
Abstract
Background
In malaria parasites (genus Plasmodium), ama-1 is a highly polymorphic locus encoding the Apical Membrane Protein-1, and there is evidence that the polymorphism at this locus is selectively maintained. We tested the hypothesis that polymorphism at the ama-1 locus reflects population history in Plasmodium vivax, which is believed to have originated in Southeast Asia and is widely geographically distributed. In particular, we tested for a signature of the introduction of P. vivax into the New World at the time of the European conquest and African slave trade and subsequent population expansion.
Results
One hundred and five ama-1 sequences were generated and analyzed from samples from six different Brazilian states and compared with database sequences from the Old World. Old World populations of P. vivax showed substantial evidence of population substructure, with high sequence divergence among localities at both synonymous and nonsynonymous sites, while Brazilian isolates showed reduced diversity and little population substructure.
Conclusion
These results show that genetic diversity in P. vivax AMA-1 reflects population history, with population substructure characterizing long-established Old World populations, whereas Brazilian populations show evidence of loss of diversity and recent population expansion.
Note
Nucleotide sequence data reported is this paper are available in the GenBank™ database under the accession numbers EF031154 – EF031216 and EF057446 – EF057487
Similar content being viewed by others


Background
Studies of the population diversity of the malaria parasites have practical significance for the development for strategies of disease control, including vaccine development [1]. Moreover, the characterization of genes responsible for resistance to therapeutic agents by both Plasmodium falciparum and Plasmodium vivax depends on a thorough knowledge of each parasite's genetic diversity in natural populations [2]. The primary factors affecting genetic diversity at such loci are natural selection [3, 4], and genetic drift. Genetic drift reflects the population history, including population bottlenecks; and it has a substantial effect on genetic diversity even at loci subject to balancing selection [5–7]. Thus, the knowledge of the parasite's population history and its genetic diversity is important for a full understanding of the epidemiology of malaria and potential response of the parasite to therapeutic strategies.
P. vivax is widely geographically distributed, being present in both tropical and temperate areas; this species is responsible for about 80 million annual cases of human malaria, especially in Latin America, Asia and Oceania [8]. It is the prevalent species in a great number of countries and territories, including Brazil, which accounted for about 81% of the approximately 460,000 cases reported in 2007 [9]. P. vivax infections rarely culminate in death of the patient but are a very important cause of morbidity and social economic loss [10]. P. vivax is believed to have first entered hominid populations in Southeast Asia and to have spread from there throughout the Old World based on its close relation to malaria parasites of non-human primates from Southeast Asia [11]. However, there is archaeological evidence supporting the hypothesis that both P. vivax and P. falciparum were absent from the New World in pre-Columbian times and were introduced after European colonization, presumably as a result of the African slave trade [12]. Thus P. vivax in the New World might be expected to have a somewhat reduced effective population size and thus reduced genetic diversity in comparison to Old World populations, as a result of founder effects in the sampling of Old World populations. Microsatellite markers have shown evidence of a substantial reduction of genetic diversity in the case of South American P. falciparum [13]. On the other hand, in P. vivax, microsatellite markers showed revealed only a rather modest reduction in genetic diversity in South America in comparison to Asia [14].
In addition to microsatellites [14–18], several polymorphic protein-coding loci have been used to examine genetic diversity of P. vivax populations, including genes encoding the merozoite surface proteins (MSP) [19–28]; the circumsporozoite protein (CSP) [21, 24, 26, 28–30]; and the locus encoding apical membrane antigen 1 (AMA-1) [20, 31–36]. AMA-1 is an immunogenic type 1 integral membrane protein [37–40] that is present in all Plasmodium species so far examined, with at least 16 cysteine residues incorporated into eight intramolecular disulfide bonds, forming the three domains of the protein [41]. This protein is synthesized late in the development of schizonts [42] during the last four hours of the erythrocytic phase [38]. AMA-1 may have a role in the beginning of the invasion process of the erythrocyte and may be directly responsible for reorientation of the merozoite; and it may initiate the junctional contact between these two cells, which is presumably dependent on Duffy binding proteins [43].
At the ama-1 locus in P. falciparum, polymorphisms occur non-randomly along the coding region, and the highest polymorphism is found in the three ectodomains, especially in domain I [44]. Moreover, the number of nonsynonymous nucleotide substitutions per nonsynonymous site (d N ) exceeds that of synonymous nucleotide substitutions per synonymous site (d S ), providing evidence that positive Darwinian selection has acted at this locus [4]. Combined with the evidence of a high level of polymorphism at this locus, this result supports the hypothesis that balancing selection has acted to maintain polymorphisms at this locus [4]. It has been proposed that host immune system pressure is responsible for this selection [4]; and, consistent with this hypothesis, there is evidence that polymorphisms at this locus are responsible for evasion of host antibody-mediated inhibition in P. falciparum [45]. In P. vivax, d N has been found to exceed d S in partial ama-1 sequences [20], suggesting that this locus is subject to balancing selection in P. vivax as well.
