
Abstract
DNA double-strand breaks are among the most serious types of DNA damage and their signaling and repair is critical for all cells and organisms. The repair of both induced and programmed DNA breaks is fundamental as demonstrated by the many human syndromes, neurodegenerative diseases, immunodeficiency and cancer associated with defective repair of these DNA lesions. Homologous recombination and non-homologous end-joining pathways are the two major DNA repair pathways responsible for mediating the repair of DNA double-strand breaks. The signaling of DNA double-strand breaks is critical for cells to orchestrate the repair pathways and maintain genomic integrity. This signaling network is highly regulated and involves a growing number of proteins and elaborated posttranslational modifications including phosphorylation and ubiquitylation. Here, we highlight the recent progress in the signaling of DNA double-strand breaks, the major proteins and posttranslational modifications involved and the diseases and syndromes associated with impaired signaling of these breaks.
Similar content being viewed by others


Background
Mammalian cells and organisms have evolved elegant ways to maintain their genomic integrity and respond to the various DNA lesions that they continuously face. DNA damage can result from exogenous stresses, such as ionizing radiation (IR), ultraviolet (UV) light and chemical compounds, or from endogenous insults such as reactive oxygen species (ROS) and DNA replication errors [1].
DNA double-strand breaks (DSBs) are among the most serious and lethal types of DNA damage, as a single DSB is sufficient to kill a cell or disturb its genomic integrity [1]. DSBs are generated in response to exogenous and endogenous DNA insults. For instance, DSBs are induced in response to oncogenic activation [2]. In human precancerous lesions, oncogene activation has been shown to lead to continuous formation of DNA DSBs [3, 4]. These DSBs activate the tumor suppressor p53 that mediate apoptosis and/or senescence to restrain the growth of the precancerous cells. In the presence of additional mutations that inactivate p53, precancerous cells become cancerous as they escape p53 mediated apoptosis and/or senescence [5, 6]. In addition to the induced DSBs, there are also programmed DSBs that are critical for physiological processes such as meiosis and T and B-cell receptor rearrangements [7, 8].
DNA damage response (DDR) to various types of DNA insults is a well orchestrated process and is required to maintain genomic integrity (Figure 1) [9–12]. In response to DSBs, a signaling process activates cell cycle checkpoints and pauses cell cycle progression, thus granting time for damaged cells to repair their DNA (Figure 2and section 2s) [13]. Two major repair pathways for DSBs exist in mammalian cells; the homologous recombination (HR) and the non-homologous end-joining (NHEJ) pathways [14]. The HR pathway is error free but requires an intact homologous template such as a sister chromatid. The NHEJ recombination pathway is the prominent pathway for DSB repair in mammalian cells; however this pathway is error prone as unlike HR pathway it does not require a long homologous sequence to guide the repair [15]. The choice of HR or NHEJ pathway for repairing DSBs is dependent on the phases of the cell cycle. HR is the main DSB repair pathway used during the S and G2 phase when sister chromatids are intact and readily available, whereas NHEJ is predominant during the G1 phase of the cell cycle [16, 17].

Mammalian DNA damage repair pathways and checkpoints. Various exogenous and endogenous sources can generate damaged DNA. In response to DNA damage cells activate the appropriate DNA damage repair and checkpoint pathways or apoptosis. Lesions of single or double-stranded DNA lead to the activation of cell cycle checkpoints and a number of DNA damage repair pathways including MMR (mismatch repair), BER (base excision repair), NER (nucleotide excision repair), TLS (translesion DNA synthesis), HR (homologous recombination) and NHEJ (non-homologous end joining). Impaired repair of damaged DNA can lead to accumulation of, mutations and genomic instability. In addition, defective repair of damaged DNA can lead to aging as well as predisposition for various genetic diseases including cancer and immunodeficiency.

Schematic representation of the DNA damage-signaling that leads to activation of cell cycle checkpoints or apoptosis. (A) Examples of proteins involved in the different steps of DNA damage signaling are shown. DNA lesions are recognized by sensors (e.g ATM), and mediators (e.g 53BP1) serve to amplify the signaling of DNA damage. Next, proteins including CHK2 serve to transduce the DNA damage signals. Finally, effectors (e.g p53) are required to trigger the appropriate DNA damage cellular responses that include apoptosis, senescence or cell cycle arrest or delay that allow cells to repair their damaged DNA. (B) In response to DSBs and/or DNA replication failure, activated ATM and/or ATR phosphorylate the CHK2 and CHK1. Activated ATM, ATR and CHK2 also phosphorylate p53, thus increasing its stability and activation. Activated p53 transactivates the p21 that inhibits the cyclin-dependent-kinases and delays the G1/S transition. In the case the damage DNA is beyond repair, p53 can promote apoptosis of the damaged cells through the transactivation of its transcriptional targets including the Bax, Puma and Noxa. CHK1 is essential for S and G2/M checkpoints activation. CDC25C inactivation and WEE1 activation through their phosphorylation by CHK1 result in the inhibition of CDC2/cyclin B activity and G2/M arrest. CDC25C dephosphorylates CDC2 leading to its activation [177]. In response to DNA damage, CHK1 phosphorylates CDC25C allowing its interaction with 14-3-3 and inhibition of its phosphatase activity [178, 179]. CHK1 also phosphorylates WEE1, affecting its distribution through interaction with 14-3-3. Finally, CHK1 also phosphorylates CDC25A leading to CDK2 inactivation and delayed intra-S phase [180].
In the situation where cells are unable to repair their DSBs, DDR pathways are activated by kinases such as ATM and ATR, thus leading to the senescence or the death of damaged cells [18–21] (Figure 2). Failure to detect, signal or repair DSBs, can result in damaged cells escaping the cell cycle checkpoints and evading death and thus these damaged cells can potentially generate a progeny that carries harmful mutations or chromosomal aberrations. Such genomic instability is a key driving force for the development of various human syndromes, immunodeficiency, aging and cancer [1, 9].
During the past few years, significant progress has been made toward better understanding of the mechanisms underlying the DSB responses in mammalian cells. Current data have demonstrated the importance of the early detection and signaling of DSBs. This signaling of DSBs has emerged as a highly regulated and complex process. Mutations in a number of genes involved in DSBs signaling have highlighted the importance of this process.
In this review we will focus on proteins that have been demonstrated to play important roles in the detection and signaling of DSBs. We will discuss a number of post-translational modifications (PTMs) including phosphorylation, ubiquitylation, SUMOylation, acetylation and methylation, that are critical for DSBs signaling. Mutations in a number of genes involved in the signaling of DSBs have been demonstrated to lead to various human pathologies including cancer and will be also discussed in this review.
Signaling of DNA Double-Strand Breaks
Role of Phosphorylation and Dephosphorylation in the Signaling of DSBs
Mammalian cells have evolved a sophisticated network of proteins to sense and signal DSBs (Figure 3). Members of the Phosphatidylinositol-3 kinase-related kinases (PIKK) family play important roles in different stages of DSB signaling through their ability to phosphorylate a number of substrates leading to the propagation of DSB signaling [22]. Members of the PIKK family consist of serine/threonine protein kinases with a conserved kinase domain (KD). PIKK family members also show conservation of three other domains that regulate their KD activity. These three domains are the FRAP-ATM-TRRAP (FAT) domain, the PIKK-regulatory domain (PRD) and the FRAP-ATM-TRRAP-C-terminal (FATC) domain [23–25]}. The FATC domain is critical for the kinase activity of the PIKK family members and mutations of this domain reduce the kinase activity of PIKK family members [23, 26]. In addition, the FATC domain was shown to serve protein-protein interactions. The C-terminal PRD is located between the KD and FATC domains and is the target of posttranslational modifications.

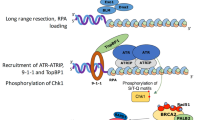
Multistep signaling of DNA double-stranded breaks. ATM and ATR, through the phosphorylation of their downstream substrates, play central roles in DSB signaling and the activation of cell cycle checkpoints. (A) Mre11/Rad50/NBS1 (MRN)-complex mainly recognizes DSBs and undergo ATM activation. Activated ATM phophorylates multiple substrates, including H2A.X and MDC1. MDC1 recognizes γ-H2A.X and bind to it. Then RNF8 recognizes the thyreonyl-phosphorylated MDC1 via its FHA domain and is recruited to DSB sites where it ubiquitylates the histone H2A. Subsequently, RNF168 is recruited to the ubiquitylated H2A, further ubiquitylating it with Ubc13 and likely leading to the modification of the chromatin structure. Through its tutor domain, 53BP1 is recruited to histone H4 di-methylated on lysine 20, while RAP80 and its protein complex are recruited to the polyubiquitylated H2A. (B) In the S and G2 phase, DSBs are recognized by the MRN-complex and CtIP is recruited to DSBs. CtIP is phosphorylated by CDK and ATM, and is ubiquitylated by BRCA1 in response to DNA damage. Collaboration of MRN and CtIP results in DSB end resection, and subsequently DNA2/BLM-complex and Exo1 promote further the resection of DSB ends leading to generation of single strand DNA (ssDNA). RPA binds to the ssDNA and induces ATR activation and HR repair [181–184]. (C) Replication fork stalling. The damaged single-stranded DNA is coated by RPA and this structure plays crucial roles in the recruitment of the helicase HARP and the ATR-ATRIP complex to sites of DNA damage[185–187]. P: phosphorylation, Ub: ubiquitin, S: SUMOylation, ATRIP: ATR interacting protein, RPA: Replication protein A. HARP: HepA-related protein.
