizes to the Golgi apparatus during lytic infection and is required for the maintenance of latency in some [189], but not other, in vitro model systems [187]. A mechanism for how UL138 may regulate latency has not been proposed. A specific HCMV-encoded microRNA, miR-UL112-1, has been proposed as a potential way that viral IE gene expression may be downregulated during latency [190, 191], although the expression of this or any other HCMV microRNA during latency has not been reported.
Reactivation of latent HCMV genomes is known to depend upon cell differentiation [162, 192–195], although the detailed molecular mechanisms behind this event are not understood. What is known is that latent viral genomes lose their heterochromatic marks and obtain marks of active euchromatin upon the differentiation of CD34+ cells into dendritic cells [162, 163]. This seems to indicate that the cellular intrinsic defense that helps establish latency is inactivated during the process of reactivation.
Animating Latent HCMV Infections
A significant obstacle to our understanding of HCMV latency is the technical difficulties inherent in using CD34+ cells as an in vitro model. These cells are heterologous in nature, are difficult or impossible to maintain in an undifferentiated state for even short periods of time, and infect inefficiently, even with clinical strain viruses. Models for how HCMV may animate from latency have included the de novo expression of cellular transcription factors specific for the MIEP upon differentiation, or the de novo expression of pp71. Both models have their deficiencies. The transactivator model cannot explain how the Daxx-mediated intrinsic defense would be overcome. In this sense it is analogous to a pp71-null virus infection of a differentiated fibroblast, where MIEP activating transcription factors are present but viral IE gene expression is still poor [2]. The de novo expression of pp71 prior to IE gene expression upon the differentiation of latently infected CD34+ cells was examined but not detected [188], indicating either that it does not occur, or was below the limit of detection of the assay used. How differentiation triggers HCMV genome animation is still unclear (Figure 3B).
Establishing Latent EBV Infections
Studies of EBV replication are significantly different from those of HSV-1 and HCMV. Latent EBV infection of B cells transforms them [196] and this represents a cancer burden in the human population [17, 30]. Thus, most work on EBV concentrates on the mechanisms through which EBV transforms cells. Consequently, molecular mechanisms that control the initiation of a lytic infection upon primary infection of a cell permissive for productive infection, or the establishment of latency upon the primary infection of a B cell, are poorly understood. For HSV-1 and HCMV, it is clear that infection of cell types fully permissive for productive infection substantially amplifies the amount of virus present. This amplification by de novo lytic infection may be important for the establishment of latency because of the accessibility of cell types that support latency. Though a similar process most likely occurs during EBV infection [197], it is currently unclear if such an initial amplification by lytic infection in fully permissive cell types such as epithelial cells is required for the efficient establishment of EBV latency [198]. It is possible that the initial infectious event is a latent infection of a circulating B cell, and that reactivation of that infection could be the source of amplified virus. Thus, it is unclear how much insight into the pathology of the virus is provided by the study of initial events occurring during primary infection of epithelial cells. Such experiments are challenging to perform because infectious stocks of EBV virions are difficult to make and transfer of virus to epithelial cells, either by free virions or by B cell associated virions is inefficient [199–201]. Furthermore, the differentiation state of the epithelial cell appears to impact the outcome of an acute infection [201]. Recent work has determined that an abortive infection of primary epithelial cells resulted in gene expression patterns that were different from infection of epithelial cell lines and primary B cells [200]. However, the gene expression pattern of a de novo initiated, productive lytic infection has not been determined, and thus the molecular mechanisms that would account for this gene expression pattern are unknown. Critically, it is not known which or even if tegument proteins activate viral gene expression under these circumstances.
Significantly more is known about the establishment of latency upon primary infection of a B cell (Figure 1C), although the molecular details have generally not been deciphered. Viral IE gene expression is not silenced upon infection of cells destined for latency. At least three lytic phase genes are expressed upon latent infection of primary B cells and are apparently required in order to efficiently establish latency. These are the Bcl-2 homologs BALF1 and BHRF1 [202] and the AP-1 homolog BZLF1 [203, 204]. BHRF1 is also expressed during some types of latency [205]. Other reports of more substantial lytic gene expression during the establishment of latency likely represent productive replication in a subset of fully permissive cells found in the peripheral blood mononuclear cells (PBMCs) used for those experiments [206]. Cellular AP-1 is a DNA binding transcription factor, and so is the Z protein (also called Zta, ZEBRA and EB1) encoded by BZLF1 [207]. Z expression reportedly drives the proliferation of quiescent naïve and memory B cells upon infection, and this significantly increases the efficiency with which EBV establishes latency [203]. However, Z-null viruses can still establish latent infections in vitro [39, 208], so this step appears not to be absolutely required. The unscheduled proliferation induced by Z may be pro-apoptotic, and this may necessitate the expression of BALF1 and BHRF1 proteins. Like their cellular counterpart Bcl-2 [209], these proteins have anti-apoptotic effects [210, 211]. Deletion of both BALF1 and BHRF1 inhibited the ability of EBV to latently infect and induce the transformation of primary B cells [203]. The BHRF1 locus also encodes four miRNAs that may enhance the establishment of latency by promoting cellular proliferation and survival [212, 213].
The role of Z during the establishment of latency is clearly different from its role during reactivation, when it drives viral early and late gene expression and activates the lytic origin of DNA replication, leading to infectious virion production [29, 214]. Early and late lytic phase genes are not expressed during the establishment of latency even though Z is [203, 204]. The differential effects of Z on viral gene expression during the establishment and reactivation phases of latency can be explained by the methylation status of the viral genome [215–217]. In the virion, and thus initially upon de novo infection, the viral genome is unmethylated. After latency is established, the EBV genome becomes extensively methylated [203]. Though DNA methylation is typically a cellular mark of transcriptional inactivity, EBV has evolved a clever way to overcome methylation-mediated silencing by the cell. The Z protein is able to bind to methylated DNA more strongly than unmethylated DNA [216, 217], and Z activates transcription from methylated promoters significantly more than from unmethylated promoters [203, 215]. Thus, while Z is expressed during the establishment of latency, the viral genome is unmethylated and so Z cannot activate the expression of early and late viral genes, and therefore under these circumstances, does not induce the lytic phase. Interestingly, another EBV IE gene product, the R protein encoded by the BRLF1 gene, is a transcription factor that preferentially activates unmethylated promoters [216]. There is speculation that R may be more important than Z in initiating de novo lytic infection (Shannon Kenney, personal communication). Thus EBV may encode two unique IE proteins with independent activities, one to promote de novo lytic infection (R), and one (Z) to promote the establishment and reactivation (see below) of latency.
It is unclear if the expression of Z, BALF1 and BHRF1 at the start of latency requires a tegument protein in a similar manner to the lytic phase IE genes of HSV-1 and HCMV. Transfected HSV-1 or HCMV DNA is capable of initiating a lytic infection, but co-transfection with an expression plasmid for VP16 or pp71 (respectively) increases the efficiency of this process by at least 10-fold, mimicking the effects of the tegument-delivered protein upon de novo infection [218, 219]. Interestingly, a virus deficient in the EBV tegument protein BNRF1 can enter cells but fails to efficiently establish latency [220]. BNRF1 has recently been shown to bind to Daxx, disrupt its association with ATRX, and stimulate viral gene expression from a co-transfected EBV bacterial artificial chromosome (BAC) construct (Paul Lieberman, personal communication). Thus, EBV tegument proteins may promote the expression of genes required for the establishment of viral latency in a homologous fashion to the manner in which HCMV pp71 promotes the expression of viral genes that initiate lytic infection.
Maintaining and Reactivating Latent EBV Infections
Latently infected B cells can be generated in vitro or isolated from infected patients. EBV transforms and immortalizes latently infected B cells, creating lymphoblastoid cell lines, or LCLs [17, 30, 196]. These cells divide, and thus EBV must ensure both the replication and the faithful partitioning of its genome during the maintenance of the latent state [221–225]. An enormous amount of literature exists concerning how EBV transforms B cells and maintains latency. As this has been extensively reviewed in several recent manuscripts [29–31, 196, 226–228], we only briefly describe the general mechanistic concepts here.
In latently infected B cells (Figure 2C) up to nine virally encoded proteins are expressed, these include the EBV nuclear antigens (EBNA-1, -2, -3A, -3B, -3C and -LP) and the latent membrane proteins (LMP-1, -2A and -2B). Different types of latency (e.g. type I, type II, or type III) display different sets of virally expressed genes [32, 227, 229, 230]. EBNA-1 is expressed in all types of latency and plays a central role in maintenance of the viral genome as it is responsible for initiation of episomal DNA replication and segregation during cell division [221, 229, 231, 232]. The other viral proteins contribute to the transformation and immortalization of the infected B cells [226, 229, 230]. In addition to these viral proteins, non-coding viral RNAs are detected in all latently infected cells, including the EBERs (EBV-encoded RNAs) and the BARTs (BamHI-A rightward transcripts). EBERs inhibit PKR-mediated apoptosis and induce expression of the cellular chemokines IL-6 and IL-10, which promote B cell growth [213, 233, 234]. BART transcripts are processed into microRNAs (miRNA) that modulate LMP-1 expression [235] and the ability of infected B cells to avoid immune detection and clearance [236].
Latent viral gene expression is regulated by differential promoter utilization and is controlled by epigenetic marks to both DNA-bound histones as well as the DNA itself [33, 237]. In general, loci that are active during latency display unmethylated DNA and acetylated histones, whereas repressed loci display methylated DNA and H3K9 trimethylated histones [238–240]. The molecular mechanisms that lead to these epigenetic marks have not been described. The binding of the chromatin insulator CTCF protein has also recently been shown to modulate viral gene expression during latency [241, 242]. As mentioned earlier, lytic viral gene expression during latency is suppressed by genome methylation. In addition, expression of the Z protein is specifically inhibited by the cellular transcription factor Zeb1 [243, 244].
