
Abstract
Background
Targeted re-sequencing is one of the most powerful and widely used strategies for population genetics studies because it allows an unbiased screening for variation that is suitable for a wide variety of organisms. Examples of studies that require re-sequencing data are evolutionary inferences, epidemiological studies designed to capture rare polymorphisms responsible for complex traits and screenings for mutations in families and small populations with high incidences of specific genetic diseases. Despite the advent of next-generation sequencing technologies, Sanger sequencing is still the most popular approach in population genetics studies because of the widespread availability of automatic sequencers based on capillary electrophoresis and because it is still less prone to sequencing errors, which is critical in population genetics studies. Two popular software applications for re-sequencing studies are Phred-Phrap-Consed-Polyphred, which performs base calling, alignment, graphical edition and genotype calling and DNAsp, which performs a set of population genetics analyses. These independent tools are the start and end points of basic analyses. In between the use of these tools, there is a set of basic but error-prone tasks to be performed with re-sequencing data.
Results
In order to assist with these intermediate tasks, we developed a pipeline that facilitates data handling typical of re-sequencing studies. Our pipeline: (1) consolidates different outputs produced by distinct Phred-Phrap-Consed contigs sharing a reference sequence; (2) checks for genotyping inconsistencies; (3) reformats genotyping data produced by Polyphred into a matrix of genotypes with individuals as rows and segregating sites as columns; (4) prepares input files for haplotype inferences using the popular software PHASE; and (5) handles PHASE output files that contain only polymorphic sites to reconstruct the inferred haplotypes including polymorphic and monomorphic sites as required by population genetics software for re-sequencing data such as DNAsp.
Conclusion
We tested the pipeline in re-sequencing studies of haploid and diploid data in humans, plants, animals and microorganisms and observed that it allowed a substantial decrease in the time required for sequencing analyses, as well as being a more controlled process that eliminates several classes of error that may occur when handling datasets. The pipeline is also useful for investigators using other tools for sequencing and population genetics analyses.
Similar content being viewed by others

Background
Targeted re-sequencing is one of the most powerful and widely used strategies for population genetics studies because it allows screening of variation in a way that is unbiased in respect to the allele frequency spectrum and because it is suitable for a wide variety of living organisms. Although there is a plethora of new opportunities from next-generation sequencing (NGS) technologies [1], re-sequencing studies are traditionally performed using Sanger DNA sequencing. This is due, in part, to the widespread availability of automatic sequencers based on capillary electrophoresis and also to the fact that Sanger sequencing is still less prone to base-calling errors [2], which is critical in population genetics studies for which the accurate identification of substitutions carried by unique chromosomes (singletons) is highly informative [3]. Examples of studies in different areas of genetics that require re-sequencing data are: (a) inferences of past demographic parameters of populations of humans [4, 5], animals [6], plants [7] and microorganisms [8], and of the action of natural selection based on ascertainment-bias-free allelic spectra [9–12]; (b) epidemiological studies designed to capture rare polymorphisms responsible for complex traits [13–15]; (c) screening for variation in populations that are not included in public databases such as HapMap, to optimally select informative single nucleotide polymorphism SNPs (tag-SNPs) for association studies [16]; (d) forensic studies or analyses based on mitochondrial DNA data [17, 18]; and (e) screenings for mutations in families or small populations with high incidences of specific genetic diseases [19]. Two of the most popular, powerful and freely available tools for re-sequencing studies are (1) the software package Phred-Phrap-Consed-Polyphred (PPCP) [20–24] that performs base calling, alignment, graphical edition and polymorphism identification and (2) the DNA Sequence Polymorphism software (DNAsp) [25], which performs a wide set of population genetics analyses through a user-friendly Windows interface. As these tools were created by different groups, they are not integrated, despite their wide combined use. Frequently, they are the start and end points of basic analyses for many population genetics re-sequencing studies. In between the use of these tools, there are a set of basic but error-prone tasks to be performed with re-sequencing data. In order to facilitate these tasks, we developed and tested a pipeline that improves the handling of sequencing data. Our pipeline was created with the wide community of investigators using PPCP and DNAsp in mind but it is also useful for investigators who use other population genetics packages, such as VariScan [26], the command-line-based version of DNAsp that is designed for large-scale datasets. Forthcoming versions of our pipeline will be integrated with forthcoming Phred-Phrap functions to analyse NGS data and with other computationally robust population genetics tools, such as the libsequence library (http://molpopgen.org/software/libsequence.html[27]).