Three members of the PIKK family are essential for the response to DSBs (Figure 2). These kinases are Ataxia-Telangiectasia-Mutated (ATM), Ataxia Telangiectasia and Rad3 Related (ATR) and DNA-dependent Protein Kinase catalytic subunit (DNA-PKcs). ATM and DNA-PKcs are critical for the signaling of DSBs while ATR is mainly involved in the response to DNA single strand breaks (SSBs) and stalled replication forks [13, 22, 27]. Remarkably, recent studies indicated that these PIKKs have a large number of substrates, and more than 900 sites of over 700 proteins have been identified as potential phosphorylation sites for ATM and ATR in response to DNA damage [28]. It has been shown that when ATM activity is compromised, DNA-PKcs can compensate to a certain degree for the defective ATM in some situations, such as V(D)J or class switch recombination in lymphocytes [29–31]. However, ATM, DNA-PKcs and ATR functions are all essential as demonstrated by the association of their mutations with a number of human syndromes discussed in section 3.
In mammalian cells, DSB ends are recognized by the MRE11/Rad50/NBS1 (MRN) and the KU70/KU80 complexes that function as sensors of DSBs but are also involved in processing the DNA break ends [32, 33]. The MRN complex recruits ATM, while KU70/80 recruits DNA-PKcs, to DNA lesion sites [34]. The MRE11 (Meiotic Recombination 11) protein is conserved from archaea to mammalian cells and is involved in HR, NHEJ and maintenance of telomeres [35]. MRE11 has single strand endonuclease activity and 3' to 5' DNA exonuclease activity. RAD50 is a homolog of the S. cerevisiae rad50 and is a member of the structural maintenance of chromosome (SMC) protein family [36]. The third partner in the MRN complex is the NBS1 (Nijmegen breakage syndrome 1) protein also known as Nibrin and p95. Interaction of MRE11 with RAD50 upregulates both its exonuclease and endonuclease activities, while its interaction with NBS1 upregulates only its endonuclease activity [37].
The MRN complex interacts with the N-terminal domain of ATM and recruits it to DSBs. The MRN complex is also required for ATM activation[38–41]. In undamaged cells, ATM forms inactive dimers or multimers; however, upon the formation of DSBs, ATM is autophosphorylated on serine 1981 leading to its dissociation into active monomers [25, 42]. In addition to serine 1981, ATM is also autophosphorylated on serine 367 and serine 1893 and mutations of these sites reduce its activity [25, 42, 43]. ATM substrates such as NBS1 are rapidly phosphorylated following ATM autophosphorylation and thus ATM autophosphorylation is considered as a hallmark for the activation of ATM [25, 44]. Autophosphorylation of ATM serine 1981 has been demonstrated to be indispensable for its monomerization and chromatin association at the DSB sites [45]. A recent study demonstrated that ATM autophosphorylation is not required for its initial localization to DSBs, but is important its stabilization at the sites of DSBs [46]. The recent finding that transgenic mice carrying alanine substituted autophosphorylation serine sites 1987, 367 and 1899 of Atm (corresponding to human ATM serine 1981, 367 and 1893) display functional Atm-dependent responses have raised questions regarding the importance of ATM autophosphorylation for its in vivo functions [47].
Three serine/threonine protein phosphatase PP2A, PP5 and WIP1, have been implicated in the control of ATM activation [48–50]. In undamaged cells, PP2A interacts with ATM to ensure that ATM is not inappropriately activated by autophosphorylation [48]. In response to DNA DSBs, PP2A dissociates from ATM, therefore minimizing the risk of competition between phosphorylation and phosphatase activities [48]. The phosphatase WIP1 is also capable of removing phosphates from serine 367 and serine 1981 autophosphorylation sites [49], while PP5 has been shown to interact with ATM and to upregulate its activity in response to DNA damage [50].
In response to DNA damage, activated ATM, or alternatively DNA-PKcs or ATR, rapidly phosphorylates H2A.X, a histone H2A variant, on its serine 139 (γ-H2A.X) [51]. The large number of substrates for activated ATM includes proteins such as the structural maintenance of chromosome-1 (SMC1), NBS1, checkpoint kinase 2 (CHK2), tumor protein 53 (P53), breast cancer early onset-1 (BRCA1) and mediator of DNA damage checkpoint protein-1 (MDC1) [25].
Tyrosine 142 residue of H2A.X is constitutively phosphorylated and its subsequent dephosphorylation in response to DNA damage may enhance MDC1 and ATM recruitment to extend and maintain γ-H2A.X phosphorylation [52]. Phosphorylation and dephosphorylation of H2A.X on its tyrosine 142 is regulated by the tyrosine kinase WSTF (Williams-Beuren syndrome transcription factor, also known as BAZ1B) and the protein phosphatase EYA (Eyes Absent) [53, 54]. Pre-existing H2A.X tyrosine 142 phosphorylation by WSTF is critical for DNA damage induced formation and retention of γ-H2A.X foci after DNA damage, while EYA removes the tyrosine phosphorylation in response to DNA damage. The presence of phosphorylated tyrosine 142 on H2A.X interferes with the MDC1 mediated recruitment of DNA damage signaling and repair factors to phosphorylated serine 139 residue of H2A.X[54].
The elimination of γ-H2A.X at DNA damage sites is required for appropriate DNA damage repair. In mammalian cells, dephosphorylation of serine 139 residue of H2A.X is regulated by the protein phosphatase PP2A and PP4 [55–57]. While PP4 dephosphorylates serine 139 of H2A.X in response to relatively low levels of DNA damage that occur during DNA replication, higher levels of damage might require both PP2A and PP4 to eliminate γ-H2AX [56]. In the absence of the catalytic subunit of PP2A (PP2Ac), γ-H2A.X foci persist and DNA repair is impaired [55]. The effect of PP2A on γ-H2A.X is independent of ATM, ATR and DNA-PKcs. PP4 dephosphorylates γ-H2A.X both at the sites of DNA damage and in undamaged chromatin, and depletion of PP4 catalytic subunit (PP4C) causes prolonged MDC1 retention at the sites of DNA lesions and results in a prolonged checkpoint arrest [57].
MDC1 recognizes γ-H2A.X and binds to it via its tandem BRCA1 C-terminal (BRCT) domains [39]. This recruitment of MDC1 to DSB sites further promotes the accumulation and retention of active ATM and MRN complexes to γ-H2A.X containing chromatin surrounding the sites of DNA damage [38, 39, 58, 59]. The MDC1 serine-aspartic acid-threonine (SDT) repeats near its N-terminus, which are phosphorylated by Caseine Kinase 2 (CK2), interact with the N-terminal fork-head associated (FHA) domain of NBS1 [60–63]. In undamaged cells, MDC1 exists in a complex with MRN [60–63]; however, following ATM activation, MDC1 and its interacting MRN complex are recruited to γ-H2A.X [60–63]. Interestingly, while the N-terminus of NBS1 interacts with MDC1, its C-terminus interacts with ATM. Hence, MDC1 bridges the interaction of MRN to γ-H2A.X and ATM. The concentrated binding of ATM to MDC1 and MRN further promotes the phosphorylation of H2A.X and triggers the accumulation and retention at the sites of DNA lesion of a number of factors essential for DSB signaling including the RING finger proteins RNF8 and RNF168, p53 binding protein 1 (53BP1) and BRCA1 [64].
These examples of phosphorylation/dephosphorylation events highlight the importance of these processes in the signaling of DSBs.
Roles of Ubiquitylation and SUMOylation in the Signal Transduction of DSBs
Recent evidence have demonstrated major roles for ubiquitylation and Sumoylation in the signaling of DSBs [65–68]. Post-translational modifications of proteins by ubiquitin or small ubiquitin-like modifier (SUMO) are ATP-dependent and are mediated by a sequential enzyme cascade that involves ubiquitin or SUMO activating enzymes (E1), conjugation enzymes (E2) and protein ligases (E3) [69–71]. Ubiquitin is activated by the E1 enzyme and then a thioester linkage is formed between the carboxyl terminal glycine of ubiquitin and the reactive cysteine in E1. Subsequently, the ubiquitin is transferred from E1 to the active site cysteine of the conjugation enzyme E2 through a trans-thioesterification reaction. The E2 then interacts with its specific E3 ligase that catalyzes an isopeptide bond between an ε-amino group of a lysine residue in the substrate and the C-terminal glycine of ubiquitin or SUMO, thus completing the post-translational modification.
E3 ligases govern the specificity of ubiquitylated substrates and fall into four major groups; the HECT (Homologous to the E6-AP Carboxyl Terminus) type E3, the RING (Really Interesting New Gene) type E3, the U-box type E3 and the A20-type C2/C2 zinc-finger [72]. In the ubiquitin pathways, one E1 enzyme is shared by all ubiquitin ligases, and the combination of 20 to 30 E2 enzymes and thousands of E3 ligases provide the specificity of the ubiquitylated substrates [71]. Ubiquitin is highly conserved among eukaryotes, and possesses seven lysine (K) residues (K6, K11, K27, K29, K33, K48 and K63) that can be conjugated to other ubiquitins to generate polyubiquitin chains and sometimes mixed-linkage ubiquitin chains [73–76]. A number of ubiquitin modifications including monoubiquitylation, multiubiquitylation and polyubiquitylation serve diverse functions and cellular processes. Monoubiquitylation is involved in a wide variety of cellular processes including membrane protein transport, viral budding, protein localization and DNA damage responses [77–79]. While different types of ubiquitin linkages exist, K48 and K63-linked polyubiquitin chains are the best characterized. K48 mediated polyubiquitylation is known to target substrates for proteasomal degradation [80] whereas K63-linked polyubiquitylation is important for signal transduction during immune responses, cell cycle and DNA damage responses [81–84].