Reactivation of natural EBV infections (Figure 3C) occurs when infected memory B cells differentiate into plasma cells in response to antigen stimulation [29, 214, 229]. This can be mimicked in vitro by cross-linking of the B cell receptor by treatment with an anti-immunoglobulin antibody [245, 246]. The activation of cellular transcription factors BLIMP1 and XBP-1 upon differentiation likely plays a role in facilitating viral lytic phase gene expression [247–249]. Reactivation is also commonly initiated in vitro by the transfection of an expression plasmid for the Z protein or by treating cells with a combination of the phorbol ester TPA and the HDAC inhibitor sodium butyrate [245, 250, 251]. Z protein function is absolutely required for reactivation, as a mutant Z protein, Z(S186A), fails to induce reactivation [252, 253]. Unlike during the establishment of EBV latency, the viral genome at the time of a reactivation event is methylated, and thus the newly expressed Z is able to efficiently activate the expression of the viral early and late genes and thereby promote the productive, lytic replication program.
Animating Latent EBV Infections
Z expression is necessary and sufficient for the reactivation of latent EBV infections [214, 254, 255]. Artificial downregulation of the cellular Zeb proteins that silence Z during latency induces Z expression and reactivation [243, 244]. Furthermore, microarray data mining indicates that Zeb mRNA levels decrease quickly and precipitously after antigen-mediated differentiation of B cells into plasma cells (Janet Mertz, personal communication). Thus, it is distinctly possible that induction of Z expression is the animating event during EBV reactivation, and that this may in part occur by the disappearance of cellular repressor proteins that silence Z expression during latency. However, recent evidence indicates that de novo gene expression is required in order to induce Z expression upon B cell differentiation [256]. Production of Z transcripts following cross-linking of the B cell receptor was prevented by protein synthesis inhibitors, leading authors of that study speculated that a newly synthesized cellular protein was responsible for turning on Z expression. However, in analogy to HSV-1 (and perhaps HCMV) animation, it is distinctly possible that the de novo expression of a tegument protein, perhaps BNRF1, is the actual event prevented by the protein synthesis inhibitors that impair EBV reactivation. It would be interesting to see if the BNRF1-null virus fails to reactivate from latently infected B cells upon receptor crosslinking, and if the Zeb proteins also modulate BNRF1 expression. If this were true, animation would likely be the only step during reactivation that requires BNRF1, because the null virus was proficient for reactivation from 293 cells upon ectopic Z expression [220].
Conclusions
Establishment of latency (Figure 1) for HSV-1 and HCMV appears to be quite similar. Viral IE genes are not expressed because the tegument transactivators required for that event are restricted from entering the nucleus. Other viral gene expression may not be required for the establishment of latency. The major difference is that the silencing of IE gene expression for HSV-1 appears to result from a lack of promoter activation, whereas for HCMV it results from both active promoter repression by cellular factors such as Daxx and HDACs as well as by an unidentified trans-dominant, HDAC-independent mechanism [153]. The establishment of EBV latency is significantly different. It requires viral gene expression, including lytic phase genes of the IE and early classes, to promote cellular proliferation and prevent apoptosis [202–204]. It is presently unclear if or how viral tegument proteins activate this gene expression.
Maintenance of latency (Figure 2) is significantly different for each virus, although assembling a repressive chromatin architecture on the promoters of lytic phase genes appears to be a common control mechanism [20–22, 27, 33, 257]. HSV-1 remains latent in a non-dividing cell and thus does not need to replicate or faithfully partition its genome. Significant control measures during latency appear to be miRNA mediated silencing of any spurious IE gene expression that may occur [103, 108], and NGF-mediated Akt phosphorylation that inhibits reactivation by an unknown mechanism [34]. Inhibition of apoptosis may also be important, as is the quelling of reactivation events in a non-cytolytic manner by interferon gamma and granzyme B mediated degradation of ICP4 [258]. HCMV appears to express at least some proteins during the maintenance of latency. Thus, not surprisingly, at least one (vIL10) appears to limit immune detection and clearance of latently infected cells [186, 187]. Roles for other viral proteins expressed during latency are not known. Likewise, it is unclear if latently infected cells divide, and thus it is also unknown if mechanisms for genome replication or partitioning exist or are required. miRNA mediated silencing of IE gene expression during latency has been proposed but not demonstrated. EBV expresses multiple genes during latency, many of which ensure cell survival and proliferation. In addition, EBNA1 promotes replication of the viral genome and equal partitioning to daughter cells during cell division. Most lytic phase genes are kept silent by DNA methylation at elements within their promoter regions.
Animation of latency (Figure 3) has been most extensively characterized for HSV-1, where de novo expression of the tegument transactivator VP16 appears to be the initiating step of the reactivation process [130]. EBV also requires de novo protein expression prior to synthesis of its IE gene encoding the Z protein during latency animation [256], but whether the required protein(s) is viral or cellular (or both) is not known. Thus, whether or not EBV and/or HCMV genomes are animated by de novo expression of tegument transactivator remains to be determined. Interestingly, recent experiments indicate that the HCMV tegument transactivator pp71 is a target of granzyme M mediated cleavage [259]. Thus it is possible that if de novo expression of pp71 is an animating event for HCMV, granzyme mediated protein cleavage may help extinguish HCMV reactivation events as it appears to do for HSV-1. Reactivation for all herpesviruses likely begins at or prior to IE gene expression, but then continues with a similar kinetic cascade of gene expression that is observed during de novo lytic infections. Interestingly, HSV-1, HCMV, and EBV each encode IE proteins (ICP0, IE1, and Z, respectively) that disrupt PML-NBs, nuclear structures that suppress the lytic replication of DNA tumor viruses [260–263], indicating that proteins that localize to these structures may also play significant roles during the establishment and/or maintenance of latency [147]. Thus, the molecular mechanisms of animation and reactivation for the individual herpesviruses, although initiated by different stimuli, may be more conserved than currently appreciated.
References
Pellet P, Roizman B: Herpesviridae: A Brief Introduction. Fields Virology. Edited by: Howley P. 2007, Philadelphia: Lippincott, 2480-2499. 5
Bresnahan WA, Shenk TE: UL82 virion protein activates expression of immediate early viral genes in human cytomegalovirus-infected cells. Proc Natl Acad Sci USA. 2000, 97: 14506-14511. 10.1073/pnas.97.26.14506.
Moriuchi H, Moriuchi M, Straus SE, Cohen JI: Varicella-zoster virus open reading frame 10 protein, the herpes simplex virus VP16 homolog, transactivates herpesvirus immediate-early gene promoters. J Virol. 1993, 67: 2739-2746.
Wysocka J, Herr W: The herpes simplex virus VP16-induced complex: the makings of a regulatory switch. Trends Biochem Sci. 2003, 28: 294-304. 10.1016/S0968-0004(03)00088-4.
Nicholson IP, Sutherland JS, Chaudry TN, Blewett EL, Barry PA, Nicholl MJ, Preston CM: Properties of virion transactivator proteins encoded by primate cytomegaloviruses. Virol J. 2009, 6: 65-10.1186/1743-422X-6-65.
Schreiber A, Harter G, Schubert A, Bunjes D, Mertens T, Michel D: Antiviral treatment of cytomegalovirus infection and resistant strains. Expert Opin Pharmacother. 2009, 10: 191-209. 10.1517/14656560802678138.
Billaud G, Thouvenot D, Morfin F: Drug targets in herpes simplex and Epstein Barr Virus infections. Infect Disord Drug Targets. 2009, 9: 117-125.
Roizman B, Knipe D, Whitley R: Herpes Simplex Viruses. Fields Virology. Edited by: Howley P. 2007, Philadelphia: Lippincott, 2501-2601. 5
Cohen J, Straus S, Arvin A: Varicella-Zoster Virus Replication, Pathogenesis, and Management. Fields Virology. Edited by: Howley P. 2007, Philadelphia: Lippincott, 2773-2818. 5
Sinclair J: Human cytomegalovirus: Latency and reactivation in the myeloid lineage. J Clin Virol. 2008, 41: 180-185. 10.1016/j.jcv.2007.11.014.
Mendelson M, Monard S, Sissons P, Sinclair J: Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J Gen Virol. 1996, 77 (Pt 12): 3099-3102. 10.1099/0022-1317-77-12-3099.
Mocarski E, Shenk T, Pass R: Cytomegaloviruses. Fields Virology. Edited by: Howley P. 2007, Philadelphia: Lippincott, 2701-2772. 5
De Bolle L, Naesens L, De Clercq E: Update on human herpesvirus 6 biology, clinical features, and therapy. Clin Microbiol Rev. 2005, 18: 217-245. 10.1128/CMR.18.1.217-245.2005.
Yamanishi K, Mori Y, Pellet P: Human Herpesviruses 6 and 7. Fields Virology. Edited by: Howley P. 2007, Philadelphia: Lippincott, 2820-2845. 5
Miyake F, Yoshikawa T, Sun H, Kakimi A, Ohashi M, Akimoto S, Nishiyama Y, Asano Y: Latent infection of human herpesvirus 7 in CD4(+) T lymphocytes. J Med Virol. 2006, 78: 112-116. 10.1002/jmv.20511.
Coleman CB, Nealy MS, Tibbetts SA: Immature and transitional B cells are latency reservoirs for a gammaherpesvirus. J Virol. 2010, 84: 13045-13052. 10.1128/JVI.01455-10.
Rickinson A, Kieff E: Epstein-Barr Virus. Fields Virology. Edited by: Howley P. 2007, Philadelphia: Lippincott, 2655-2700. 5
Ganem D: Kaposi's Sarcoma-associated Herpesvirus. Fields Virology. Edited by: Howley P. 2007, Philadelphia: Lippincott, 2848-2888. 5
Efstathiou S, Preston CM: Towards an understanding of the molecular basis of herpes simplex virus latency. Virus Res. 2005, 111: 108-119. 10.1016/j.virusres.2005.04.017.