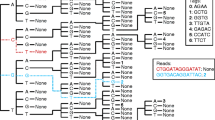
We assume the case of an investigator who is partially or totally re-sequencing a specific genomic region in a set of individuals and that a reference sequence is available for this targeted region (Figure 1). After experimentally obtaining the re-sequencing data (usually with a minimal individual coverage of 2× using forward and reverse primers), the sequencing analyses are performed with software such as PPC. For our purposes (population genetics studies), we define a contig as set of aligned sequences obtained from a set of individuals using the same sequencing primer or a pair of forward/reverse sequencing primers (Figure 1) with a minimum individual coverage of 2× for each sequenced base. In conjunction with PPC, Polyphred is frequently used to automatically identify polymorphic sites and to call genotypes for each read but, in our experience [9, 28–30], visual inspection of peaks is necessary to ensure high quality data. After data production and application of quality control (QC) filters (for example, based on Phred scores), the following information should be available for entry into the pipeline: (1) the sequenced regions defined by their coordinates with respect to the reference sequence; and (2) for these regions, the coordinates of the observed segregating sites and their observed genotypes for each read. The pipeline assumes that this information is available in the output format of Polyphred (the Polyphred output file generated for each contig).

Overview of the re-sequencing data analysis process integrated with the pipeline. An example of the entire process of re-sequencing analysis for a specific genomic region from a set of individuals. (A) Re-sequencing steps, base calling, alignment and heterozygous site identification for an entire region sharing a reference sequence. (B) PolyPhred output files of discontinuous sub-regions, such as exons, re-sequenced independently. (C) Consolidation of different exons of a gene that were independently amplified and re-sequenced using the same reference genomic sequence. (D) Files formats that can be handled by the pipeline either as input or output files.
Method
Design and building
The pipeline was developed as an online system using the Perl programming language for handling dynamic scripting. The current version runs on a Linux/Apache Web server. In order to guarantee portability and accessibility, the system was fully tested in different operating systems and web browsers (see Availability and requirements section).
An overview of the web-based system's architecture is shown in Figure 2. The arrows represent the flow of data and controls across the system's modules (boxes in Figure 2) and are labelled according to their order of execution. The system starts by receiving the user's choice of start and end points for the pipeline which represent, respectively, the type of input file that the user has and the format into which the user wants to transform the original file. In accordance with the combination of these start and end points, the system determines the input files (module 'Determine Input') that the user needs to provide in order to complete the chosen path through the pipeline. The required input files are presented to the user as a Web page tailored by the 'Generate HTML' module. The user can then upload the input files that he or she wants to convert to the format required for a specific population genetics program. These files are received by the system's 'Coordination Module', which controls the execution of all required steps through the pipeline, including a verification step (the 'Verification module') for checking whether the provided input files are in their correct formats. Depending on the combination chosen by the user for start and end points, different scripts are invoked by the 'Coordination Module' (as illustrated in Figure 2). Each script has a specific functionality that is related to a determined file transformation procedure. It is important to note that the modular design of the system's architecture is intended to facilitate future extensions of the pipeline to include other functionalities.

The system's background operation and user interaction. The arrows represent the flow of data and controls across the system's modules (boxes) and are labelled according to their order of execution. The system starts by receiving the user's choice of start and end points for the pipeline which represent, respectively, the type of input file that the user has and the format into which the user wants to transform the original file. In accordance with the combination of these start and end points, the system determines the input files (module 'Determine Input') that the user needs to provide in order to complete the chosen path through the pipeline. The required input files are presented to the user as a Web page tailored by the 'Generate HTML' module. The user can then upload the input files that need to be converted to the format required for a specific population genetics program. These files are received by the system's 'Coordination Module', which controls the execution of all required steps through the pipeline, including a verification step (the 'Verification module') for checking whether the provided input files are in their correct formats. Depending on the combination chosen by the user for start and end points, different scripts are invoked by the 'Coordination Module'. These scripts generate outputs that are presented to the user through the 'Format Output Module'.