In the SUMOylation pathway, three mammalian SUMO isoforms can be used for covalent modifications of protein substrates. SUMO-1 conjugates only a single SUMO moiety to a protein substrate, while SUMO-2 and SUMO-3 which are similar, can form poly-SUMO chains. SUMO isoforms are initially cleaved by SUMO-specific proteases (SNEPs). Subsequently the cleaved SUMOs are activated by the E1 enzyme that consists of SUMO1 activating enzyme subunit (SAE) 1 and 2 also known as Uba2 and Aos1. SUMO is then transferred to the single E2 enzyme UBC9 and subsequently ligated to the target substrates by a few E3 ligases known as Protein Inhibitor of Activated STAT (PIAS1, 2,3 and 4) [71, 85].
Similar to other cellular processes, different ubiquitin E3 ligases including RNF8, RNF168 and BRCA1/BARD1 have been demonstrated to play central roles in the early signaling of DSBs (Figure 3). RNF8 and RNF168 function in collaboration with the E2 enzyme UBC13 [84], while the ligase BRCA1/BARD1 requires the E2 enzyme Ubch5c for its functions [86].
In response to DSBs, the three conserved threonine/glutamine/X/phenylalanine (T-Q-X-F) clusters of MDC1 are phosphorylated by ATM [28, 87–89]. The threonyl-phosphorylated MDC1 interacts with the FHA domain of the E3 ligase RNF8 and recruits it to DSB sites [87–89]. In addition to its N-terminal FHA domain, RNF8 also contains a C-terminal RING finger domain important for its E3 ligase activities. Once RNF8 is recruited by MDC1 to DSB sites, it collaborates with UBC13 to mediate K63-linked ubiquitylation of the histones H2A and H2A.X at the flanking sites of the DNA lesion [90]. The histone H2A has been previously demonstrated to be mono-ubiquitylated at K119 by the E3 ligases RING1B (RING2/RNF2), RING1A/RING1 and 2A-HUB/hRUL138 [91–95]. In addition, 5 to 15% of H2A is estimated to be constitutively ubiquitylated [96, 97].
RNF8 ubiquitylated H2A serves to recruit the E3 ligase RNF168 through its Motif Interacting with Ubiquitin 2 (MIU2) [98–100]. RNF168 and UBC13 mediate K63-linked polyubiquitination of H2A and this polyubiquitylation has been proposed to modulate the chromatin structure facilitating the recruitment of 53BP1 to DSB sites. K63-linked polyubiquitylation of H2A also serves to recruit Receptor-Associated Protein 80 (RAP80) via its tandem Ubiquitin Interaction Motifs (UIMs) [101–105]. Subsequent to its interaction with ubiquitylated histones, RAP80 facilitates the recruitment to the DSB flanking sites of the BRCA1-A complex that in addition to RAP80, contains BRCA1, BARD1, ABRAXAS/FAM175A, BRCC3/BRCC36, BRE/BRCC45 and MERIT40/NBA1 [101].
Interestingly, the heterodimer BRCA1/BARD1 was reported to form K6-linked ubiquitin chain during DNA replication or repair [106, 107]. BRCA1, in collaboration with BARD1, has been shown to mediate ubiquitylation of several substrates in vitro; however, it has been difficult to identify its in vivo ubiquitylation substrates. γ-tubulin [108], RPB8 [109], Topoisomerase IIα [110], and CtIP [111] are among the established in vivo ubiquitylation substrates for BRCA1.
While the importance of SUMOylation in DNA repair and replication has been demonstrated earlier [71, 112–115], recent studies have highlighted its close relationship with ubiquitylation during DSB signaling [66, 68]. The SUMO E3 ligases PIAS1 and PIAS4 were shown to be recruited to DSBs and to be required for the DSB-induced ubiquitylation mediated by RNF8 and RNF168. PIAS4 is required for the accumulation of RNF168 on DSB sites, through regulation of RNF8 and/or RNF168 E3 ligase activities or through amplification of protein-protein interactions. Furthermore, these studies demonstrated the SUMOylation of RAP80 and indicated that RNF168 SUMOylation is mediated by PIAS4 or PIAS1 [67, 116]. Interestingly, in response to DSBs, PIAS4 also mediates SUMO1 modification of 53BP1 and BRCA1 while SUMO2/3 modification of BRCA1 is also mediated by PIAS1. These studies also indicated that SUMOylation of BRCA1 is critical for its ubiquitin E3 ligase activity [66].
Thus, in addition to phosphorylation, increasing evidence indicates the requirement for other posttranslational modifications including ubiquitylation and SUMOylation for the signaling of DSBs.
DNA Damage Response Proteins and Hereditary Human Diseases
A number of hereditary human diseases/syndromes have been associated with mutations that target genes involved in the signaling of DSBs. Some of these genes and the human diseases and syndromes associated with their mutations are discussed here.
MRE11, RAD50 and NBS1
The MRN complex is recruited to the DNA damage site and activates ATM. In contrast to Atm, knockout mouse models for Mre11, Rad50 and Nbs1 are embryonic lethal [117–119]. Hypomorphic mutant mice for Nbs1 or Mre11 exhibit increased radiosensitivity, defective cell cycle checkpoints, chromosome instability and immunodeficiency [120–123]. In contrast to Nbs1 or Mre11 hypomorphic mutant mice, Rad50 hypomorphic mutants show partial embryonic lethality and exhibit progressive hematopoietic stem cell failure without increased radiosensitivity or defective cell cycle checkpoints [124].
In human, NBS1 mutations have been associated with the Nijmegen breakage syndrome (MIM #251260) characterized by microcephaly, radiosensitivity, growth delay, ovarian dysgenesis, immunodeficiency and marked cancer predisposition [125]. Patients with this syndrome are prone to infectious diseases owing to humoral and cellular immune deficiency. At least 10 different NBS1 mutations, most of them leading to truncated NBS1 proteins or amino-acid substitutions that might affect NBS1 protein-protein interactions, have been reported for patients with the Nijmegen breakage syndrome. These NBS1 mutants with truncations or amino-acid substitutions are likely to retain partial NBS1 activities, thus explaining the milder defects observed in these patients compared to Nbs1 null mutation in mice that leads to embryonic lethality [118, 125].
Hypomorphic mutations of human MRE11 gene result in ataxia telangiectagia like disorder (ATLD; MIM #604391) characterized by cerebellar atrophy and radiosensitivity without marked immunodeficiency, cancer predisposition or telangiectagia [126]. ATLD has slower progression and is a milder condition compared to ataxia telangiectagia (A-T) syndrome that associates with ATM mutations. In contrast to NBS and A-T, patients with ATLD have almost normal immune responses. While the limited number of ATLD patients does not allow determination of whether this syndrome associates with increased cancer risk, MreATLD1/ATLD1 mice exhibited impaired Atm functions but showed no increased cancer susceptibility [121].
RAD50 deficiency (MIM #613078) was reported for a patient with NBS-like disorder characterized by microcephaly, growth retardation and radiosensitivity without marked immunodeficiency or cancer predisposition [127]. Cells from this patient exhibit cellular radio sensitivity and defective activation of cell cycle checkpoints in response to DSBs.
ATM
ATM plays important roles in the responses to induced DSBs as well as to programmed DSBs generated during V(D)J and meiotic recombinations. Atm deficiency in mice results in defective activation of cell cycle checkpoints, impaired rearrangement and expression of T-cell receptors, reduced class switch recombination of immunoglobulins, sterility and tumorigenesis [22, 128–133].
In human, mutations of ATM result in ataxia-telangiectagia (A-T; MIM #208900) characterized by progressive cerebellar ataxia, oculocutaneous telangiectagia, immune defects and lymphoid tumors [134]. In addition, primary immunodeficiency, radiosensitivity and progressive neurodegeneration are hallmarks of patients with A-T. The cause of neurodegeneration in A-T patients remains unknown. While, one-third of A-T patients develop malignancies including lymphoid and breast cancers, some A-T patients also develop insulin-resistant diabetes and metabolic syndromes [25, 135–137].
Somatic mutations of ATM have been also associated with human cancers. Analysis of coding exons of 518 protein kinases in 210 different human cancers, indicated that ATM gene ranked third in terms of mutation frequency, behind Titin and BRAF (B-Raf proto-oncogene serine/threonine-protein kinase) [138].
ATR
In humans, the autosomal recessive disorder known as Seckel syndrome (MIM #210600) is associated with ATR mutation [139, 140]. Features of this syndrome include microcephaly, developmental delay, mental retardation, mild sensitivity to ultraviolet radiation, characteristic facial features but no marked radiosensitivity and immunodeficiency. ATR is a phosphoinositol 3-kinase-like kinase that is activated by single-strand regions of DNA. Several proteins are required for the ATR-signaling response and defects of these proteins can result in Seckel-like clinical features [141]. Thus, haploinsufficiency of replication protein A1 (RPA1) and replication factor C2 (RFC2) has been associated with the Miller-Dieker lissencephaly syndrome (MIM #247200) and the William-Beuren syndrome (MIM #194050), respectively. These syndromes are also characterized by microcephaly, growth retardation and facial abnormality [142, 143].