Bloom DC, Giordani NV, Kwiatkowski DL: Epigenetic regulation of latent HSV-1 gene expression. Biochim Biophys Acta. 2010, 1799: 246-256.
Knipe DM, Cliffe A: Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol. 2008, 6: 211-221. 10.1038/nrmicro1794.
Kutluay SB, Triezenberg SJ: Role of chromatin during herpesvirus infections. Biochim Biophys Acta. 2009, 1790: 456-466.
Perng GC, Jones C: Towards an understanding of the herpes simplex virus type 1 latency-reactivation cycle. Interdiscip Perspect Infect Dis. 2010, 2010: 262415-
Sissons JG, Bain M, Wills MR: Latency and reactivation of human cytomegalovirus. J Infect. 2002, 44: 73-77. 10.1053/jinf.2001.0948.
Bego MG, St Jeor S: Human cytomegalovirus infection of cells of hematopoietic origin: HCMV-induced immunosuppression, immune evasion, and latency. Exp Hematol. 2006, 34: 555-570. 10.1016/j.exphem.2005.11.012.
Slobedman B, Cao JZ, Avdic S, Webster B, McAllery S, Cheung AK, Tan JC, Abendroth A: Human cytomegalovirus latent infection and associated viral gene expression. Future Microbiol. 2010, 5: 883-900. 10.2217/fmb.10.58.
Sinclair J: Chromatin structure regulates human cytomegalovirus gene expression during latency, reactivation and lytic infection. Biochim Biophys Acta. 2010, 1799: 286-295.
Sinclair J, Sissons P: Latency and reactivation of human cytomegalovirus. J Gen Virol. 2006, 87: 1763-1779. 10.1099/vir.0.81891-0.
Amon W, Farrell PJ: Reactivation of Epstein-Barr virus from latency. Rev Med Virol. 2005, 15: 149-156. 10.1002/rmv.456.
Young LS, Rickinson AB: Epstein-Barr virus: 40 years on. Nat Rev Cancer. 2004, 4: 757-768. 10.1038/nrc1452.
Tsurumi T, Fujita M, Kudoh A: Latent and lytic Epstein-Barr virus replication strategies. Rev Med Virol. 2005, 15: 3-15. 10.1002/rmv.441.
Young LS, Dawson CW, Eliopoulos AG: The expression and function of Epstein-Barr virus encoded latent genes. Mol Pathol. 2000, 53: 238-247. 10.1136/mp.53.5.238.
Tempera I, Lieberman PM: Chromatin organization of gammaherpesvirus latent genomes. Biochim Biophys Acta. 2010, 1799: 236-245.
Camarena V, Kobayashi M, Kim JY, Roehm P, Perez R, Gardner J, Wilson AC, Mohr I, Chao MV: Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe. 2010, 8: 320-330. 10.1016/j.chom.2010.09.007.
Danaher RJ, Jacob RJ, Miller CS: Establishment of a quiescent herpes simplex virus type 1 infection in neurally-differentiated PC12 cells. J Neurovirol. 1999, 5: 258-267. 10.3109/13550289909015812.
Jamieson DR, Robinson LH, Daksis JI, Nicholl MJ, Preston CM: Quiescent viral genomes in human fibroblasts after infection with herpes simplex virus type 1 Vmw65 mutants. J Gen Virol. 1995, 76 (Pt 6): 1417-1431. 10.1099/0022-1317-76-6-1417.
Russell J, Preston CM: An in vitro latency system for herpes simplex virus type 2. J Gen Virol. 1986, 67 (Pt 2): 397-403. 10.1099/0022-1317-67-2-397.
Wilcox CL, Johnson EM: Nerve growth factor deprivation results in the reactivation of latent herpes simplex virus in vitro. J Virol. 1987, 61: 2311-2315.
Ma SD, Hegde S, Young KH, Sullivan R, Rajesh D, Zhou Y, Jankowska-Gan E, Burlingham WJ, Sun X, Gulley ML, et al: A new model of EBV infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J Virol. 2010, 85: 165-177. 10.1128/JVI.01512-10.
Smith MS, Goldman DC, Bailey AS, Pfaffle DL, Kreklywich CN, Spencer DB, Othieno FA, Streblow DN, Garcia JV, Fleming WH, Nelson JA: Granulocyte-colony stimulating factor reactivates human cytomegalovirus in a latently infected humanized mouse model. Cell Host Microbe. 2010, 8: 284-291. 10.1016/j.chom.2010.08.001.
Ace CI, McKee TA, Ryan JM, Cameron JM, Preston CM: Construction and characterization of a herpes simplex virus type 1 mutant unable to transinduce immediate-early gene expression. J Virol. 1989, 63: 2260-2269.
Imai Y, Apakupakul K, Krause PR, Halford WP, Margolis TP: Investigation of the mechanism by which herpes simplex virus type 1 LAT sequences modulate preferential establishment of latent infection in mouse trigeminal ganglia. J Virol. 2009, 83: 7873-7882. 10.1128/JVI.00043-09.
Margolis TP, Imai Y, Yang L, Vallas V, Krause PR: Herpes simplex virus type 2 (HSV-2) establishes latent infection in a different population of ganglionic neurons than HSV-1: role of latency-associated transcripts. J Virol. 2007, 81: 1872-1878. 10.1128/JVI.02110-06.
Proenca JT, Coleman HM, Connor V, Winton DJ, Efstathiou S: A historical analysis of herpes simplex virus promoter activation in vivo reveals distinct populations of latently infected neurones. J Gen Virol. 2008, 89: 2965-2974. 10.1099/vir.0.2008/005066-0.
Margolis TP, Dawson CR, LaVail JH: Herpes simplex viral infection of the mouse trigeminal ganglion. Immunohistochemical analysis of cell populations. Invest Ophthalmol Vis Sci. 1992, 33: 259-267.
Yang L, Voytek CC, Margolis TP: Immunohistochemical analysis of primary sensory neurons latently infected with herpes simplex virus type 1. J Virol. 2000, 74: 209-217. 10.1128/JVI.74.1.209-217.2000.
Sawtell NM, Thompson RL: Herpes simplex virus type 1 latency-associated transcription unit promotes anatomical site-dependent establishment and reactivation from latency. J Virol. 1992, 66: 2157-2169.
Speck PG, Simmons A: Synchronous appearance of antigen-positive and latently infected neurons in spinal ganglia of mice infected with a virulent strain of herpes simplex virus. J Gen Virol. 1992, 73 (Pt 5): 1281-1285. 10.1099/0022-1317-73-5-1281.
Lachmann RH, Sadarangani M, Atkinson HR, Efstathiou S: An analysis of herpes simplex virus gene expression during latency establishment and reactivation. J Gen Virol. 1999, 80 (Pt 5): 1271-1282.
Sawtell NM: Comprehensive quantification of herpes simplex virus latency at the single-cell level. J Virol. 1997, 71: 5423-5431.
Thompson RL, Sawtell NM: Replication of herpes simplex virus type 1 within trigeminal ganglia is required for high frequency but not high viral genome copy number latency. J Virol. 2000, 74: 965-974. 10.1128/JVI.74.2.965-974.2000.
Coen DM, Kosz-Vnenchak M, Jacobson JG, Leib DA, Bogard CL, Schaffer PA, Tyler KL, Knipe DM: Thymidine kinase-negative herpes simplex virus mutants establish latency in mouse trigeminal ganglia but do not reactivate. Proc Natl Acad Sci USA. 1989, 86: 4736-4740. 10.1073/pnas.86.12.4736.
Speck PG, Simmons A: Divergent molecular pathways of productive and latent infection with a virulent strain of herpes simplex virus type 1. J Virol. 1991, 65: 4001-4005.
Katan M, Haigh A, Verrijzer CP, van der Vliet PC, O'Hare P: Characterization of a cellular factor which interacts functionally with Oct-1 in the assembly of a multicomponent transcription complex. Nucleic Acids Res. 1990, 18: 6871-6880. 10.1093/nar/18.23.6871.
Wilson AC, LaMarco K, Peterson MG, Herr W: The VP16 accessory protein HCF is a family of polypeptides processed from a large precursor protein. Cell. 1993, 74: 115-125. 10.1016/0092-8674(93)90299-6.
Xiao P, Capone JP: A cellular factor binds to the herpes simplex virus type 1 transactivator Vmw65 and is required for Vmw65-dependent protein-DNA complex assembly with Oct-1. Mol Cell Biol. 1990, 10: 4974-4977.
Stern S, Tanaka M, Herr W: The Oct-1 homoeodomain directs formation of a multiprotein-DNA complex with the HSV transactivator VP16. Nature. 1989, 341: 624-630. 10.1038/341624a0.
O'Hare P, Goding CR, Haigh A: Direct combinatorial interaction between a herpes simplex virus regulatory protein and a cellular octamer-binding factor mediates specific induction of virus immediate-early gene expression. Embo J. 1988, 7: 4231-4238.
O'Hare P, Goding CR: Herpes simplex virus regulatory elements and the immunoglobulin octamer domain bind a common factor and are both targets for virion transactivation. Cell. 1988, 52: 435-445.
La Boissiere S, Hughes T, O'Hare P: HCF-dependent nuclear import of VP16. Embo J. 1999, 18: 480-489. 10.1093/emboj/18.2.480.
Preston CM, Frame MC, Campbell ME: A complex formed between cell components and an HSV structural polypeptide binds to a viral immediate early gene regulatory DNA sequence. Cell. 1988, 52: 425-434. 10.1016/S0092-8674(88)80035-7.
Herrera FJ, Triezenberg SJ: VP16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J Virol. 2004, 78: 9689-9696. 10.1128/JVI.78.18.9689-9696.2004.