Web interface
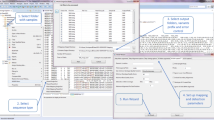
The system's external shell, behind which lies the described architecture, is the web interface illustrated in Figure 3. The grey rectangles in Figure 3 represent the steps of the pipeline that are not automated, such as PHASE and DNAsp executions. The light coloured rectangles represent the modules or functionalities provided by the pipeline, which can be combined in order to reach the desired output. The user-friendly interface allows the user to select the desired start and end points of the pipeline by clicking within the rectangles (or modules) composing the pipeline. Whenever the user clicks on one of the rectangles, a brief explanation of the type of input file that it accepts and the output file that it generates is shown. The system then indicates the input files that need to be provided by the user in order to run the chosen path through the pipeline. This is performed dynamically depending on the user's choice of start and end points. After the selection of the start and end points, no user intervention is needed until the final output is presented.

A snapshot of the main pipeline's Web interface. The user-friendly interface allows the user to select the desired input and output file formats by clicking within the rectangles (modules) composing the pipeline. The Web page includes a brief introduction to the pipeline with access to end-user documentation and a description of the basic steps needed to run the pipeline. A contact email address is provided in order to guarantee permanent user support.
Results
The web interface of the pipeline is shown in Figure 3. The pipeline allows the procedures described below to be performed using a web page with a graphical and user-friendly interface http://www.cebio.org/pipelineldgh. Step 1 integrates different outputs produced by different PPCP contigs that share a reference sequence (Figure 1B). For instance, this step can combine different exons of a gene that have been independently amplified and re-sequenced, so that they might be analysed using a shared reference genomic sequence (Figure 1C). Step 2 reformats genotypes from reads in Polyphred output file format into a user-friendly rectangular matrix of genotypes with individuals as rows and segregating sites as columns (for example, SDAT format; Figure 1D). In this step, the pipeline consolidates reads from the same individuals (sharing the same identifier) by checking for genotype inconsistencies among different reads of the same individual (for example, forward and reverse reads of the same amplicon). In the case of diploid data, if the investigator prefers to infer haplotypes using the popular software PHASE [31], which requires multiple runs with specific parameters, the pipeline prepares the input files for PHASE (Step 3; Figure 1D). PHASE output files contain the inferred haplotypes for each individual but only include the segregating sites. For some population genetics analyses using re-sequencing data (for example, DNAsp), it is necessary to reconstruct the entire sequence, including both monomorphic and polymorphic sites. Step 4 of the pipeline uses the reference sequence and the information from PHASE output files (positions of segregating sites in relation to the reference sequences and inferred haplotypes for each individual) to reconstruct for the targeted region the two DNA sequences corresponding to the two inferred haplotypes of each individual. The pipeline generates a FASTA file that may be used as input for DNAsp or other population genetics tools (Figure 1D).
Discussion
The following are features of the pipeline that deserve additional commentary.
Data production and the use of the pipeline
There are different experimental approaches to the generation data for a re-sequencing population genetics project. It is possible to continuously re-sequence an entire region or to target specific discontinuous subregions, such as exons (Figure 1A). In order to achieve these goals, different strategies that combine polymerase chain reaction (PCR) and re-sequencing are available. For instance, it is possible to amplify regions of ~400-600 bps that will be independently re-sequenced [32]. It is also possible to amplify larger regions consisting of a few kilobases by long-PCR [28] and to perform more than one re-sequencing reaction on each amplicon. In our experience, independent of the wet-lab strategy, two procedures are advisable to analyse the sequencing data. First, we recommend the use of a unique reference sequence for the entire genomic region, which allows unambiguous determination of the position of variable sites independently of their position on each read. Second, each set of reads that is re-sequenced using the same sequencing primers (or with forward and reverse primers) should be aligned separately (such as, in different Phrap-Consed contigs). These procedures minimize the mix of good and bad quality calls for a specific position in the same contig, which facilitates both automatic and visual genotype calls.
When using PPCP to analyse reads in small- to medium-scale re-sequencing studies, we perform visual verification of the chromatograms. Although Polyphred genotype calls are very useful, the process is prone to mistakes, particularly for heterozygous genotypes. We observed that this miscalling happens in around 2.5% of genotype calls (in 15% of the inferred SNPs), considering good quality reads (phred scores > 30) and data generated with Applied Biosystems BigDye v.3.1 and run in a 3730 or 3100 Applied Biosystems sequencer (calculated from unpublished data from ETS and SJC on the basis of ~7 Mb re-sequenced in a population genetics study). For this reason, we visually check all Consed chromatogram peaks that are both monomorphic (called by Phred) and polymorphic (called by Polyphred).