Extensive data on ama-1 polymorphism in P. vivax (pvama-1) have been obtained from Asia, Oceania and Africa [20, 31–33, 35, 36], but there is a relative lack of data from South America, including Brazil. The only sequence data from Brazil involves domain I of 20 isolates; 13 polymorphic sites and eight haplotypes were reported in three Brazilian states [34].
The intention of the present study was to characterize the worldwide genetic diversity of the polymorphic domain of pvama-1. In addition to published sequences from throughout the world, we obtained sequences from patients in different endemic areas in the Brazilian Amazon. By examining polymorphism at this locus in Brazil and comparing it to other populations throughout the world, we tested the hypothesis that the pattern of genetic diversity at pvama-1 reflects population history, in particular a reduction of the effective population size of P. vivax in the New World. Theoretically, it is expected that effective population size will be the major factor determining gene diversity even at a locus under balancing selection, if the mutation rate and selection coefficient are constant [5–7]. A more complete understanding of the parasite's history in the New World in turn has implications for the epidemiology and control of this parasite in Brazil, where it has become a major public health problem in recent years due to the rapid peopling of the Brazilian Amazon [46–49].
Results
We obtained 105 Brazilian pvama-1 sequences, covering bases 274–759 of the PH-84 isolate, corresponding to amino acids 92–253 (GenBank accession nos. EF031154 – EF031216 and EF057446 – EF057487 – see Additional file 1). The Brazilian isolates included 28 polymorphic nucleotide sites, leading to 26 amino acids replacements. Eight polymorphic sites were not previously described, including a synonymous substitution and seven non-synonymous substitutions (see Additional file 2). From 93 unique sequences, 27 haplotypes were identified (see Additional file 3). The nucleotide and haplotype diversities among Brazilian samples were 0.016620 ± 0.00073 and 0.91800 ± 0.00019, respectively.
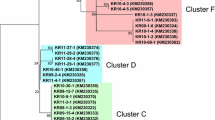
A phylogenetic tree of Brazilian and worldwide sequences (Figure 1) showed no tendency toward geographic clustering of isolates. Rather, isolates from different parts of the world were found throughout the phylogenetic tree (Figure 1). The Brazilian sequences thus appeared to represent a sample from worldwide genetic diversity, rather than from any particular lineage of worldwide pvama-1 sequences.

Phylogenetic tree of unique Brazilian and worldwide pvama-1 sequences. Green = South America; Red = Old Word. Numbers on the branches are percentages of bootstrap samples supporting the branch; only values ≥ 50% are shown.
In order to compare nucleotide diversity within geographic regions, we computed π, π S , and π N for all pairwise comparisons within Brazilian states and within and worldwide regions (Figure 2). Likewise, we computed mean d, d S , and d N for all pairwise comparisons between Brazilian states and between worldwide regions. In worldwide comparisons, mean π π S , and π N within regions were always significantly lower than the corresponding values of d, d S , and d N between region (Figure 2). By contrast, mean π, π S , and π N within Brazilian states were not significantly different from the corresponding values of d, d S , and d N between states (Figure 2). Thus, these results show that pvama-1 did not show the degree of sequence divergence among the Brazilian states that was seen among different regions in the world. Mean π, π S , and π N within Brazilian states were significantly lower than the corresponding values within world regions (Figure 2). Likewise mean d, d S , and d N between Brazilian states were significantly lower than corresponding values between world regions (Figure 2). These results show that sequence divergence in pvama-1 among states in Brazil was low than that in comparisons of different Old World populations.

Means of (A) π, (B) π S , and (C) π N within Brazilian states and within worldwide regions; and of (A) d , (B) d S , and (C) d N between Brazilian states and between worldwide regions. Test of the hypothesis that a value for Brazil equals the corresponding value for worldwide comparisons: * P < 0.05; *** P < 0.001. Tests of the hypothesis that mean value within regions equals the corresponding value between regions: +++ < 0.001.
Similar results were obtained from estimation of pairwise F ST , which provides an index of the genetic differentiation between populations. F ST values among different world regions were often significantly greater than zero, indicating genetic differentiation between populations (Table 1). By contrast, estimates of F ST among Brazilian states were never significantly different from zero, indicating a lack of genetic differentiation among the Brazilian states (Table 2).