Triezenberg SJ, Kingsbury RC, McKnight SL: Functional dissection of VP16, the trans-activator of herpes simplex virus immediate early gene expression. Genes Dev. 1988, 2: 718-729. 10.1101/gad.2.6.718.
Goodrich JA, Hoey T, Thut CJ, Admon A, Tjian R: Drosophila TAFII40 interacts with both a VP16 activation domain and the basal transcription factor TFIIB. Cell. 1993, 75: 519-530. 10.1016/0092-8674(93)90386-5.
Ingles CJ, Shales M, Cress WD, Triezenberg SJ, Greenblatt J: Reduced binding of TFIID to transcriptionally compromised mutants of VP16. Nature. 1991, 351: 588-590. 10.1038/351588a0.
Klemm RD, Goodrich JA, Zhou S, Tjian R: Molecular cloning and expression of the 32-kDa subunit of human TFIID reveals interactions with VP16 and TFIIB that mediate transcriptional activation. Proc Natl Acad Sci USA. 1995, 92: 5788-5792. 10.1073/pnas.92.13.5788.
Lin YS, Ha I, Maldonado E, Reinberg D, Green MR: Binding of general transcription factor TFIIB to an acidic activating region. Nature. 1991, 353: 569-571. 10.1038/353569a0.
Uesugi M, Nyanguile O, Lu H, Levine AJ, Verdine GL: Induced alpha helix in the VP16 activation domain upon binding to a human TAF. Science. 1997, 277: 1310-1313. 10.1126/science.277.5330.1310.
Tal-Singer R, Pichyangkura R, Chung E, Lasner TM, Randazzo BP, Trojanowski JQ, Fraser NW, Triezenberg SJ: The transcriptional activation domain of VP16 is required for efficient infection and establishment of latency by HSV-1 in the murine peripheral and central nervous systems. Virology. 1999, 259: 20-33. 10.1006/viro.1999.9756.
Hancock MH, Cliffe AR, Knipe DM, Smiley JR: Herpes simplex virus VP16, but not ICP0, is required to reduce histone occupancy and enhance histone acetylation on viral genomes in U2OS osteosarcoma cells. J Virol. 2010, 84: 1366-1375. 10.1128/JVI.01727-09.
Peng H, Nogueira ML, Vogel JL, Kristie TM: Transcriptional coactivator HCF-1 couples the histone chaperone Asf1b to HSV-1 DNA replication components. Proc Natl Acad Sci USA. 2010, 107: 2461-2466. 10.1073/pnas.0911128107.
Oh J, Fraser NW: Temporal association of the herpes simplex virus genome with histone proteins during a lytic infection. J Virol. 2008, 82: 3530-3537. 10.1128/JVI.00586-07.
Leinbach SS, Summers WC: The structure of herpes simplex virus type 1 DNA as probed by micrococcal nuclease digestion. J Gen Virol. 1980, 51: 45-59. 10.1099/0022-1317-51-1-45.
Placek BJ, Berger SL: Chromatin dynamics during herpes simplex virus-1 lytic infection. Biochim Biophys Acta. 2010, 1799: 223-227.
Paulus C, Nitzsche A, Nevels M: Chromatinisation of herpesvirus genomes. Rev Med Virol. 2010, 20: 34-50. 10.1002/rmv.632.
Kent JR, Zeng PY, Atanasiu D, Gardner J, Fraser NW, Berger SL: During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J Virol. 2004, 78: 10178-10186. 10.1128/JVI.78.18.10178-10186.2004.
Huang J, Kent JR, Placek B, Whelan KA, Hollow CM, Zeng PY, Fraser NW, Berger SL: Trimethylation of histone H3 lysine 4 by Set1 in the lytic infection of human herpes simplex virus 1. J Virol. 2006, 80: 5740-5746. 10.1128/JVI.00169-06.
Memedula S, Belmont AS: Sequential recruitment of HAT and SWI/SNF components to condensed chromatin by VP16. Curr Biol. 2003, 13: 241-246. 10.1016/S0960-9822(03)00048-4.
Neely KE, Hassan AH, Wallberg AE, Steger DJ, Cairns BR, Wright AP, Workman JL: Activation domain-mediated targeting of the SWI/SNF complex to promoters stimulates transcription from nucleosome arrays. Mol Cell. 1999, 4: 649-655. 10.1016/S1097-2765(00)80216-6.
Utley RT, Ikeda K, Grant PA, Cote J, Steger DJ, Eberharter A, John S, Workman JL: Transcriptional activators direct histone acetyltransferase complexes to nucleosomes. Nature. 1998, 394: 498-502. 10.1038/28886.
Kundu TK, Palhan VB, Wang Z, An W, Cole PA, Roeder RG: Activator-dependent transcription from chromatin in vitro involving targeted histone acetylation by p300. Mol Cell. 2000, 6: 551-561. 10.1016/S1097-2765(00)00054-X.
Kraus WL, Manning ET, Kadonaga JT: Biochemical analysis of distinct activation functions in p300 that enhance transcription initiation with chromatin templates. Mol Cell Biol. 1999, 19: 8123-8135.
Barlev NA, Candau R, Wang L, Darpino P, Silverman N, Berger SL: Characterization of physical interactions of the putative transcriptional adaptor, ADA2, with acidic activation domains and TATA-binding protein. J Biol Chem. 1995, 270: 19337-19344. 10.1074/jbc.270.33.19337.
Kutluay SB, DeVos SL, Klomp JE, Triezenberg SJ: Transcriptional coactivators are not required for herpes simplex virus type 1 immediate-early gene expression in vitro. J Virol. 2009, 83: 3436-3449. 10.1128/JVI.02349-08.
Roizman B, Sears AE: An inquiry into the mechanisms of herpes simplex virus latency. Annu Rev Microbiol. 1987, 41: 543-571. 10.1146/annurev.mi.41.100187.002551.
Kristie TM, Vogel JL, Sears AE: Nuclear localization of the C1 factor (host cell factor) in sensory neurons correlates with reactivation of herpes simplex virus from latency. Proc Natl Acad Sci USA. 1999, 96: 1229-1233. 10.1073/pnas.96.4.1229.
LaBoissiere S, O'Hare P: Analysis of HCF, the cellular cofactor of VP16, in herpes simplex virus-infected cells. J Virol. 2000, 74: 99-109. 10.1128/JVI.74.1.99-109.2000.
Lu R, Misra V: Zhangfei: a second cellular protein interacts with herpes simplex virus accessory factor HCF in a manner similar to Luman and VP16. Nucleic Acids Res. 2000, 28: 2446-2454. 10.1093/nar/28.12.2446.
Akhova O, Bainbridge M, Misra V: The neuronal host cell factor-binding protein Zhangfei inhibits herpes simplex virus replication. J Virol. 2005, 79: 14708-14718. 10.1128/JVI.79.23.14708-14718.2005.
Lu R, Yang P, O'Hare P, Misra V: Luman, a new member of the CREB/ATF family, binds to herpes simplex virus VP16-associated host cellular factor. Mol Cell Biol. 1997, 17: 5117-5126.
Lu R, Misra V: Potential role for luman, the cellular homologue of herpes simplex virus VP16 (alpha gene trans-inducing factor), in herpesvirus latency. J Virol. 2000, 74: 934-943. 10.1128/JVI.74.2.934-943.2000.
Kolb G, Kristie TM: Association of the cellular coactivator HCF-1 with the Golgi apparatus in sensory neurons. J Virol. 2008, 82: 9555-9563. 10.1128/JVI.01174-08.
Cliffe AR, Garber DA, Knipe DM: Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol. 2009, 83: 8182-8190. 10.1128/JVI.00712-09.
Wang QY, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM: Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc Natl Acad Sci USA. 2005, 102: 16055-16059. 10.1073/pnas.0505850102.
Deshmane SL, Fraser NW: During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J Virol. 1989, 63: 943-947.
Nogueira ML, Wang VE, Tantin D, Sharp PA, Kristie TM: Herpes simplex virus infections are arrested in Oct-1-deficient cells. Proc Natl Acad Sci USA. 2004, 101: 1473-1478. 10.1073/pnas.0307300101.
He X, Treacy MN, Simmons DM, Ingraham HA, Swanson LW, Rosenfeld MG: Expression of a large family of POU-domain regulatory genes in mammalian brain development. Nature. 1989, 340: 35-41. 10.1038/340035a0.
Stevens JG, Wagner EK, Devi-Rao GB, Cook ML, Feldman LT: RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science. 1987, 235: 1056-1059. 10.1126/science.2434993.
Batchelor AH, O'Hare P: Regulation and cell-type-specific activity of a promoter located upstream of the latency-associated transcript of herpes simplex virus type 1. J Virol. 1990, 64: 3269-3279.
Zwaagstra JC, Ghiasi H, Slanina SM, Nesburn AB, Wheatley SC, Lillycrop K, Wood J, Latchman DS, Patel K, Wechsler SL: Activity of herpes simplex virus type 1 latency-associated transcript (LAT) promoter in neuron-derived cells: evidence for neuron specificity and for a large LAT transcript. J Virol. 1990, 64: 5019-5028.
Kubat NJ, Amelio AL, Giordani NV, Bloom DC: The herpes simplex virus type 1 latency-associated transcript (LAT) enhancer/rcr is hyperacetylated during latency independently of LAT transcription. J Virol. 2004, 78: 12508-12518. 10.1128/JVI.78.22.12508-12518.2004.
Drolet BS, Perng GC, Cohen J, Slanina SM, Yukht A, Nesburn AB, Wechsler SL: The region of the herpes simplex virus type 1 LAT gene involved in spontaneous reactivation does not encode a functional protein. Virology. 1998, 242: 221-232. 10.1006/viro.1997.9020.
Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR: MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature. 2008, 454: 780-783.