Haploid data
Our pipeline was developed keeping in mind the more general case of diploid data. However, it may be easily used with haploid data. There is an option to specify if the data to be analysed are haploid or diploid and conveniently adapting outputs to this information. We recommend that users interested in analysing haploid data follow the same procedures specified for the analysis of diploid data, assuming that all genotypes are homozygous.
Haplotype inferences using PHASE
Although the latest version of DNAsp (v. 5.0) incorporates the algorithm implemented in the PHASE software [31], investigators may prefer running PHASE separately for several reasons: the need to use different parameters for burn-in and length of the runs; the possibility of performing the computationally demanding haplotype inferences in a more powerful computer; or the preference for the PHASE for Linux/Unix platforms, which bypasses the limitations of the Windows version. We developed the pipeline with the user who prefers to run PHASE separately in mind. However, for large datasets, inferences using PHASE may be computationally prohibitive. In this case, a faster, although less accurate method, was implemented using the software fastPHASE [33]. As input files for fastPHASE and PHASE are the same, our pipeline is compatible with both programs.
QC procedures of the pipeline
In order to save time preparing input files, our pipeline has a set of QC procedures that are executed before any of the file formatting steps is performed. This includes the identification of inconsistent genotype calls for different reads of the same individuals and the verification of the different input/output files' formats.
Future developments
We will continue to expand the functions of our pipeline, so that it will include: (a) reformatting of SDAT files to generate a Haploview input file for linkage disequilibrium analysis; (b) the option of reformatting files in both directions (for example, being able to generate the Polyphred output from the SDAT format; and (c) the possibility of generating either the SDAT file format or the DNAsp FASTA file for diploid organisms using the International Union of Pure and Applied Chemistry ambiguity nomenclature for heterozygous genotypes.
Conclusions
Our pipeline is designed to handle re-sequencing data and is complementary to resources such as FORMATOMATIC [34] and CONVERT http://www.agriculture.purdue.edu/fnr/html/faculty/rhodes/students%20and%20staff/glaubitz/software.htm, which are useful for analysing microsatellites and SNPs but not for sequencing data. We tested our pipeline with several users who were performing re-sequencing studies of haploid and diploid loci in humans, plants, animals and microorganisms. We verified that our pipeline is robust and substantially decreases the time required for re-sequencing data analyses. Also, our pipeline allows for a more controlled process that eliminates several classes of error that may occur in population genetics, epidemiological, clinical and forensic studies when handling such data.
Availability and requirements
The sequencing pipeline is available at http://www.cebio.org/pipelineldgh.
The web-based system will be freely available for academic purposes.
Operating systems: Windows, 32-bit Linux, 64-bit Linux, MAC-OS.
Programming languages: Perl, HTML and JavaScript.
Browsers: Internet Explorer (Windows), Firefox (Linux, Windows), Safari (MAC-OS)
Abbreviations
- DNAsp:
-
DNA sequence polymorphism
- NGS:
-
next generation sequencing
- PCR:
-
polymerase chain reaction
- PPCP:
-
Phred-Phrap-Consed-Polyphred
- QC:
-
quality control
- SNP:
-
single nucleotide polymorphism.
References
Mardis ER, Wilson RK: Cancer genome sequencing: a review. Human Molec Genetics. 2009, 18: R163-R168. 10.1093/hmg/ddp396.
Harismendy O, Ng PC, Strausberg RL, Wang XY, Stockwell TB, Beeson KY, Schork NJ, Murray SS, Topol EJ, Levy S, Frazer KA: Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009, 10: R32-10.1186/gb-2009-10-3-r32.
Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD: Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. Plos Genetics. 2009, 5: e1000695-10.1371/journal.pgen.1000695.
Fagundes NJR, Ray N, Beaumont M, Neuenschwander S, Salzano FM, Bonatto SL, Excoffier L: Statistical evaluation of alternative models of human evolution. Proc Natl Acad Sci USA. 2007, 104: 17614-17619. 10.1073/pnas.0708280104.
Nielsen R, Hubisz MJ, Hellmann I, Torgerson D, Andres AM, Albrechtsen A, Gutenkunst R, Adams MD, Cargill M, Boyko A: Darwinian and demographic forces affecting human protein coding genes. Genome Res. 2009, 19: 838-849. 10.1101/gr.088336.108.