In contrast to Atm, null Atr mutation leads to early embryonic lethality in mice [144]. Interestingly, a mouse model carrying the ATR mutation (A2101G) associated with the Seckel syndrome in patients, recapitulated the symptoms of the human disease [145].
DNA-PKcs
DNA-PKcs-null mice are viable but exhibit radiosensitivity, immunodeficiency, and hyperplasia and dysplasia of the intestinal mucosa. However, these mutant mice are not growth retarded [146].
In humans, DNA-PKcs mutations result in radiosensitive severe combined immunodeficiency (RS-SCID) characterized by radiosensitivity and immunodeficiency without microcephaly and mental retardation [147]. The B-cell development defects of patients with this RS-SCID are reminiscent of Artemis deficient SCID patients and RAG (recombination activating genes) deficient SCID patients [148, 149]. No increased cancer predisposition has been reported for patients with the RS-SCID associated with DNA-PKcs mutations.
RNF168
The recently identified RIDDLE syndrome (MIM #611943) has been associated with homozygous RNF168 mutations [99, 150]. This syndrome is very rare as only one patient has been identified to date. Characteristics of the RIDDLE syndrome include radiosensitivity, immunodeficiency, dysmorphic features and learning difficulties. The identified RIDDLE patient has been reported to have low concentrations of serum immunoglobulin while the number of his peripheral lymphocytes remained within normal ranges. Analysis of skin fibroblasts from this patient showed a mildly increased radiosensitivity, defective intra-S and G2/M phase checkpoints, and impaired recruitment of 53BP1, RAP80 and BRCA1 to DSB sites. More patients with the RIDDLE syndrome are needed to determine the spectrum of pathologies associated with this syndrome and whether it is associated with increased cancer predisposition.
Mouse models carrying mutations of Rnf8, an E3 ligase required for the recruitment of Rnf168 to sites of DNA damage, have been reported. While no human syndrome/disease have been reported yet to associate with RNF8 mutations, Rnf8-/- mice, similar to the RIDDLE syndrome patient, suffer from defective IgH Class Switch Recombination and are immunodeficient [151, 152]. Rnf8-/- males also exhibit impaired spermatogenesis and are infertile [151–153]. Remarkably, Rnf8-/- mice are growth retarded and display increased cancer predisposition, demonstrating that Rnf8 is a novel tumor suppressor [151]. Studies of mouse models for Rnf168 mutations are required to determine whether they reproduce characteristics of the RIDDLE syndrome and whether, similar to Rnf8, Rnf168 plays a role in cancer.
BRCA1
BRCA1 is important for a number of cellular functions including activation of cell cycle checkpoints and repair of DSBs through HR pathway [154]. Other functions that have been ascribed to BRCA1 include transcription, ubiquitylation, estrogen receptor signaling, and chromatin remodeling [155–160].
Mutations of the tumor suppressor BRCA1 (MIM #113705) predispose women for breast and ovarian cancer [161, 162]. Typically, a germline mutation in one of the BRCA1 alleles is inherited from one of the parents and loss of heterozygosity (LOH) of the Wild-type BRCA1 allele is required in order for tumors to develop. Women with inherited inactivating mutation of BRCA1 gene have about a 65-80% lifetime risk of developing breast cancer [163]. The familial breast cancer associated with BRCA1 inactivation develops at younger ages and is frequently bilateral compared to the sporadic types of breast cancer. In addition, women with inherited mutations of BRCA1 gene are also predisposed for ovarian cancer with a lifetime risk about 37-62% compared to less than 2% for women that do not carry BRCA1 or BRCA2 mutations [163]. BRCA1 forms an E3 ligase with its partner BARD1 and its RING finger domain is essential for this E3 ligase activity. Mutation of cysteine 61 in the RING finger domain of BRCA1 (BRCA1C61G) has been observed in patients with the familial breast cancer.
A remarkable therapeutic opportunity has been discovered recently for cancer patients that carry mutations of the familial breast cancer genes BRCA1 or BRCA2. Both genes play important roles in the HR repair pathway. Inactivation of either BRCA1 or BRCA2 has been shown to drastically decreases HR activities [164, 165]. Interestingly, recent studies indicated that inhibition of the poly (ADP-ribose) polymerase (PARP) leads to synthetic lethality of tumors deficient for either BRCA1 or BRCA2 [166–169]. PARP1 and PARP2 facilitate the repair of SSBs by recruiting DNA repair proteins to the damaged sites and promoting the restart of stalled replication forks [170]. In the absence of PARP activity, SSBs can be converted to DSBs during replication. As BRCA1 and BRCA2 deficient tumors are defective in HR mediated repair of DSBs, inactivation of PARP in these tumors leads to collapsed replication folks, generation of DSBs, cell cycle arrest and cell death [166, 167]. Several PARP inhibitors, including the third generation PARP inhibitors with high potency and specificity, are in clinical trial development worldwide [170]. There is currently great hope that these PARP inhibitors may represent a great opportunity to improve the therapy for BRCA1 and BRCA2 cancer patients.
Several mouse models for Brca1 mutation have been reported[171]. While Brca1 null mutations lead to early embryonic lethality [172], targeted Brca1 mutations to mammary epithelial cells lead to mammary tumorigenesis [173, 174]. In addition, inactivation of the Chk2-p53 pathway was found to synergize the development of mammary tumors associated with Brca1 deficiency [174].
Interestingly, recent studies demonstrated that inactivation of 53bp1 rescued the proliferation defect and hypersensitivity to DNA damaging agents, and partially restored the HR defects of Brca1 deficient cells [175, 176]. Inactivation of 53bp1 suppressed the development of mammary tumors in mice carrying hypomorphic Brca1 mutations (Brca1Δ11/Δ11 ) [175]. 53BP1 expression was recently found frequently decreased in a subset of basal-like/triple-negative breast cancers and in BRCA1 or BRCA2 negative breast tumors [176]. Further investigations are needed to clarify the precise effects of 53BP1 inactivation on the development of BRCA1 associated breast cancers, the responses of these cancers to therapies and the mechanisms and significance of the reduced 53BP1 expression in breast tumors with BRCA1 or BRCA2 mutations.
Conclusion
Remarkable progress has been made over the past few years regarding the signaling and repair mechanisms of DNA double-strand breaks. Post-translational modifications have emerged as key factors that regulate these processes. Although more studies are required to identify missing players in these repair/signaling pathways and the mechanisms that control these processes, it is likely that the considerable amount of knowledge accumulated over the years will facilitate the development of better therapies for human diseases including cancer.
References
Jackson SP, Bartek J: The DNA-damage response in human biology and disease. Nature. 2009, 461 (7267): 1071-1078. 10.1038/nature08467.
Halazonetis TD, Gorgoulis VG, Bartek J: An oncogene-induced DNA damage model for cancer development. Science. 2008, 319 (5868): 1352-1355. 10.1126/science.1140735.
Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Kastrinakis NG, Levy B, et al: Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005, 434 (7035): 907-913. 10.1038/nature03485.
Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al: DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005, 434 (7035): 864-870. 10.1038/nature03482.
Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al: Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006, 444 (7119): 633-637. 10.1038/nature05268.
Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, et al: Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006, 444 (7119): 638-642. 10.1038/nature05327.
Soulas-Sprauel P, Rivera-Munoz P, Malivert L, Le Guyader G, Abramowski V, Revy P, de Villartay JP: V(D)J and immunoglobulin class switch recombinations: a paradigm to study the regulation of DNA end-joining. Oncogene. 2007, 26 (56): 7780-7791. 10.1038/sj.onc.1210875.
Longhese MP, Bonetti D, Guerini I, Manfrini N, Clerici M: DNA double-strand breaks in meiosis: checking their formation, processing and repair. DNA Repair (Amst). 2009, 8 (9): 1127-1138. 10.1016/j.dnarep.2009.04.005.
Hakem R: DNA-damage repair; the good, the bad, and the ugly. Embo J. 2008, 27 (4): 589-605. 10.1038/emboj.2008.15.
Rouse J, Jackson SP: Interfaces between the detection, signaling, and repair of DNA damage. Science. 2002, 297 (5581): 547-551. 10.1126/science.1074740.
Harper JW, Elledge SJ: The DNA damage response: ten years after. Mol Cell. 2007, 28 (5): 739-745. 10.1016/j.molcel.2007.11.015.
Harrison JC, Haber JE: Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet. 2006, 40: 209-235. 10.1146/annurev.genet.40.051206.105231.
Bartek J, Lukas J: DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol. 2007, 19 (2): 238-245. 10.1016/j.ceb.2007.02.009.
Shrivastav M, De Haro LP, Nickoloff JA: Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18 (1): 134-147. 10.1038/cr.2007.111.
Guirouilh-Barbat J, Huck S, Bertrand P, Pirzio L, Desmaze C, Sabatier L, Lopez BS: Impact of the KU80 pathway on NHEJ-induced genome rearrangements in mammalian cells. Mol Cell. 2004, 14 (5): 611-623. 10.1016/j.molcel.2004.05.008.
Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A, Takeda S: Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. Embo J. 1998, 17 (18): 5497-5508. 10.1093/emboj/17.18.5497.
Delacote F, Lopez BS: Importance of the cell cycle phase for the choice of the appropriate DSB repair pathway, for genome stability maintenance: the trans-S double-strand break repair model. Cell Cycle. 2008, 7 (1): 33-38. 10.4161/cc.7.1.5149.