Umbach JL, Nagel MA, Cohrs RJ, Gilden DH, Cullen BR: Analysis of human alphaherpesvirus microRNA expression in latently infected human trigeminal ganglia. J Virol. 2009, 83: 10677-10683. 10.1128/JVI.01185-09.
Jurak I, Kramer MF, Mellor JC, van Lint AL, Roth FP, Knipe DM, Coen DM: Numerous conserved and divergent microRNAs expressed by herpes simplex viruses 1 and 2. J Virol. 2010, 84: 4659-4672. 10.1128/JVI.02725-09.
Cui C, Griffiths A, Li G, Silva LM, Kramer MF, Gaasterland T, Wang XJ, Coen DM: Prediction and identification of herpes simplex virus 1-encoded microRNAs. J Virol. 2006, 80: 5499-5508. 10.1128/JVI.00200-06.
Tang S, Bertke AS, Patel A, Wang K, Cohen JI, Krause PR: An acutely and latently expressed herpes simplex virus 2 viral microRNA inhibits expression of ICP34.5, a viral neurovirulence factor. Proc Natl Acad Sci USA. 2008, 105: 10931-10936. 10.1073/pnas.0801845105.
Tang S, Patel A, Krause PR: Novel less-abundant viral microRNAs encoded by herpes simplex virus 2 latency-associated transcript and their roles in regulating ICP34.5 and ICP0 mRNAs. J Virol. 2009, 83: 1433-1442. 10.1128/JVI.01723-08.
Everett RD: Trans activation of transcription by herpes virus products: requirement for two HSV-1 immediate-early polypeptides for maximum activity. Embo J. 1984, 3: 3135-3141.
Gelman IH, Silverstein S: Identification of immediate early genes from herpes simplex virus that transactivate the virus thymidine kinase gene. Proc Natl Acad Sci USA. 1985, 82: 5265-5269. 10.1073/pnas.82.16.5265.
O'Hare P, Hayward GS: Evidence for a direct role for both the 175,000- and 110,000-molecular-weight immediate-early proteins of herpes simplex virus in the transactivation of delayed-early promoters. J Virol. 1985, 53: 751-760.
Quinlan MP, Knipe DM: Stimulation of expression of a herpes simplex virus DNA-binding protein by two viral functions. Mol Cell Biol. 1985, 5: 957-963.
Grewal SI, Jia S: Heterochromatin revisited. Nat Rev Genet. 2007, 8: 35-46. 10.1038/nrg2008.
Lomonte P, Thomas J, Texier P, Caron C, Khochbin S, Epstein AL: Functional interaction between class II histone deacetylases and ICP0 of herpes simplex virus type 1. J Virol. 2004, 78: 6744-6757. 10.1128/JVI.78.13.6744-6757.2004.
Gu H, Liang Y, Mandel G, Roizman B: Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc Natl Acad Sci USA. 2005, 102: 7571-7576. 10.1073/pnas.0502658102.
Gu H, Roizman B: Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc Natl Acad Sci USA. 2007, 104: 17134-17139. 10.1073/pnas.0707266104.
Perng GC, Jones C, Ciacci-Zanella J, Stone M, Henderson G, Yukht A, Slanina SM, Hofman FM, Ghiasi H, Nesburn AB, Wechsler SL: Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science. 2000, 287: 1500-1503. 10.1126/science.287.5457.1500.
Thompson RL, Sawtell NM: The herpes simplex virus type 1 latency-associated transcript gene regulates the establishment of latency. J Virol. 1997, 71: 5432-5440.
Leib DA, Bogard CL, Kosz-Vnenchak M, Hicks KA, Coen DM, Knipe DM, Schaffer PA: A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J Virol. 1989, 63: 2893-2900.
Kosz-Vnenchak M, Jacobson J, Coen DM, Knipe DM: Evidence for a novel regulatory pathway for herpes simplex virus gene expression in trigeminal ganglion neurons. J Virol. 1993, 67: 5383-5393.
Cai WZ, Schaffer PA: Herpes simplex virus type 1 ICP0 plays a critical role in the de novo synthesis of infectious virus following transfection of viral DNA. J Virol. 1989, 63: 4579-4589.
Leib DA, Coen DM, Bogard CL, Hicks KA, Yager DR, Knipe DM, Tyler KL, Schaffer PA: Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. J Virol. 1989, 63: 759-768.
Jordan R, Schaffer PA: Activation of gene expression by herpes simplex virus type 1 ICP0 occurs at the level of mRNA synthesis. J Virol. 1997, 71: 6850-6862.
Pesola JM, Zhu J, Knipe DM, Coen DM: Herpes simplex virus 1 immediate-early and early gene expression during reactivation from latency under conditions that prevent infectious virus production. J Virol. 2005, 79: 14516-14525. 10.1128/JVI.79.23.14516-14525.2005.
Cai W, Astor TL, Liptak LM, Cho C, Coen DM, Schaffer PA: The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J Virol. 1993, 67: 7501-7512.
Halford WP, Schaffer PA: ICP0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. J Virol. 2001, 75: 3240-3249. 10.1128/JVI.75.7.3240-3249.2001.
Stow ND, Stow EC: Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. J Gen Virol. 1986, 67 (Pt 12): 2571-2585. 10.1099/0022-1317-67-12-2571.
Sacks WR, Schaffer PA: Deletion mutants in the gene encoding the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in cell culture. J Virol. 1987, 61: 829-839.
Thompson RL, Sawtell NM: Evidence that the herpes simplex virus type 1 ICP0 protein does not initiate reactivation from latency in vivo. J Virol. 2006, 80: 10919-10930. 10.1128/JVI.01253-06.
Thompson RL, Preston CM, Sawtell NM: De novo synthesis of VP16 coordinates the exit from HSV latency in vivo. PLoS Pathog. 2009, 5: e1000352-10.1371/journal.ppat.1000352.
Steiner I, Spivack JG, Deshmane SL, Ace CI, Preston CM, Fraser NW: A herpes simplex virus type 1 mutant containing a nontransinducing Vmw65 protein establishes latent infection in vivo in the absence of viral replication and reactivates efficiently from explanted trigeminal ganglia. J Virol. 1990, 64: 1630-1638.
Ecob-Prince MS, Rixon FJ, Preston CM, Hassan K, Kennedy PG: Reactivation in vivo and in vitro of herpes simplex virus from mouse dorsal root ganglia which contain different levels of latency-associated transcripts. J Gen Virol. 1993, 74 (Pt 6): 995-1002. 10.1099/0022-1317-74-6-995.
Sawtell NM: Quantitative analysis of herpes simplex virus reactivation in vivo demonstrates that reactivation in the nervous system is not inhibited at early times postinoculation. J Virol. 2003, 77: 4127-4138. 10.1128/JVI.77.7.4127-4138.2003.
Sawtell NM, Thompson RL, Haas RL: Herpes simplex virus DNA synthesis is not a decisive regulatory event in the initiation of lytic viral protein expression in neurons in vivo during primary infection or reactivation from latency. J Virol. 2006, 80: 38-50. 10.1128/JVI.80.1.38-50.2006.
Petrucelli A, Rak M, Grainger L, Goodrum F: Characterization of a novel Golgi apparatus-localized latency determinant encoded by human cytomegalovirus. J Virol. 2009, 83: 5615-5629. 10.1128/JVI.01989-08.
Cheung AK, Abendroth A, Cunningham AL, Slobedman B: Viral gene expression during the establishment of human cytomegalovirus latent infection in myeloid progenitor cells. Blood. 2006, 108: 3691-3699. 10.1182/blood-2005-12-026682.
Jenkins C, Abendroth A, Slobedman B: A novel viral transcript with homology to human interleukin-10 is expressed during latent human cytomegalovirus infection. J Virol. 2004, 78: 1440-1447. 10.1128/JVI.78.3.1440-1447.2004.
Bego M, Maciejewski J, Khaiboullina S, Pari G, St Jeor S: Characterization of an antisense transcript spanning the UL81-82 locus of human cytomegalovirus. J Virol. 2005, 79: 11022-11034. 10.1128/JVI.79.17.11022-11034.2005.
Goodrum FD, Jordan CT, High K, Shenk T: Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proc Natl Acad Sci USA. 2002, 99: 16255-16260. 10.1073/pnas.252630899.
Schierling K, Stamminger T, Mertens T, Winkler M: Human cytomegalovirus tegument proteins ppUL82 (pp71) and ppUL35 interact and cooperatively activate the major immediate-early enhancer. J Virol. 2004, 78: 9512-9523. 10.1128/JVI.78.17.9512-9523.2004.
Stamminger T, Gstaiger M, Weinzierl K, Lorz K, Winkler M, Schaffner W: Open reading frame UL26 of human cytomegalovirus encodes a novel tegument protein that contains a strong transcriptional activation domain. J Virol. 2002, 76: 4836-4847. 10.1128/JVI.76.10.4836-4847.2002.
Cristea IM, Moorman NJ, Terhune SS, Cuevas CD, O'Keefe ES, Rout MP, Chait BT, Shenk T: Human cytomegalovirus pUL83 stimulates activity of the viral immediate-early promoter through its interaction with the cellular IFI16 protein. J Virol. 2010, 84: 7803-7814. 10.1128/JVI.00139-10.
Terhune SS, Moorman NJ, Cristea IM, Savaryn JP, Cuevas-Bennett C, Rout MP, Chait BT, Shenk T: Human cytomegalovirus UL29/28 protein interacts with components of the NuRD complex which promote accumulation of immediate-early RNA. PLoS Pathog. 2010, 6: e1000965-10.1371/journal.ppat.1000965.
Liu B, Stinski MF: Human cytomegalovirus contains a tegument protein that enhances transcription from promoters with upstream ATF and AP-1 cis-acting elements. J Virol. 1992, 66: 4434-4444.