Vargas SM, Araujo FCF, Monteiro DS, Estima SC, Almeida AP, Soares LS, Santos FR: Genetic diversity and origin of leatherback turtles (Dermochelys coriacea) from the Brazilian coast. J Heredity. 2008, 99: 215-220. 10.1093/jhered/esm120.
Novaes RML, De Lemos JP, Ribeiro RA, Lovato MB: Phylogeography of Plathymenia reticulata (Leguminosae) reveals patterns of recent range expansion towards northeastern Brazil and southern Cerrados in Eastern Tropical South America. Molec Ecology. 2010, 19: 985-998. 10.1111/j.1365-294X.2010.04530.x.
Grynberg P, Fontes CJF, Hughes AL, Braga EM: Polymorphism at the apical membrane antigen 1 locus reflects the world population history of Plasmodium vivax. BMC Evol Biol. 2008, 8: 123-10.1186/1471-2148-8-123.
Tarazona-Santos E, Fabbri C, Yeager M, Magalhães WCS, Burdett L, Crenshaw A, Pettener D, Chanock SJ: Diversity in the glucose transporter-4 gene (SLC2A4) in Humans reflects the action of natural selection along the old-world primates evolution. PloS One. 2010, 5: e9827-10.1371/journal.pone.0009827.
Nielsen R, Williamson S, Kim Y, Hubisz MJ, Clark AG, Bustamante C: Genomic scans for selective sweeps using SNP data. Genome Res. 2005, 15: 1566-1575. 10.1101/gr.4252305.
Andres AM, Hubisz MJ, Indap A, Torgerson DG, Degenhardt JD, Boyko AR, Gutenkunst RN, White TJ, Green ED, Bustamante CD: Targets of balancing selection in the human genome. Molec Biol Evol. 2009, 26: 2755-2764. 10.1093/molbev/msp190.
Fuselli S, de Filippo C, Mona S, Sistonen J, Fariselli P, Destro-Bisol G, Barbujani G, Bertorelle G, Sajantila A: Evolution of detoxifying systems: the role of environment and population history in shaping genetic diversity at human CYP2D6 locus. Pharmacogen Genomics. 2010, 20: 485-499. 10.1097/FPC.0b013e32833bba25.
Parikh H, Deng ZM, Yeager M, Boland J, Matthews C, Jia JP, Collins I, White A, Burdett L, Hutchinson A: A comprehensive resequence analysis of the KLK15-KLK3-KLK2 locus on chromosome 19q13.33. Human Genetics. 2010, 127: 91-99. 10.1007/s00439-009-0751-5.
Petersen GM, Amundadottir L, Fuchs CS, Kraft P, Stolzenberg-Solomon RZ, Jacobs KB, Arslan AA, Bueno-de-Mesquita HB, Gallinger S, Gross M: A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nature Genetics. 2010, 42: 224-U229. 10.1038/ng.522.
Bhangale TR, Rieder MJ, Nickerson DA: Estimating coverage and power for genetic association studies using near-complete variation data. Nature Genetics. 2008, 40: 841-843. 10.1038/ng.180.
Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA: Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Human Genetics. 2004, 74: 106-120. 10.1086/381000.
Budowle B, Ge JY, Aranda XG, Planz JV, Eisenberg AJ, Chakraborty R: Texas population substructure and its impact on estimating the rarity of Y STR haplotypes from DNA evidence. J Forensic Sci. 2009, 54: 1016-1021. 10.1111/j.1556-4029.2009.01105.x.
Budowle B, van Daal A: Extracting evidence from forensic DNA analyses: future molecular biology directions. Biotechniques. 2009, 46: 339-40. 10.2144/000113136.
Souza CP, Valadares ER, Trindade ALC, Rocha VL, Oliveira LR, Godard ALB: Mutation in intron 5 of GTP cyclohydrolase 1 gene causes dopa-responsive dystonia (Segawa syndrome) in a Brazilian family. Genetics Molec Res. 2008, 7: 687-694. 10.4238/vol7-3gmr467.
Ewing B, Hillier L, Wendl MC, Green P: Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8: 175-185.
Ewing B, Green P: Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8: 186-194.
Gordon D, Abajian C, Green P: Consed: a graphical tool for sequence finishing. Genome Res. 1998, 8: 195-202.