Zhivotovsky B, Kroemer G: Apoptosis and genomic instability. Nat Rev Mol Cell Biol. 2004, 5 (9): 752-762. 10.1038/nrm1443.
Bartek J, Lukas J, Bartkova J: DNA damage response as an anti-cancer barrier: damage threshold and the concept of 'conditional haploinsufficiency'. Cell Cycle. 2007, 6 (19): 2344-2347. 10.4161/cc.6.19.4754.
Mallette FA, Ferbeyre G: The DNA damage signaling pathway connects oncogenic stress to cellular senescence. Cell Cycle. 2007, 6 (15): 1831-1836. 10.4161/cc.6.15.4516.
Mallette FA, Gaumont-Leclerc MF, Ferbeyre G: The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007, 21 (1): 43-48. 10.1101/gad.1487307.
Shiloh Y: ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003, 3 (3): 155-168. 10.1038/nrc1011.
Lempiainen H, Halazonetis TD: Emerging common themes in regulation of PIKKs and PI3Ks. Embo J. 2009, 28 (20): 3067-3073. 10.1038/emboj.2009.281.
Mordes DA, Glick GG, Zhao R, Cortez D: TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev. 2008, 22 (11): 1478-1489. 10.1101/gad.1666208.
Lavin MF: Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008, 9 (10): 759-769. 10.1038/nrm2514.
Sun Y, Jiang X, Chen S, Fernandes N, Price BD: A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci USA. 2005, 102 (37): 13182-13187. 10.1073/pnas.0504211102.
Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP: ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006, 8 (1): 37-45. 10.1038/ncb1337.
Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al: ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007, 316 (5828): 1160-1166. 10.1126/science.1140321.
Callen E, Jankovic M, Wong N, Zha S, Chen HT, Difilippantonio S, Di Virgilio M, Heidkamp G, Alt FW, Nussenzweig A, et al: Essential role for DNA-PKcs in DNA double-strand break repair and apoptosis in ATM-deficient lymphocytes. Mol Cell. 2009, 34 (3): 285-297. 10.1016/j.molcel.2009.04.025.
Tomimatsu N, Mukherjee B, Burma S: Distinct roles of ATR and DNA-PKcs in triggering DNA damage responses in ATM-deficient cells. EMBO Rep. 2009, 10 (6): 629-635. 10.1038/embor.2009.60.
Bredemeyer AL, Huang CY, Walker LM, Bassing CH, Sleckman BP: Aberrant V(D)J recombination in ataxia telangiectasia mutated-deficient lymphocytes is dependent on nonhomologous DNA end joining. J Immunol. 2008, 181 (4): 2620-2625.
Difilippantonio S, Celeste A, Fernandez-Capetillo O, Chen HT, Reina San Martin B, Van Laethem F, Yang YP, Petukhova GV, Eckhaus M, Feigenbaum L, et al: Role of Nbs1 in the activation of the Atm kinase revealed in humanized mouse models. Nat Cell Biol. 2005, 7 (7): 675-685. 10.1038/ncb1270.
Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD: The Mre11 complex is required for ATM activation and the G2/M checkpoint. Embo J. 2003, 22 (24): 6610-6620. 10.1093/emboj/cdg630.
Mordes DA, Cortez D: Activation of ATR and related PIKKs. Cell Cycle. 2008, 7 (18): 2809-2812.
Dimitrova N, de Lange T: Cell cycle-dependent role of MRN at dysfunctional telomeres: ATM signaling-dependent induction of nonhomologous end joining (NHEJ) in G1 and resection-mediated inhibition of NHEJ in G2. Mol Cell Biol. 2009, 29 (20): 5552-5563. 10.1128/MCB.00476-09.
Kinoshita E, van der Linden E, Sanchez H, Wyman C: RAD50, an SMC family member with multiple roles in DNA break repair: how does ATP affect function?. Chromosome Res. 2009, 17 (2): 277-288. 10.1007/s10577-008-9018-6.
Williams RS, Williams JS, Tainer JA: Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem Cell Biol. 2007, 85 (4): 509-520. 10.1139/O07-069.
Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ: MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003, 421 (6926): 961-966. 10.1038/nature01446.
Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP: MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005, 123 (7): 1213-1226. 10.1016/j.cell.2005.09.038.
Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y: Requirement of the MRN complex for ATM activation by DNA damage. Embo J. 2003, 22 (20): 5612-5621. 10.1093/emboj/cdg541.
Lee JH, Paull TT: ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005, 308 (5721): 551-554. 10.1126/science.1108297.
Bakkenist CJ, Kastan MB: DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003, 421 (6922): 499-506. 10.1038/nature01368.
Kozlov SV, Graham ME, Peng C, Chen P, Robinson PJ, Lavin MF: Involvement of novel autophosphorylation sites in ATM activation. Embo J. 2006, 25 (15): 3504-3514. 10.1038/sj.emboj.7601231.
Kastan MB, Lim DS, Kim ST, Yang D: ATM--a key determinant of multiple cellular responses to irradiation. Acta Oncol. 2001, 40 (6): 686-688. 10.1080/02841860152619089.
Berkovich E, Monnat RJ, Kastan MB: Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol. 2007, 9 (6): 683-690. 10.1038/ncb1599.
So S, Davis AJ, Chen DJ: Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J Cell Biol. 2009, 187 (7): 977-990. 10.1083/jcb.200906064.
Daniel JA, Pellegrini M, Lee JH, Paull TT, Feigenbaum L, Nussenzweig A: Multiple autophosphorylation sites are dispensable for murine ATM activation in vivo. J Cell Biol. 2008, 183 (5): 777-783. 10.1083/jcb.200805154.
Goodarzi AA, Jonnalagadda JC, Douglas P, Young D, Ye R, Moorhead GB, Lees-Miller SP, Khanna KK: Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. Embo J. 2004, 23 (22): 4451-4461. 10.1038/sj.emboj.7600455.
Shreeram S, Demidov ON, Hee WK, Yamaguchi H, Onishi N, Kek C, Timofeev ON, Dudgeon C, Fornace AJ, Anderson CW, et al: Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol Cell. 2006, 23 (5): 757-764. 10.1016/j.molcel.2006.07.010.
Ali A, Zhang J, Bao S, Liu I, Otterness D, Dean NM, Abraham RT, Wang XF: Requirement of protein phosphatase 5 in DNA-damage-induced ATM activation. Genes Dev. 2004, 18 (3): 249-254. 10.1101/gad.1176004.
Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM: DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998, 273 (10): 5858-5868. 10.1074/jbc.273.10.5858.
Xiao A, Li H, Shechter D, Ahn SH, Fabrizio LA, Erdjument-Bromage H, Ishibe-Murakami S, Wang B, Tempst P, Hofmann K, et al: WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature. 2009, 457 (7225): 57-62. 10.1038/nature07668.
Krishnan N, Jeong DG, Jung SK, Ryu SE, Xiao A, Allis CD, Kim SJ, Tonks NK: Dephosphorylation of the C-terminal tyrosyl residue of the DNA damage-related histone H2A.X is mediated by the protein phosphatase eyes absent. J Biol Chem. 2009, 284 (24): 16066-16070. 10.1074/jbc.C900032200.
Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG: Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009, 458 (7238): 591-596. 10.1038/nature07849.
Chowdhury D, Keogh MC, Ishii H, Peterson CL, Buratowski S, Lieberman J: gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol Cell. 2005, 20 (5): 801-809. 10.1016/j.molcel.2005.10.003.
Chowdhury D, Xu X, Zhong X, Ahmed F, Zhong J, Liao J, Dykxhoorn DM, Weinstock DM, Pfeifer GP, Lieberman J: A PP4-phosphatase complex dephosphorylates gamma-H2AX generated during DNA replication. Mol Cell. 2008, 31 (1): 33-46. 10.1016/j.molcel.2008.05.016.
Nakada S, Chen GI, Gingras AC, Durocher D: PP4 is a gamma H2AX phosphatase required for recovery from the DNA damage checkpoint. EMBO Rep. 2008, 9 (10): 1019-1026. 10.1038/embor.2008.162.
Goldberg M, Stucki M, Falck J, D'Amours D, Rahman D, Pappin D, Bartek J, Jackson SP: MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature. 2003, 421 (6926): 952-956. 10.1038/nature01445.
Lukas C, Melander F, Stucki M, Falck J, Bekker-Jensen S, Goldberg M, Lerenthal Y, Jackson SP, Bartek J, Lukas J: Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. Embo J. 2004, 23 (13): 2674-2683. 10.1038/sj.emboj.7600269.
Chapman JR, Jackson SP: Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep. 2008, 9 (8): 795-801. 10.1038/embor.2008.103.
Melander F, Bekker-Jensen S, Falck J, Bartek J, Mailand N, Lukas J: Phosphorylation of SDT repeats in the MDC1 N terminus triggers retention of NBS1 at the DNA damage-modified chromatin. J Cell Biol. 2008, 181 (2): 213-226. 10.1083/jcb.200708210.
Spycher C, Miller ES, Townsend K, Pavic L, Morrice NA, Janscak P, Stewart GS, Stucki M: Constitutive phosphorylation of MDC1 physically links the MRE11-RAD50-NBS1 complex to damaged chromatin. J Cell Biol. 2008, 181 (2): 227-240. 10.1083/jcb.200709008.
Wu L, Luo K, Lou Z, Chen J: MDC1 regulates intra-S-phase checkpoint by targeting NBS1 to DNA double-strand breaks. Proc Natl Acad Sci USA. 2008, 105 (32): 11200-11205. 10.1073/pnas.0802885105.