Saffert RT, Kalejta RF: Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J Virol. 2006, 80: 3863-3871. 10.1128/JVI.80.8.3863-3871.2006.
Preston CM, Nicholl MJ: Role of the cellular protein hDaxx in human cytomegalovirus immediate-early gene expression. J Gen Virol. 2006, 87: 1113-1121. 10.1099/vir.0.81566-0.
Saffert R, Kalejta R: Promyelocytic leukemia-nuclear body proteins: herpesvirus enemies, accomplices, or both?. Future Virology. 2008, 3: 265-277. 10.2217/17460794.3.3.265.
Tavalai N, Papior P, Rechter S, Leis M, Stamminger T: Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J Virol. 2006, 80: 8006-8018. 10.1128/JVI.00743-06.
Tavalai N, Papior P, Rechter S, Stamminger T: Nuclear domain 10 components promyelocytic leukemia protein and hDaxx independently contribute to an intrinsic antiviral defense against human cytomegalovirus infection. J Virol. 2008, 82: 126-137. 10.1128/JVI.01685-07.
Tavalai N, Stamminger T: Intrinsic cellular defense mechanisms targeting human cytomegalovirus. Virus Res.
Ishov AM, Vladimirova OV, Maul GG: Daxx-mediated accumulation of human cytomegalovirus tegument protein pp71 at ND10 facilitates initiation of viral infection at these nuclear domains. J Virol. 2002, 76: 7705-7712. 10.1128/JVI.76.15.7705-7712.2002.
Saffert RT, Kalejta RF: Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J Virol. 2007, 81: 9109-9120. 10.1128/JVI.00827-07.
Saffert RT, Penkert RR, Kalejta RF: Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34+ cells. J Virol. 2010, 84: 5594-5604. 10.1128/JVI.00348-10.
Cantrell SR, Bresnahan WA: Interaction between the human cytomegalovirus UL82 gene product (pp71) and hDaxx regulates immediate-early gene expression and viral replication. J Virol. 2005, 79: 7792-7802. 10.1128/JVI.79.12.7792-7802.2005.
Hwang J, Kalejta RF: Proteasome-dependent, ubiquitin-independent degradation of Daxx by the viral pp71 protein in human cytomegalovirus-infected cells. Virology. 2007, 367: 334-338. 10.1016/j.virol.2007.05.037.
Hwang J, Kalejta RF: Human cytomegalovirus protein pp71 induces Daxx SUMOylation. J Virol. 2009, 83: 6591-6598. 10.1128/JVI.02639-08.
Woodhall DL, Groves IJ, Reeves MB, Wilkinson G, Sinclair JH: Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J Biol Chem. 2006, 281: 37652-37660. 10.1074/jbc.M604273200.
Maxwell KL, Frappier L: Viral proteomics. Microbiol Mol Biol Rev. 2007, 71: 398-411. 10.1128/MMBR.00042-06.
Groves IJ, Reeves MB, Sinclair JH: Lytic infection of permissive cells with human cytomegalovirus is regulated by an intrinsic 'pre-immediate-early' repression of viral gene expression mediated by histone post-translational modification. J Gen Virol. 2009, 90: 2364-2374. 10.1099/vir.0.012526-0.
Nitzsche A, Paulus C, Nevels M: Temporal dynamics of cytomegalovirus chromatin assembly in productively infected human cells. J Virol. 2008, 82: 11167-11180. 10.1128/JVI.01218-08.
Murphy JC, Fischle W, Verdin E, Sinclair JH: Control of cytomegalovirus lytic gene expression by histone acetylation. Embo J. 2002, 21: 1112-1120. 10.1093/emboj/21.5.1112.
Reeves MB, Lehner PJ, Sissons JG, Sinclair JH: An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J Gen Virol. 2005, 86: 2949-2954. 10.1099/vir.0.81161-0.
Reeves MB, MacAry PA, Lehner PJ, Sissons JG, Sinclair JH: Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc Natl Acad Sci USA. 2005, 102: 4140-4145. 10.1073/pnas.0408994102.
Ioudinkova E, Arcangeletti MC, Rynditch A, De Conto F, Motta F, Covan S, Pinardi F, Razin SV, Chezzi C: Control of human cytomegalovirus gene expression by differential histone modifications during lytic and latent infection of a monocytic cell line. Gene. 2006, 384: 120-128. 10.1016/j.gene.2006.07.021.
Cuevas-Bennett C, Shenk T: Dynamic histone H3 acetylation and methylation at human cytomegalovirus promoters during replication in fibroblasts. J Virol. 2008, 82: 9525-9536. 10.1128/JVI.00946-08.
Dosa R, Burian K, Gonczol E: Human cytomegalovirus latency is associated with the state of differentiation of the host cells: an in vitro model in teratocarcinoma cells. Acta Microbiol Immunol Hung. 2005, 52: 397-406. 10.1556/AMicr.52.2005.3-4.11.
Lukashchuk V, McFarlane S, Everett RD, Preston CM: Human cytomegalovirus protein pp71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J Virol. 2008, 82: 12543-12554. 10.1128/JVI.01215-08.
Gibbons RJ, McDowell TL, Raman S, O'Rourke DM, Garrick D, Ayyub H, Higgs DR: Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation. Nat Genet. 2000, 24: 368-371. 10.1038/74191.
Xue Y, Gibbons R, Yan Z, Yang D, McDowell TL, Sechi S, Qin J, Zhou S, Higgs D, Wang W: The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc Natl Acad Sci USA. 2003, 100: 10635-10640. 10.1073/pnas.1937626100.
Yuan J, Liu X, Wu AW, McGonagill PW, Keller MJ, Galle CS, Meier JL: Breaking human cytomegalovirus major immediate-early gene silence by vasoactive intestinal peptide stimulation of the protein kinase A-CREB-TORC2 signaling cascade in human pluripotent embryonal NTera2 cells. J Virol. 2009, 83: 6391-6403. 10.1128/JVI.00061-09.
Liu X, Yuan J, Wu AW, McGonagill PW, Galle CS, Meier JL: Phorbol ester-induced human cytomegalovirus major immediate-early (MIE) enhancer activation through PKC-delta, CREB, and NF-kappaB desilences MIE gene expression in quiescently infected human pluripotent NTera2 cells. J Virol. 2010, 84: 8495-8508. 10.1128/JVI.00416-10.
Penkert RR, Kalejta RF: Nuclear localization of tegument-delivered pp71 in human cytomegalovirus-infected cells is facilitated by one or more factors present in terminally differentiated fibroblasts. J Virol. 2010, 84: 9853-9863. 10.1128/JVI.00500-10.
Cha TA, Tom E, Kemble GW, Duke GM, Mocarski ES, Spaete RR: Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J Virol. 1996, 70: 78-83.
Dolan A, Cunningham C, Hector RD, Hassan-Walker AF, Lee L, Addison C, Dargan DJ, McGeoch DJ, Gatherer D, Emery VC, et al: Genetic content of wild-type human cytomegalovirus. J Gen Virol. 2004, 85: 1301-1312. 10.1099/vir.0.79888-0.
Murphy E, Yu D, Grimwood J, Schmutz J, Dickson M, Jarvis MA, Hahn G, Nelson JA, Myers RM, Shenk TE: Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc Natl Acad Sci USA. 2003, 100: 14976-14981. 10.1073/pnas.2136652100.
Morissette G, Flamand L: Herpesviruses and chromosomal integration. J Virol. 2010, 84: 12100-12109. 10.1128/JVI.01169-10.
Luppi M, Barozzi P, Morris CM, Merelli E, Torelli G: Integration of human herpesvirus 6 genome in human chromosomes. Lancet. 1998, 352: 1707-1708. 10.1016/S0140-6736(05)61483-3.
Arbuckle JH, Medveczky MM, Luka J, Hadley SH, Luegmayr A, Ablashi D, Lund TC, Tolar J, De Meirleir K, Montoya JG, et al: The latent human herpesvirus-6A genome specifically integrates in telomeres of human chromosomes in vivo and in vitro. Proc Natl Acad Sci USA. 2010, 107: 5563-5568. 10.1073/pnas.0913586107.
Bolovan-Fritts CA, Mocarski ES, Wiedeman JA: Peripheral blood CD14(+) cells from healthy subjects carry a circular conformation of latent cytomegalovirus genome. Blood. 1999, 93: 394-398.
Kondo K, Xu J, Mocarski ES: Human cytomegalovirus latent gene expression in granulocyte-macrophage progenitors in culture and in seropositive individuals. Proc Natl Acad Sci USA. 1996, 93: 11137-11142. 10.1073/pnas.93.20.11137.
Kondo K, Mocarski ES: Cytomegalovirus latency and latency-specific transcription in hematopoietic progenitors. Scand J Infect Dis Suppl. 1995, 99: 63-67.
Landini MP, Lazzarotto T, Xu J, Geballe AP, Mocarski ES: Humoral immune response to proteins of human cytomegalovirus latency-associated transcripts. Biol Blood Marrow Transplant. 2000, 6: 100-108. 10.1016/S1083-8791(00)70072-3.
White KL, Slobedman B, Mocarski ES: Human cytomegalovirus latency-associated protein pORF94 is dispensable for productive and latent infection. J Virol. 2000, 74: 9333-9337. 10.1128/JVI.74.19.9333-9337.2000.
Chee MS, Bankier AT, Beck S, Bohni R, Brown CM, Cerny R, Horsnell T, Hutchison CA, Kouzarides T, Martignetti JA, et al: Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr Top Microbiol Immunol. 1990, 154: 125-169.
Zipeto D, Bodaghi B, Laurent L, Virelizier JL, Michelson S: Kinetics of transcription of human cytomegalovirus chemokine receptor US28 in different cell types. J Gen Virol. 1999, 80 (Pt 3): 543-547.