We plotted F ST against the geographical distance between the sites where samples were collected separately for data from Brazil and data from Asia and Oceania (Figure 3). In the data from Asia and Oceania, there was not a significant correlation between F ST and geographical distance (r = -0.196; n.s.; randomization test; Figure 3). By contrast, in Brazil, there was a strong positive correlation between F ST and geographical distance (r = 0.780; P < 0.01; randomization test; Figure 3). The correlation coefficient for the Brazilian data was significantly different from that for the Asian and Oceanian data (p < 0.01; randomization test).

Plot of F ST vs. geographical distance for Brazilian samples ( green ) and samples from Asia and Oceania ( red ). In the data from Brazil, there was a significant correlation between F ST and geographical distance (r = 0.780; P < 0.01; randomization test). In the data from Asia and Oceania, there was not a significant correlation between F ST and geographical distance (r = -0.196; n.s.; randomization test).
The range of geographical distances among the Brazilian samples (450–2340 km) overlapped only with the lower nine values from the Asian and Oceanian sample (range 600–2240; Figure 3). If we considered only the nine data points in the Asian and Oceanian sample that overlapped the Brazilian data, there was again no significant correlation between F ST and geographical distance (r = 0.091; n.s.; randomization test; Figure 3). Moreover, for the nine Asian and Oceanian comparisons with geographical distances comparable to those in Brazil, mean F ST (0.111) was significantly greater than that for the 10 Brazilian comparisons (mean F ST = -0.011; randomization test; P < 0.01).
Discussion
Here, we characterized the polymorphic gene pvama-1 domain I in Plasmodium vivax isolates from patients in the Brazilian Amazon, where this species poses an important public health problem and compared those sequences with previously published sequences from the Old World. Although most branches in a phylogeny of pvama-1 sequences were not well resolved, it was clear that Brazilian sequences did not cluster separately from Old World sequences (Figure 1). This pattern supports the hypothesis that any reduction in population size accompanying the invasion of the Americas [12] was not so severe that only one or a few lineages of pvama-1 alleles survived in the New World. Rather, pvama-1 sequences from Brazil were found throughout the phylogenetic tree, consistent with the hypothesis that the alleles that became established in the New World represented a sample of worldwide genetic diversity at this locus. There was evidence of reduced genetic diversity at the pvama-1 locus in Brazil, consistent with some reduction in effective population size of P. vivax in the New World after its introduction.
There were very low values of F ST among the Brazilian states, with none being significantly greater than zero. The latter was in marked contrast to the Old World, particularly Southeast Asia, where high F ST values were consistently observed. Of course, the geographical distances among the Brazilian states sampled were low in comparison to many of the geographical distances among samples from Asia and Oceania (Figure 3). Nonetheless, mean F ST among the Brazilian samples was much lower than that among those samples from Asia and Oceania taken from comparable geographical distances. Thus, pvama-1 shows strikingly less geographical differentiation in Brazil than in Southeast Asia, consistent with a recent and rapid spread of the parasite in Brazil.
There is evidence that the polymorphism at the ama-1 locus is selectively maintained in P. falciparum, with host immune recognition likely being responsible for that selection [44]. Given the high polymorphism and prevalence of nonsynonymous polymorphisms at the pvama-1 locus, it seems likely that the same is true in P. vivax [20]. An alternative hypothesis to account for the reduced polymorphism in Brazilian pvama-1 sequences might be that selection at this locus has been relaxed in the New World. In the Brazilian Amazon, P. vivax has achieved high levels of infection in an ethnically diverse and rapidly growing host population [46]. If the selection on pvama-1 arises primarily from interaction with the human host immune system, it seems unlikely that selection would be relaxed under such circumstances. However, as long as the basis of natural selection on pvama-1 remains poorly known, it is impossible to rule out some role of natural selection in the pattern of sequence diversity observed in the New World.
In spite of the overall low F ST values in the Brazilian samples, there was evidence in Brazil of a strong positive relationship between F ST and geographical distance. By contrast, in the Old World, even though F ST values were high, there was no correlation between F ST and geographical distance. The latter was observed both in an extensive sample of populations from Asia and Oceania and when we examined only populations whose geographical distances were comparable to those among Brazilian states. The results from Brazil can be explained as reflecting effects of recent spread of the parasite, whereas those from the Old World appear to reflect a very ancient selectively maintained polymorphism. In the latter case, different populations are expected to show substantially different allelic frequency distributions due to divergent population histories, including the effects of genetic drift. Such a pattern is seen, for example, in the case of vertebrate major histocompatibility complex loci [50], at which high levels of polymorphism are maintained by balancing selection [51, 52].