Soutoglou E, Misteli T: Activation of the cellular DNA damage response in the absence of DNA lesions. Science. 2008, 320 (5882): 1507-1510. 10.1126/science.1159051.
Bartek J, Hodny Z: SUMO boosts the DNA damage response barrier against cancer. Cancer Cell. 2010, 17 (1): 9-11. 10.1016/j.ccr.2009.12.030.
Morris JR, Boutell C, Keppler M, Densham R, Weekes D, Alamshah A, Butler L, Galanty Y, Pangon L, Kiuchi T, et al: The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature. 2009, 462 (7275): 886-890. 10.1038/nature08593.
Morris JR: SUMO in the mammalian response to DNA damage. Biochem Soc Trans. 2010, 38 (Pt 1): 92-97. 10.1042/BST0380092.
Galanty Y, Belotserkovskaya R, Coates J, Polo S, Miller KM, Jackson SP: Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature. 2009, 462 (7275): 935-939. 10.1038/nature08657.
Hershko A: Ubiquitin: roles in protein modification and breakdown. Cell. 1983, 34 (1): 11-12. 10.1016/0092-8674(83)90131-9.
Kerscher O, Felberbaum R, Hochstrasser M: Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 2006, 22: 159-180. 10.1146/annurev.cellbio.22.010605.093503.
Bergink S, Jentsch S: Principles of ubiquitin and SUMO modifications in DNA repair. Nature. 2009, 458 (7237): 461-467. 10.1038/nature07963.
Wertz IE, Dixit VM: Regulation of death receptor signaling by the ubiquitin system. Cell Death Differ. 17 (1): 14-24. 10.1038/cdd.2009.168.
Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi SP: A proteomics approach to understanding protein ubiquitination. Nat Biotechnol. 2003, 21 (8): 921-926. 10.1038/nbt849.
Xu P, Peng J: Characterization of polyubiquitin chain structure by middle-down mass spectrometry. Anal Chem. 2008, 80 (9): 3438-3444. 10.1021/ac800016w.
Kirisako T, Kamei K, Murata S, Kato M, Fukumoto H, Kanie M, Sano S, Tokunaga F, Tanaka K, Iwai K: A ubiquitin ligase complex assembles linear polyubiquitin chains. Embo J. 2006, 25 (20): 4877-4887. 10.1038/sj.emboj.7601360.
Kim HT, Kim KP, Lledias F, Kisselev AF, Scaglione KM, Skowyra D, Gygi SP, Goldberg AL: Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J Biol Chem. 2007, 282 (24): 17375-17386. 10.1074/jbc.M609659200.
Hicke L: Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol. 2001, 2 (3): 195-201. 10.1038/35056583.
Di Fiore PP, Polo S, Hofmann K: When ubiquitin meets ubiquitin receptors: a signalling connection. Nat Rev Mol Cell Biol. 2003, 4 (6): 491-497. 10.1038/nrm1124.
Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I: Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat Cell Biol. 2003, 5 (5): 461-466. 10.1038/ncb983.
Hershko A, Ciechanover A: The ubiquitin system. Annu Rev Biochem. 1998, 67: 425-479. 10.1146/annurev.biochem.67.1.425.
Haglund K, Dikic I: Ubiquitylation and cell signaling. Embo J. 2005, 24 (19): 3353-3359. 10.1038/sj.emboj.7600808.
Hayden MS, Ghosh S: Shared principles in NF-kappaB signaling. Cell. 2008, 132 (3): 344-362. 10.1016/j.cell.2008.01.020.
Bennett EJ, Harper JW: DNA damage: ubiquitin marks the spot. Nat Struct Mol Biol. 2008, 15 (1): 20-22. 10.1038/nsmb0108-20.
Panier S, Durocher D: Regulatory ubiquitylation in response to DNA double-strand breaks. DNA Repair (Amst). 2009, 8 (4): 436-443. 10.1016/j.dnarep.2009.01.013.
Rytinki MM, Kaikkonen S, Pehkonen P, Jaaskelainen T, Palvimo JJ: PIAS proteins: pleiotropic interactors associated with SUMO. Cell Mol Life Sci. 2009, 66 (18): 3029-3041. 10.1007/s00018-009-0061-z.
Polanowska J, Martin JS, Garcia-Muse T, Petalcorin MI, Boulton SJ: A conserved pathway to activate BRCA1-dependent ubiquitylation at DNA damage sites. Embo J. 2006, 25 (10): 2178-2188. 10.1038/sj.emboj.7601102.
Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, et al: Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007, 318 (5856): 1637-1640. 10.1126/science.1150034.
Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J: RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007, 131 (5): 887-900. 10.1016/j.cell.2007.09.040.
Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J: RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007, 131 (5): 901-914. 10.1016/j.cell.2007.09.041.
Brooks L, Heimsath EG, Loring GL, Brenner C: FHA-RING ubiquitin ligases in cell division cycle control. Cell Mol Life Sci. 2008, 65 (21): 3458-3466. 10.1007/s00018-008-8220-1.
de Napoles M, Mermoud JE, Wakao R, Tang YA, Endoh M, Appanah R, Nesterova TB, Silva J, Otte AP, Vidal M, et al: Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and × inactivation. Dev Cell. 2004, 7 (5): 663-676. 10.1016/j.devcel.2004.10.005.
Fang J, Chen T, Chadwick B, Li E, Zhang Y: Ring1b-mediated H2A ubiquitination associates with inactive X chromosomes and is involved in initiation of X inactivation. J Biol Chem. 2004, 279 (51): 52812-52815. 10.1074/jbc.C400493200.
Cao R, Tsukada Y, Zhang Y: Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell. 2005, 20 (6): 845-854. 10.1016/j.molcel.2005.12.002.
Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y: Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004, 431 (7010): 873-878. 10.1038/nature02985.
Zhou W, Zhu P, Wang J, Pascual G, Ohgi KA, Lozach J, Glass CK, Rosenfeld MG: Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol Cell. 2008, 29 (1): 69-80. 10.1016/j.molcel.2007.11.002.
Matsui SI, Seon BK, Sandberg AA: Disappearance of a structural chromatin protein A24 in mitosis: implications for molecular basis of chromatin condensation. Proc Natl Acad Sci USA. 1979, 76 (12): 6386-6390. 10.1073/pnas.76.12.6386.
Goldknopf IL, Sudhakar S, Rosenbaum F, Busch H: Timing of ubiquitin synthesis and conjugation into protein A24 during the HeLa cell cycle. Biochem Biophys Res Commun. 1980, 95 (3): 1253-1260. 10.1016/0006-291X(80)91608-3.
Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, et al: RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009, 136 (3): 435-446. 10.1016/j.cell.2008.12.041.
Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, Nakada S, Ylanko J, Olivarius S, Mendez M, et al: The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009, 136 (3): 420-434. 10.1016/j.cell.2008.12.042.
Pinato S, Scandiuzzi C, Arnaudo N, Citterio E, Gaudino G, Penengo L: RNF168, a new RING finger, MIU-containing protein that modifies chromatin by ubiquitination of histones H2A and H2AX. BMC Mol Biol. 2009, 10: 55-10.1186/1471-2199-10-55.
Wang B, Elledge SJ: Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc Natl Acad Sci USA. 2007, 104 (52): 20759-20763. 10.1073/pnas.0710061104.
Wang B, Hurov K, Hofmann K, Elledge SJ: NBA1, a new player in the Brca1 A complex, is required for DNA damage resistance and checkpoint control. Genes Dev. 2009, 23 (6): 729-739. 10.1101/gad.1770309.
Feng L, Huang J, Chen J: MERIT40 facilitates BRCA1 localization and DNA damage repair. Genes Dev. 2009, 23 (6): 719-728. 10.1101/gad.1770609.
Shao G, Lilli DR, Patterson-Fortin J, Coleman KA, Morrissey DE, Greenberg RA: The Rap80-BRCC36 de-ubiquitinating enzyme complex antagonizes RNF8-Ubc13-dependent ubiquitination events at DNA double strand breaks. Proc Natl Acad Sci USA. 2009, 106 (9): 3166-3171. 10.1073/pnas.0807485106.
Wu J, Huen MS, Lu LY, Ye L, Dou Y, Ljungman M, Chen J, Yu X: Histone ubiquitination associates with BRCA1-dependent DNA damage response. Mol Cell Biol. 2009, 29 (3): 849-860. 10.1128/MCB.01302-08.
Wu-Baer F, Lagrazon K, Yuan W, Baer R: The BRCA1/BARD1 heterodimer assembles polyubiquitin chains through an unconventional linkage involving lysine residue K6 of ubiquitin. J Biol Chem. 2003, 278 (37): 34743-34746. 10.1074/jbc.C300249200.
Morris JR, Pangon L, Boutell C, Katagiri T, Keep NH, Solomon E: Genetic analysis of BRCA1 ubiquitin ligase activity and its relationship to breast cancer susceptibility. Hum Mol Genet. 2006, 15 (4): 599-606. 10.1093/hmg/ddi476.
Starita LM, Machida Y, Sankaran S, Elias JE, Griffin K, Schlegel BP, Gygi SP, Parvin JD: BRCA1-dependent ubiquitination of gamma-tubulin regulates centrosome number. Mol Cell Biol. 2004, 24 (19): 8457-8466. 10.1128/MCB.24.19.8457-8466.2004.