Jenkins C, Garcia W, Godwin MJ, Spencer JV, Stern JL, Abendroth A, Slobedman B: Immunomodulatory properties of a viral homolog of human interleukin-10 expressed by human cytomegalovirus during the latent phase of infection. J Virol. 2008, 82: 3736-3750. 10.1128/JVI.02173-07.
Cheung AK, Gottlieb DJ, Plachter B, Pepperl-Klindworth S, Avdic S, Cunningham AL, Abendroth A, Slobedman B: The role of the human cytomegalovirus UL111A gene in down-regulating CD4+ T-cell recognition of latently infected cells: implications for virus elimination during latency. Blood. 2009, 114: 4128-4137. 10.1182/blood-2008-12-197111.
Reeves MB, Sinclair JH: Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J Gen Virol. 2010, 91: 599-604. 10.1099/vir.0.015602-0.
Goodrum F, Reeves M, Sinclair J, High K, Shenk T: Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood. 2007, 110: 937-945. 10.1182/blood-2007-01-070078.
Murphy E, Vanicek J, Robins H, Shenk T, Levine AJ: Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: implications for latency. Proc Natl Acad Sci USA. 2008, 105: 5453-5458. 10.1073/pnas.0711910105.
Grey F, Meyers H, White EA, Spector DH, Nelson J: A human cytomegalovirus-encoded microRNA regulates expression of multiple viral genes involved in replication. PLoS Pathog. 2007, 3: e163-10.1371/journal.ppat.0030163.
Hertel L, Lacaille VG, Strobl H, Mellins ED, Mocarski ES: Susceptibility of immature and mature Langerhans cell-type dendritic cells to infection and immunomodulation by human cytomegalovirus. J Virol. 2003, 77: 7563-7574. 10.1128/JVI.77.13.7563-7574.2003.
Soderberg-Naucler C, Fish KN, Nelson JA: Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell. 1997, 91: 119-126. 10.1016/S0092-8674(01)80014-3.
Maciejewski JP, St Jeor SC: Human cytomegalovirus infection of human hematopoietic progenitor cells. Leuk Lymphoma. 1999, 33: 1-13.
Soderberg-Naucler C, Streblow DN, Fish KN, Allan-Yorke J, Smith PP, Nelson JA: Reactivation of latent human cytomegalovirus in CD14(+) monocytes is differentiation dependent. J Virol. 2001, 75: 7543-7554. 10.1128/JVI.75.16.7543-7554.2001.
Kuppers R: B cells under influence: transformation of B cells by Epstein-Barr virus. Nat Rev Immunol. 2003, 3: 801-812. 10.1038/nri1201.
Jiang R, Scott RS, Hutt-Fletcher LM: Epstein-Barr virus shed in saliva is high in B-cell-tropic glycoprotein gp42. J Virol. 2006, 80: 7281-7283. 10.1128/JVI.00497-06.
Cohen JI: Epstein-Barr virus infection. N Engl J Med. 2000, 343: 481-492. 10.1056/NEJM200008173430707.
Shannon-Lowe CD, Neuhierl B, Baldwin G, Rickinson AB, Delecluse HJ: Resting B cells as a transfer vehicle for Epstein-Barr virus infection of epithelial cells. Proc Natl Acad Sci USA. 2006, 103: 7065-7070. 10.1073/pnas.0510512103.
Shannon-Lowe C, Adland E, Bell AI, Delecluse HJ, Rickinson AB, Rowe M: Features distinguishing Epstein-Barr virus infections of epithelial cells and B cells: viral genome expression, genome maintenance, and genome amplification. J Virol. 2009, 83: 7749-7760. 10.1128/JVI.00108-09.
Li QX, Young LS, Niedobitek G, Dawson CW, Birkenbach M, Wang F, Rickinson AB: Epstein-Barr virus infection and replication in a human epithelial cell system. Nature. 1992, 356: 347-350. 10.1038/356347a0.
Altmann M, Hammerschmidt W: Epstein-Barr virus provides a new paradigm: a requirement for the immediate inhibition of apoptosis. PLoS Biol. 2005, 3: e404-10.1371/journal.pbio.0030404.
Kalla M, Schmeinck A, Bergbauer M, Pich D, Hammerschmidt W: AP-1 homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the epigenetic state of the viral genome. Proc Natl Acad Sci USA. 2010, 107: 850-855. 10.1073/pnas.0911948107.
Wen W, Iwakiri D, Yamamoto K, Maruo S, Kanda T, Takada K: Epstein-Barr virus BZLF1 gene, a switch from latency to lytic infection, is expressed as an immediate-early gene after primary infection of B lymphocytes. J Virol. 2007, 81: 1037-1042. 10.1128/JVI.01416-06.
Kelly GL, Long HM, Stylianou J, Thomas WA, Leese A, Bell AI, Bornkamm GW, Mautner J, Rickinson AB, Rowe M: An Epstein-Barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in burkitt lymphomagenesis: the Wp/BHRF1 link. PLoS Pathog. 2009, 5: e1000341-10.1371/journal.ppat.1000341.
Halder S, Murakami M, Verma SC, Kumar P, Yi F, Robertson ES: Early events associated with infection of Epstein-Barr virus infection of primary B-cells. PLoS One. 2009, 4: e7214-10.1371/journal.pone.0007214.
Farrell PJ, Rowe DT, Rooney CM, Kouzarides T: Epstein-Barr virus BZLF1 trans-activator specifically binds to a consensus AP-1 site and is related to c-fos. Embo J. 1989, 8: 127-132.
Hong GK, Gulley ML, Feng WH, Delecluse HJ, Holley-Guthrie E, Kenney SC: Epstein-Barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J Virol. 2005, 79: 13993-14003. 10.1128/JVI.79.22.13993-14003.2005.
Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR: The BCL-2 family reunion. Mol Cell. 2010, 37: 299-310. 10.1016/j.molcel.2010.01.025.
Bellows DS, Howell M, Pearson C, Hazlewood SA, Hardwick JM: Epstein-Barr virus BALF1 is a BCL-2-like antagonist of the herpesvirus antiapoptotic BCL-2 proteins. J Virol. 2002, 76: 2469-2479. 10.1128/jvi.76.5.2469-2479.2002.
Oudejans JJ, van den Brule AJ, Jiwa NM, de Bruin PC, Ossenkoppele GJ, van der Valk P, Walboomers JM, Meijer CJ: BHRF1, the Epstein-Barr virus (EBV) homologue of the BCL-2 protooncogene, is transcribed in EBV-associated B-cell lymphomas and in reactive lymphocytes. Blood. 1995, 86: 1893-1902.
Seto E, Moosmann A, Gromminger S, Walz N, Grundhoff A, Hammerschmidt W: Micro RNAs of Epstein-Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS Pathog. 2010, 6: e1001063-10.1371/journal.ppat.1001063. pii
Swaminathan S: Noncoding RNAs produced by oncogenic human herpesviruses. J Cell Physiol. 2008, 216: 321-326. 10.1002/jcp.21480.
Speck SH, Chatila T, Flemington E: Reactivation of Epstein-Barr virus: regulation and function of the BZLF1 gene. Trends Microbiol. 1997, 5: 399-405. 10.1016/S0966-842X(97)01129-3.
Bergbauer M, Kalla M, Schmeinck A, Gobel C, Rothbauer U, Eck S, Benet-Pages A, Strom TM, Hammerschmidt W: CpG-methylation regulates a class of Epstein-Barr virus promoters. PLoS Pathog. 2010, 6: e1001114-10.1371/journal.ppat.1001114. pii
Bhende PM, Seaman WT, Delecluse HJ, Kenney SC: The EBV lytic switch protein, Z, preferentially binds to and activates the methylated viral genome. Nat Genet. 2004, 36: 1099-1104. 10.1038/ng1424.
Dickerson SJ, Xing Y, Robinson AR, Seaman WT, Gruffat H, Kenney SC: Methylation-dependent binding of the epstein-barr virus BZLF1 protein to viral promoters. PLoS Pathog. 2009, 5: e1000356-10.1371/journal.ppat.1000356.
Baldick CJ, Marchini A, Patterson CE, Shenk T: Human cytomegalovirus tegument protein pp71 (ppUL82) enhances the infectivity of viral DNA and accelerates the infectious cycle. J Virol. 1997, 71: 4400-4408.
Werstuck G, Bilan P, Capone JP: Enhanced infectivity of herpes simplex virus type 1 viral DNA in a cell line expressing the trans-inducing factor Vmw65. J Virol. 1990, 64: 984-991.
Feederle R, Neuhierl B, Baldwin G, Bannert H, Hub B, Mautner J, Behrends U, Delecluse HJ: Epstein-Barr virus BNRF1 protein allows efficient transfer from the endosomal compartment to the nucleus of primary B lymphocytes. J Virol. 2006, 80: 9435-9443. 10.1128/JVI.00473-06.
Yates JL, Warren N, Sugden B: Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature. 1985, 313: 812-815. 10.1038/313812a0.
Kirchmaier AL, Sugden B: Plasmid maintenance of derivatives of oriP of Epstein-Barr virus. J Virol. 1995, 69: 1280-1283.
Lindner SE, Sugden B: The plasmid replicon of Epstein-Barr virus: mechanistic insights into efficient, licensed, extrachromosomal replication in human cells. Plasmid. 2007, 58: 1-12. 10.1016/j.plasmid.2007.01.003.
Nanbo A, Sugden A, Sugden B: The coupling of synthesis and partitioning of EBV's plasmid replicon is revealed in live cells. Embo J. 2007, 26: 4252-4262. 10.1038/sj.emboj.7601853.
Wang J, Lindner SE, Leight ER, Sugden B: Essential elements of a licensed, mammalian plasmid origin of DNA synthesis. Mol Cell Biol. 2006, 26: 1124-1134. 10.1128/MCB.26.3.1124-1134.2006.