Nickerson DA, Tobe VO, Taylor SL: PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Nucleic Acids Res. 1997, 25: 2745-2751. 10.1093/nar/25.14.2745.
Montgomery KTIO, Li L, Loomis S, Obourn V, Kucherlapati R: PolyPhred analysis software for mutation detection from fluorescence-based sequence data. Curr Protocol Human Genetics. 2008, Chap 7: Unit 7.16
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R: DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003, 19: 2496-2497. 10.1093/bioinformatics/btg359.
Vilella AJ, Blanco-Garcia A, Hutter S, Rozas J: VariScan: analysis of evolutionary patterns from large-scale DNA sequence polymorphism data. Bioinformatics. 2005, 21: 2791-2793. 10.1093/bioinformatics/bti403.
Thornton K: Libsequence: a C++ class library for evolutionary genetic analysis. Bioinformatics. 2003, 19: 2325-2327. 10.1093/bioinformatics/btg316.
Tarazona-Santos E, Tishkoff SA: Divergent patterns of linkage disequilibrium and haplotype structure across global populations at the interleukin-13 (IL13) locus. Genes Immunity. 2005, 6: 53-65.
Tarazona-Santos E, Bernig T, Burdett L, Magalhaes WCS, Fabbri C, Liao J, Redondo RA, Welch R, Yeager M, Chanock SJ: CYBB, an NADPH-oxidase gene: restricted diversity in humans and evidence for differential long-term purifying selection on transmembrane and cytosolic domains. Hum Mutat. 2008, 29: 623-632. 10.1002/humu.20667.
Fuselli S, Gilman RH, Chanock SJ, Bonatto SL, De Stefano G, Evans CA, Labuda D, Luiselli D, Salzano FM, Soto G: Analysis of nucleotide diversity of NAT2 coding region reveals homogeneity across Native American populations and high intra-population diversity. Pharmacogenomics J. 2007, 7: 144-152. 10.1038/sj.tpj.6500407.
Stephens M, Smith NJ, Donnelly P: A new statistical method for haplotype reconstruction from population data. Am J Human Genetics. 2001, 68: 978-989. 10.1086/319501.
Packer BR, Yeager M, Burdett L, Welch R, Beerman M, Qi LQ, Sicotte H, Staats B, Acharya M, Crenshaw A, Eckert A, Puri V, Gerhard DS, Chanock SJ: SNP500Cancer: a public resource for sequence validation, assay development, and frequency analysis for genetic variation in candidate genes. Nucleic Acids Res. 2006, 34: D617-D621. 10.1093/nar/gkj151.
Scheet P, Stephens M: A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am J Human Genetics. 2006, 78: 629-644. 10.1086/502802.
Manoukis NC: FORMATOMATIC: a program for converting diploid allelic data between common formats for population genetic analysis. Molec Ecology Notes. 2007, 7: 5 92-593.
Acknowledgements
We are grateful to Flavia Siqueira, Rodrigo Redondo, Renata Acacio, Sharon Savage and Charles Chung for helping us test the pipeline and to the Bioinformatics group of the Core Genotyping Facilities of NCI for their participation in discussions about the pipeline. This work is supported by the National Institutes of Health - Fogarty International Center (1R01TW007894-01 to ETS), Brazilian National Research Council (CNPq), Brazilian Ministry of Education (CAPES Agency) and Minas Gerais State Foundation in Aid of Research (FAPEMIG - CBB-1181/08, PPM-00439-10), CNPq (306879/2009-3) and NIH-Fogarty (TW007012).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
The first two authors MM and WCSM contributed equally to the paper. ETS conceived the project. WCSM, AS, ETS and BA developed the scripts used in this work. MM tested different versions and parts of the pipeline, interacted with several investigators and research groups and wrote the Web service documentation. AS, BA, WCSM and MR designed and integrated the pipeline modules and developed the Web interface. ETS and MR supervised the project. SJC provided resources and participated during the early parts of the project. LS provided the resources for hosting and maintaining the Web interface under the supervision of GO. ETS, MR and WCSM wrote the manuscript. All the authors read and approved the final manuscript.
Moara Machado, Wagner CS Magalhães contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Machado, M., Magalhães, W.C., Sene, A. et al. Phred-Phrap package to analyses tools: a pipeline to facilitate population genetics re-sequencing studies. Investig Genet 2, 3 (2011). https://doi.org/10.1186/2041-2223-2-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2041-2223-2-3