Wu W, Nishikawa H, Hayami R, Sato K, Honda A, Aratani S, Nakajima T, Fukuda M, Ohta T: BRCA1 ubiquitinates RPB8 in response to DNA damage. Cancer Res. 2007, 67 (3): 951-958. 10.1158/0008-5472.CAN-06-3187.
Lou Z, Minter-Dykhouse K, Chen J: BRCA1 participates in DNA decatenation. Nat Struct Mol Biol. 2005, 12 (7): 589-593. 10.1038/nsmb953.
Yu X, Fu S, Lai M, Baer R, Chen J: BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev. 2006, 20 (13): 1721-1726. 10.1101/gad.1431006.
Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S: RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002, 419 (6903): 135-141. 10.1038/nature00991.
Arakawa H, Moldovan GL, Saribasak H, Saribasak NN, Jentsch S, Buerstedde JM: A role for PCNA ubiquitination in immunoglobulin hypermutation. PLoS Biol. 2006, 4 (11): e366-10.1371/journal.pbio.0040366.
Frampton J, Irmisch A, Green CM, Neiss A, Trickey M, Ulrich HD, Furuya K, Watts FZ, Carr AM, Lehmann AR: Postreplication repair and PCNA modification in Schizosaccharomyces pombe. Mol Biol Cell. 2006, 17 (7): 2976-2985. 10.1091/mbc.E05-11-1008.
Leach CA, Michael WM: Ubiquitin/SUMO modification of PCNA promotes replication fork progression in Xenopus laevis egg extracts. J Cell Biol. 2005, 171 (6): 947-954. 10.1083/jcb.200508100.
Yan J, Yang XP, Kim YS, Joo JH, Jetten AM: RAP80 interacts with the SUMO-conjugating enzyme UBC9 and is a novel target for sumoylation. Biochem Biophys Res Commun. 2007, 362 (1): 132-138. 10.1016/j.bbrc.2007.07.158.
Luo G, Yao MS, Bender CF, Mills M, Bladl AR, Bradley A, Petrini JH: Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc Natl Acad Sci USA. 1999, 96 (13): 7376-7381. 10.1073/pnas.96.13.7376.
Zhu J, Petersen S, Tessarollo L, Nussenzweig A: Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr Biol. 2001, 11 (2): 105-109. 10.1016/S0960-9822(01)00019-7.
Buis J, Wu Y, Deng Y, Leddon J, Westfield G, Eckersdorff M, Sekiguchi JM, Chang S, Ferguson DO: Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell. 2008, 135 (1): 85-96. 10.1016/j.cell.2008.08.015.
Williams BR, Mirzoeva OK, Morgan WF, Lin J, Dunnick W, Petrini JH: A murine model of Nijmegen breakage syndrome. Curr Biol. 2002, 12 (8): 648-653. 10.1016/S0960-9822(02)00763-7.
Theunissen JW, Kaplan MI, Hunt PA, Williams BR, Ferguson DO, Alt FW, Petrini JH: Checkpoint failure and chromosomal instability without lymphomagenesis in Mre11(ATLD1/ATLD1) mice. Mol Cell. 2003, 12 (6): 1511-1523. 10.1016/S1097-2765(03)00455-6.
Reina-San-Martin B, Nussenzweig MC, Nussenzweig A, Difilippantonio S: Genomic instability, endoreduplication, and diminished Ig class-switch recombination in B cells lacking Nbs1. Proc Natl Acad Sci USA. 2005, 102 (5): 1590-1595. 10.1073/pnas.0406289102.
Dinkelmann M, Spehalski E, Stoneham T, Buis J, Wu Y, Sekiguchi JM, Ferguson DO: Multiple functions of MRN in end-joining pathways during isotype class switching. Nat Struct Mol Biol. 2009, 16 (8): 808-813. 10.1038/nsmb.1639.
Bender CF, Sikes ML, Sullivan R, Huye LE, Le Beau MM, Roth DB, Mirzoeva OK, Oltz EM, Petrini JH: Cancer predisposition and hematopoietic failure in Rad50(S/S) mice. Genes Dev. 2002, 16 (17): 2237-2251. 10.1101/gad.1007902.
Digweed M, Sperling K: Nijmegen breakage syndrome: clinical manifestation of defective response to DNA double-strand breaks. DNA Repair (Amst). 2004, 3 (8-9): 1207-1217. 10.1016/j.dnarep.2004.03.004.
Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, Raams A, Byrd PJ, Petrini JH, Taylor AM: The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999, 99 (6): 577-587. 10.1016/S0092-8674(00)81547-0.
Waltes R, Kalb R, Gatei M, Kijas AW, Stumm M, Sobeck A, Wieland B, Varon R, Lerenthal Y, Lavin MF, et al: Human RAD50 deficiency in a Nijmegen breakage syndrome-like disorder. Am J Hum Genet. 2009, 84 (5): 605-616. 10.1016/j.ajhg.2009.04.010.
Lavin MF, Kozlov S: DNA damage-induced signalling in ataxia-telangiectasia and related syndromes. Radiother Oncol. 2007, 83 (3): 231-237. 10.1016/j.radonc.2007.04.032.
Vacchio MS, Olaru A, Livak F, Hodes RJ: ATM deficiency impairs thymocyte maturation because of defective resolution of T cell receptor alpha locus coding end breaks. Proc Natl Acad Sci USA. 2007, 104 (15): 6323-6328. 10.1073/pnas.0611222104.
Reina-San-Martin B, Chen HT, Nussenzweig A, Nussenzweig MC: ATM is required for efficient recombination between immunoglobulin switch regions. J Exp Med. 2004, 200 (9): 1103-1110. 10.1084/jem.20041162.
Lumsden JM, McCarty T, Petiniot LK, Shen R, Barlow C, Wynn TA, Morse HC, Gearhart PJ, Wynshaw-Boris A, Max EE, et al: Immunoglobulin class switch recombination is impaired in Atm-deficient mice. J Exp Med. 2004, 200 (9): 1111-1121. 10.1084/jem.20041074.
Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D: Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev. 1996, 10 (19): 2411-2422. 10.1101/gad.10.19.2411.
Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, et al: Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996, 86 (1): 159-171. 10.1016/S0092-8674(00)80086-0.
Chun HH, Gatti RA: Ataxia-telangiectasia, an evolving phenotype. DNA Repair (Amst). 2004, 3 (8-9): 1187-1196. 10.1016/j.dnarep.2004.04.010.
Schalch DS, McFarlin DE, Barlow MH: An unusual form of diabetes mellitus in ataxia telangiectasia. N Engl J Med. 1970, 282 (25): 1396-1402. 10.1056/NEJM197006182822503.
Bar RS, Levis WR, Rechler MM, Harrison LC, Siebert C, Podskalny J, Roth J, Muggeo M: Extreme insulin resistance in ataxia telangiectasia: defect in affinity of insulin receptors. N Engl J Med. 1978, 298 (21): 1164-1171. 10.1056/NEJM197805252982103.
Schneider JG, Finck BN, Ren J, Standley KN, Takagi M, Maclean KH, Bernal-Mizrachi C, Muslin AJ, Kastan MB, Semenkovich CF: ATM-dependent suppression of stress signaling reduces vascular disease in metabolic syndrome. Cell Metab. 2006, 4 (5): 377-389. 10.1016/j.cmet.2006.10.002.
Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al: Patterns of somatic mutation in human cancer genomes. Nature. 2007, 446 (7132): 153-158. 10.1038/nature05610.
Majewski F, Goecke T: Studies of microcephalic primordial dwarfism I: approach to a delineation of the Seckel syndrome. Am J Med Genet. 1982, 12 (1): 7-21. 10.1002/ajmg.1320120103.
O'Driscoll M, Gennery AR, Seidel J, Concannon P, Jeggo PA: An overview of three new disorders associated with genetic instability: LIG4 syndrome, RS-SCID and ATR-Seckel syndrome. DNA Repair (Amst). 2004, 3 (8-9): 1227-1235. 10.1016/j.dnarep.2004.03.025.
O'Driscoll M, Dobyns WB, van Hagen JM, Jeggo PA: Cellular and clinical impact of haploinsufficiency for genes involved in ATR signaling. Am J Hum Genet. 2007, 81 (1): 77-86. 10.1086/518696.
Cardoso C, Leventer RJ, Ward HL, Toyo-Oka K, Chung J, Gross A, Martin CL, Allanson J, Pilz DT, Olney AH, et al: Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3. Am J Hum Genet. 2003, 72 (4): 918-930. 10.1086/374320.
Wu YQ, Sutton VR, Nickerson E, Lupski JR, Potocki L, Korenberg JR, Greenberg F, Tassabehji M, Shaffer LG: Delineation of the common critical region in Williams syndrome and clinical correlation of growth, heart defects, ethnicity, and parental origin. Am J Med Genet. 1998, 78 (1): 82-89. 10.1002/(SICI)1096-8628(19980616)78:1<82::AID-AJMG17>3.0.CO;2-K.
Brown EJ, Baltimore D: ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000, 14 (4): 397-402.
Murga M, Bunting S, Montana MF, Soria R, Mulero F, Canamero M, Lee Y, McKinnon PJ, Nussenzweig A, Fernandez-Capetillo O: A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat Genet. 2009, 41 (8): 891-898. 10.1038/ng.420.