Speck S: Regulation of EBV Latency-Associated Gene Expression. Epstein-Barr Virus. Edited by: Robertson E. 2005, Portland: Caister Academic Press, 403-427.
Speck SH, Ganem D: Viral latency and its regulation: lessons from the gamma-herpesviruses. Cell Host Microbe. 2010, 8: 100-115. 10.1016/j.chom.2010.06.014.
Thorley-Lawson DA, Duca KA, Shapiro M: Epstein-Barr virus: a paradigm for persistent infection - for real and in virtual reality. Trends Immunol. 2008, 29: 195-201. 10.1016/j.it.2008.01.006.
Kieff E, Rickinson A: Epstein-Barr Virus and Its Replication. Fields Virology. Edited by: Howley P. 2007, Philadelphia: Lippincott, 2603-2654.
Bornkamm GW, Hammerschmidt W: Molecular virology of Epstein-Barr virus. Philos Trans R Soc Lond B Biol Sci. 2001, 356: 437-459. 10.1098/rstb.2000.0781.
Reisman D, Yates J, Sugden B: A putative origin of replication of plasmids derived from Epstein-Barr virus is composed of two cis-acting components. Mol Cell Biol. 1985, 5: 1822-1832.
Sears J, Ujihara M, Wong S, Ott C, Middeldorp J, Aiyar A: The amino terminus of Epstein-Barr Virus (EBV) nuclear antigen 1 contains AT hooks that facilitate the replication and partitioning of latent EBV genomes by tethering them to cellular chromosomes. J Virol. 2004, 78: 11487-11505. 10.1128/JVI.78.21.11487-11505.2004.
Iwakiri D, Takada K: Role of EBERs in the pathogenesis of EBV infection. Adv Cancer Res. 2010, 107: 119-136. full_text.
Nanbo A, Takada K: The role of Epstein-Barr virus-encoded small RNAs (EBERs) in oncogenesis. Rev Med Virol. 2002, 12: 321-326. 10.1002/rmv.363.
Lo AK, To KF, Lo KW, Lung RW, Hui JW, Liao G, Hayward SD: Modulation of LMP1 protein expression by EBV-encoded microRNAs. Proc Natl Acad Sci USA. 2007, 104: 16164-16169. 10.1073/pnas.0702896104.
Xia T, O'Hara A, Araujo I, Barreto J, Carvalho E, Sapucaia JB, Ramos JC, Luz E, Pedroso C, Manrique M, et al: EBV microRNAs in primary lymphomas and targeting of CXCL-11 by ebv-mir-BHRF1-3. Cancer Res. 2008, 68: 1436-1442. 10.1158/0008-5472.CAN-07-5126.
Takacs M, Banati F, Koroknai A, Segesdi J, Salamon D, Wolf H, Niller HH, Minarovits J: Epigenetic regulation of latent Epstein-Barr virus promoters. Biochim Biophys Acta. 2010, 1799: 228-235.
Chau CM, Lieberman PM: Dynamic chromatin boundaries delineate a latency control region of Epstein-Barr virus. J Virol. 2004, 78: 12308-12319. 10.1128/JVI.78.22.12308-12319.2004.
Day L, Chau CM, Nebozhyn M, Rennekamp AJ, Showe M, Lieberman PM: Chromatin profiling of Epstein-Barr virus latency control region. J Virol. 2007, 81: 6389-6401. 10.1128/JVI.02172-06.
Alazard N, Gruffat H, Hiriart E, Sergeant A, Manet E: Differential hyperacetylation of histones H3 and H4 upon promoter-specific recruitment of EBNA2 in Epstein-Barr virus chromatin. J Virol. 2003, 77: 8166-8172. 10.1128/JVI.77.14.8166-8172.2003.
Tempera I, Wiedmer A, Dheekollu J, Lieberman PM: CTCF prevents the epigenetic drift of EBV latency promoter Qp. PLoS Pathog. 2010, 6: e1001048-10.1371/journal.ppat.1001048. pii
Chau CM, Zhang XY, McMahon SB, Lieberman PM: Regulation of Epstein-Barr virus latency type by the chromatin boundary factor CTCF. J Virol. 2006, 80: 5723-5732. 10.1128/JVI.00025-06.
Yu X, Wang Z, Mertz JE: ZEB1 regulates the latent-lytic switch in infection by Epstein-Barr virus. PLoS Pathog. 2007, 3: e194-10.1371/journal.ppat.0030194.
Kraus RJ, Perrigoue JG, Mertz JE: ZEB negatively regulates the lytic-switch BZLF1 gene promoter of Epstein-Barr virus. J Virol. 2003, 77: 199-207. 10.1128/JVI.77.1.199-207.2003.
Rodriguez A, Jung EJ, Flemington EK: Cell cycle analysis of Epstein-Barr virus-infected cells following treatment with lytic cycle-inducing agents. J Virol. 2001, 75: 4482-4489. 10.1128/JVI.75.10.4482-4489.2001.
Takada K: Cross-linking of cell surface immunoglobulins induces Epstein-Barr virus in Burkitt lymphoma lines. Int J Cancer. 1984, 33: 27-32. 10.1002/ijc.2910330106.
Bhende PM, Dickerson SJ, Sun X, Feng WH, Kenney SC: X-box-binding protein 1 activates lytic Epstein-Barr virus gene expression in combination with protein kinase D. J Virol. 2007, 81: 7363-7370. 10.1128/JVI.00154-07.
Sun CC, Thorley-Lawson DA: Plasma cell-specific transcription factor XBP-1s binds to and transactivates the Epstein-Barr virus BZLF1 promoter. J Virol. 2007, 81: 13566-13577. 10.1128/JVI.01055-07.
Leucci E, Onnis A, Cocco M, De Falco G, Imperatore F, Giuseppina A, Costanzo V, Cerino G, Mannucci S, Cantisani R, et al: B-cell differentiation in EBV-positive Burkitt lymphoma is impaired at posttranscriptional level by miRNA-altered expression. Int J Cancer. 2010, 126: 1316-1326.
Biggin M, Bodescot M, Perricaudet M, Farrell P: Epstein-Barr virus gene expression in P3HR1-superinfected Raji cells. J Virol. 1987, 61: 3120-3132.
Laux G, Freese UK, Fischer R, Polack A, Kofler E, Bornkamm GW: TPA-inducible Epstein-Barr virus genes in Raji cells and their regulation. Virology. 1988, 162: 503-507. 10.1016/0042-6822(88)90496-5.
Adamson AL, Kenney SC: Rescue of the Epstein-Barr virus BZLF1 mutant, Z(S186A), early gene activation defect by the BRLF1 gene product. Virology. 1998, 251: 187-197. 10.1006/viro.1998.9396.
Francis AL, Gradoville L, Miller G: Alteration of a single serine in the basic domain of the Epstein-Barr virus ZEBRA protein separates its functions of transcriptional activation and disruption of latency. J Virol. 1997, 71: 3054-3061.
Rooney CM, Rowe DT, Ragot T, Farrell PJ: The spliced BZLF1 gene of Epstein-Barr virus (EBV) transactivates an early EBV promoter and induces the virus productive cycle. J Virol. 1989, 63: 3109-3116.
Shimizu N, Takada K: Analysis of the BZLF1 promoter of Epstein-Barr virus: identification of an anti-immunoglobulin response sequence. J Virol. 1993, 67: 3240-3245.
Ye J, Gradoville L, Miller G: Cellular immediate-early gene expression occurs kinetically upstream of Epstein-Barr virus bzlf1 and brlf1 following cross-linking of the B cell antigen receptor in the Akata Burkitt lymphoma cell line. J Virol. 2010, 84: 12405-12418. 10.1128/JVI.01415-10.
Reeves MB: Chromatin-mediated regulation of cytomegalovirus gene expression. Virus Res.
Knickelbein JE, Khanna KM, Yee MB, Baty CJ, Kinchington PR, Hendricks RL: Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science. 2008, 322: 268-271. 10.1126/science.1164164.
van Domselaar R, Philippen LE, Quadir R, Wiertz EJ, Kummer JA, Bovenschen N: Noncytotoxic Inhibition of Cytomegalovirus Replication through NK Cell Protease Granzyme M-Mediated Cleavage of Viral Phosphoprotein 71. J Immunol. 2010, 185: 7605-7613. 10.4049/jimmunol.1001503.
Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A: PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J Virol. 2006, 80: 7995-8005. 10.1128/JVI.00734-06.
Wilkinson GW, Kelly C, Sinclair JH, Rickards C: Disruption of PML-associated nuclear bodies mediated by the human cytomegalovirus major immediate early gene product. J Gen Virol. 1998, 79 (Pt 5): 1233-1245.
Korioth F, Maul GG, Plachter B, Stamminger T, Frey J: The nuclear domain 10 (ND10) is disrupted by the human cytomegalovirus gene product IE1. Exp Cell Res. 1996, 229: 155-158. 10.1006/excr.1996.0353.
Adamson AL, Kenney S: Epstein-barr virus immediate-early protein BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J Virol. 2001, 75: 2388-2399. 10.1128/JVI.75.5.2388-2399.2001.
Acknowledgements and Funding
We thank Leanne Olds (University of Wisconsin-Madison) for the figure illustrations and Paul Lieberman (Wistar Institute) for communicating results prior to publication. We also thank Curtis Brandt, Shannon Kenney and Bill Sugden (University of Wisconsin-Madison) for helpful comments and suggestions. Our work described here and the writing of this review was supported by a grant from the National Institutes of Health (NIH) of the United States of America (AI074984). RRP is supported by NIH training grant T32 GM07215-33. RFK is a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
RRP and RFK wrote the manuscript. All authors have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Penkert, R.R., Kalejta, R.F. Tegument protein control of latent herpesvirus establishment and animation. Herpesviridae 2, 3 (2011). https://doi.org/10.1186/2042-4280-2-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2042-4280-2-3