Kurimasa A, Ouyang H, Dong LJ, Wang S, Li X, Cordon-Cardo C, Chen DJ, Li GC: Catalytic subunit of DNA-dependent protein kinase: impact on lymphocyte development and tumorigenesis. Proc Natl Acad Sci USA. 1999, 96 (4): 1403-1408. 10.1073/pnas.96.4.1403.
van der Burg M, Ijspeert H, Verkaik NS, Turul T, Wiegant WW, Morotomi-Yano K, Mari PO, Tezcan I, Chen DJ, Zdzienicka MZ, et al: A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous end-joining. J Clin Invest. 2009, 119 (1): 91-98.
Noordzij JG, de Bruin-Versteeg S, Verkaik NS, Vossen JM, de Groot R, Bernatowska E, Langerak AW, van Gent DC, van Dongen JJ: The immunophenotypic and immunogenotypic B-cell differentiation arrest in bone marrow of RAG-deficient SCID patients corresponds to residual recombination activities of mutated RAG proteins. Blood. 2002, 100 (6): 2145-2152.
Noordzij JG, Verkaik NS, van der Burg M, van Veelen LR, de Bruin-Versteeg S, Wiegant W, Vossen JM, Weemaes CM, de Groot R, Zdzienicka MZ, et al: Radiosensitive SCID patients with Artemis gene mutations show a complete B-cell differentiation arrest at the pre-B-cell receptor checkpoint in bone marrow. Blood. 2003, 101 (4): 1446-1452. 10.1182/blood-2002-01-0187.
Stewart GS, Stankovic T, Byrd PJ, Wechsler T, Miller ES, Huissoon A, Drayson MT, West SC, Elledge SJ, Taylor AM: RIDDLE immunodeficiency syndrome is linked to defects in 53BP1-mediated DNA damage signaling. Proc Natl Acad Sci USA. 2007, 104 (43): 16910-16915. 10.1073/pnas.0708408104.
Li L, Halaby MJ, Hakem A, Cardoso R, El Ghamrasni S, Harding S, Chan N, Bristow R, Sanchez O, Durocher D, et al: Rnf8 deficiency impairs class switch recombination, spermatogenesis, and genomic integrity and predisposes for cancer. J Exp Med. 2010, 207 (5): 983-997. 10.1084/jem.20092437.
Santos MA, Huen MS, Jankovic M, Chen HT, Lopez-Contreras AJ, Klein IA, Wong N, Barbancho JL, Fernandez-Capetillo O, Nussenzweig MC, et al: Class switching and meiotic defects in mice lacking the E3 ubiquitin ligase RNF8. J Exp Med. 2010
Lu LY, Wu J, Ye L, Gavrilina GB, Saunders TL, Yu X: RNF8-dependent histone modifications regulate nucleosome removal during spermatogenesis. Dev Cell. 2010, 18 (3): 371-384. 10.1016/j.devcel.2010.01.010.
Huen MS, Sy SM, Chen J: BRCA1 and its toolbox for the maintenance of genome integrity. Nat Rev Mol Cell Biol. 2010, 11 (2): 138-148. 10.1038/nrm2831.
Rosen EM, Fan S, Ma Y: BRCA1 regulation of transcription. Cancer Lett. 2006, 236 (2): 175-185. 10.1016/j.canlet.2005.04.037.
Hashizume R, Fukuda M, Maeda I, Nishikawa H, Oyake D, Yabuki Y, Ogata H, Ohta T: The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J Biol Chem. 2001, 276 (18): 14537-14540. 10.1074/jbc.C000881200.
Xia Y, Pao GM, Chen HW, Verma IM, Hunter T: Enhancement of BRCA1 E3 ubiquitin ligase activity through direct interaction with the BARD1 protein. J Biol Chem. 2003, 278 (7): 5255-5263. 10.1074/jbc.M204591200.
Starita LM, Parvin JD: The multiple nuclear functions of BRCA1: transcription, ubiquitination and DNA repair. Curr Opin Cell Biol. 2003, 15 (3): 345-350. 10.1016/S0955-0674(03)00042-5.
Fan S, Ma YX, Wang C, Yuan RQ, Meng Q, Wang JA, Erdos M, Goldberg ID, Webb P, Kushner PJ, et al: Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene. 2001, 20 (1): 77-87. 10.1038/sj.onc.1204073.
Fan S, Wang J, Yuan R, Ma Y, Meng Q, Erdos MR, Pestell RG, Yuan F, Auborn KJ, Goldberg ID, et al: BRCA1 inhibition of estrogen receptor signaling in transfected cells. Science. 1999, 284 (5418): 1354-1356. 10.1126/science.284.5418.1354.
King MC, Marks JH, Mandell JB: Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003, 302 (5645): 643-646. 10.1126/science.1088759.
Narod SA, Foulkes WD: BRCA1 and BRCA2: 1994 and beyond. Nat Rev Cancer. 2004, 4 (9): 665-676. 10.1038/nrc1431.
Balmana J, Diez O, Castiglione M: BRCA in breast cancer: ESMO clinical recommendations. Ann Oncol. 2009, 20 (Suppl 4): 19-20.
Moynahan ME, Chiu JW, Koller BH, Jasin M: Brca1 controls homology-directed DNA repair. Mol Cell. 1999, 4 (4): 511-518. 10.1016/S1097-2765(00)80202-6.
Moynahan ME, Cui TY, Jasin M: Homology-directed dna repair, mitomycin-c resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res. 2001, 61 (12): 4842-4850.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T: Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005, 434 (7035): 913-917. 10.1038/nature03443.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al: Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005, 434 (7035): 917-921. 10.1038/nature03445.
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, et al: Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009, 361 (2): 123-134. 10.1056/NEJMoa0900212.
Lord CJ, Ashworth A: Targeted therapy for cancer using PARP inhibitors. Curr Opin Pharmacol. 2008, 8 (4): 363-369. 10.1016/j.coph.2008.06.016.
Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG: PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010, 10 (4): 293-301. 10.1038/nrc2812.
Evers B, Jonkers J: Mouse models of BRCA1 and BRCA2 deficiency: past lessons, current understanding and future prospects. Oncogene. 2006, 25 (43): 5885-5897. 10.1038/sj.onc.1209871.
Hakem R, de la Pompa JL, Sirard C, Mo R, Woo M, Hakem A, Wakeham A, Potter J, Reitmair A, Billia F, et al: The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell. 1996, 85 (7): 1009-1023. 10.1016/S0092-8674(00)81302-1.
Xu X, Wagner KU, Larson D, Weaver Z, Li C, Ried T, Hennighausen L, Wynshaw-Boris A, Deng CX: Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet. 1999, 22 (1): 37-43. 10.1038/8743.
McPherson JP, Lemmers B, Hirao A, Hakem A, Abraham J, Migon E, Matysiak-Zablocki E, Tamblyn L, Sanchez-Sweatman O, Khokha R, et al: Collaboration of Brca1 and Chk2 in tumorigenesis. Genes Dev. 2004, 18 (10): 1144-1153. 10.1101/gad.1192704.
Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al: 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010, 141 (2): 243-254. 10.1016/j.cell.2010.03.012.
Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al: 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010, 17: 688-695. 10.1038/nsmb.1831.
O'Connell MJ, Cimprich KA: G2 damage checkpoints: what is the turn-on?. J Cell Sci. 2005, 118 (Pt 1): 1-6. 10.1242/jcs.01626.
Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H: Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997, 277 (5331): 1501-1505. 10.1126/science.277.5331.1501.
Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ: Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997, 277 (5331): 1497-1501. 10.1126/science.277.5331.1497.
Mailand N, Falck J, Lukas C, Syljuasen RG, Welcker M, Bartek J, Lukas J: Rapid destruction of human Cdc25A in response to DNA damage. Science. 2000, 288 (5470): 1425-1429. 10.1126/science.288.5470.1425.
You Z, Bailis JM: DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol. 2010, 20 (7): 402-409. 10.1016/j.tcb.2010.04.002.
Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP: Human CtIP promotes DNA end resection. Nature. 2007, 450 (7169): 509-514. 10.1038/nature06337.
Takeda S, Nakamura K, Taniguchi Y, Paull TT: Ctp1/CtIP and the MRN complex collaborate in the initial steps of homologous recombination. Mol Cell. 2007, 28 (3): 351-352. 10.1016/j.molcel.2007.10.016.
You Z, Bailis JM, Johnson SA, Dilworth SM, Hunter T: Rapid activation of ATM on DNA flanking double-strand breaks. Nat Cell Biol. 2007, 9 (11): 1311-1318. 10.1038/ncb1651.
Zou L, Elledge SJ: Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003, 300 (5625): 1542-1548. 10.1126/science.1083430.
Shechter D, Costanzo V, Gautier J: Regulation of DNA replication by ATR: signaling in response to DNA intermediates. DNA Repair (Amst). 2004, 3 (8-9): 901-908. 10.1016/j.dnarep.2004.03.020.
Yang XH, Zou L: Recruitment of ATR-ATRIP, Rad17, and 9-1-1 complexes to DNA damage. Methods Enzymol. 2006, 409: 118-131. 10.1016/S0076-6879(05)09007-5.
Acknowledgements
We thank MJ. Halaby, A. Hakem and J. Li for helpful discussions. The Hakem laboratory is supported by the Cancer Research Society, the Canadian Institute of Health Research, the Canadian Cancer Society, and the Ontario MOHLTC.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
TB, MB and RH wrote the manuscript. All authors read and approved the final manuscript.
Toshiyuki Bohgaki, Miyuki Bohgaki contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Bohgaki, T., Bohgaki, M. & Hakem, R. DNA double-strand break signaling and human disorders. Genome Integrity 1, 15 (2010). https://doi.org/10.1186/2041-9414-1-15
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2041-9414-1-15