Conclusion
Our results are consistent with the hypothesis that patterns of genetic diversity at highly polymorphic protein-coding loci of malaria parasites can show the effects of population history. Polymorphism at loci such as pvama-1 that are evidently subject to immune-driven selection may be an important factor in the epidemiology of infection by P. vivax. Understanding the factors governing the extent and pattern of polymorphism at such loci may thus have implications for the development of effective control strategies [53].
Methods
Study population
One hundred and five blood samples were collected from patients resident at Cuiabá district, capital of the state of Mato Grosso, northwestern Brazil (S 15°36'36", W 56°05'24"), where active malaria transmission does not occur. Patients had acquired malaria infection in different areas of the Brazilian Amazon comprising six different Brazilian states between April and August 1996 and between May 2001 and January 2006: 8 samples originated from Acre; 13 from Amazonas; 31 from Mato Grosso; 20 from Pará; 32 from Rondônia; and one from Roraima. Patient infections were confirmed by microscopic analysis of conventional thick smear method in a health facility (Hospital Júlio Müller, Universidade Federal de Mato Grosso). The age of patients ranged between 4 and 78 years old, with mean age of 37.6 ± 14.1.
DNA extraction and amplification of pvama-1
Blood samples were stored in guanidine 4 M and kept at -20°C. The manufacturer's instructions for 300 μL whole blood extraction from Genomic DNA Purification Kit (Puregene®) were followed. The pvama-1 gene was amplified following a previously described protocol [20]. We added 0.26 pmoles of each primer [PvAR11 (5-TCC TAA ATT TTT ACG GGG GCA3) and PvAF11 (5-AGA ATT CCA GCT CCA AGA TG-3)], 0.2 mM of dNTPs, Taq buffer 1× (Phoneutria, MG, Brazil), 1.5 mM of MgCl2, 1.25 U of Taq polymerase (Phoneutria) and 5 μL of DNA totalizing 50 μL of mixture for the first round of amplification. One cycle of 95°C for 5 min, 2 cycles of 95°C for 30 s, 45°C for 50 s and 72°C for 40 s, 33 cycles of 95°C for 30 s, 55°C for 50 s and 72°C for 40 s, followed by 72°C for 10 min. The second round of amplification used the same conditions as the first one, except for the primers [PvAR11 and PvAF05 (5-GTA TCG TCA TAG AGA ATT CCG-3')] and quantity of DNA used (2 μL of the first round amplification product). The amplification conditions included one cycle of 95°C for 5 min, 2 cycles of 95°C for 30 s, 45°C for 50 s and 72°C for 40 s, 23 cycles of 95°C for 30 s, 55°C for 50 s and 72°C for 40 s, followed by 72°C for 10 min. The fragment size of approximately 400 bp was visualized on a 6% polyacrilamide gel.
Purification of PCR products and pvama-1sequencing
PCR products were purified using the GFX PCR DNA and Gel Band Purification Kit (Amersham Biosciences®, Little Chalfont, UK), following the manufacturer's instructions and were visualized on a 1% agarose gel to determine the DNA concentration. For the sequencing reaction, we used 4 μL of Dyenamic ET Dye Terminator Cycle Sequencing kit (Amersham Biosciences®) for MegaBace DNA Analysis Systems, 1 μL of primer and 5 μL of purified DNA. For each DNA sample, we created two sequences using both AR11 and AF05 primers.
Sequence analysis
New sequences from Brazil were combined with a database of 215 sequences from Asia, Africa, and Oceania (see Additional file 1) and aligned with the ClustalW Software [54]. A 399-bp region was analyzed, corresponding to bases 322–720 (amino acid residues 108–240) of Genbank accession L27503. We used the MEGA 3.1 program [55] to estimate nucleotide diversity and evolutionary distances and to build phylogenetic trees by the neighbor-joining method [56], using the Jukes-Cantor distance [57]. The reliability of clustering patterns in the phylogenetic trees was assessed by bootstrapping [58]: 1000 bootstrap pseudo-samples were used. Before conducting the phylogenetic analysis, we tested for inter-allelic recombination using the maximum chi-square method [59] as implemented in the RDP2 program [60]. No recombination events were detected. The number of nucleotide substitutions per site (d) was estimated by Jukes and Cantor's method [57]. The numbers of synonymous nucleotide substitutions per synonymous site (d S ) and the number of nonsynonymous substitutions per nonsynonymous site (d N ) were estimated by Nei and Gojobori's method [61].
In order to examine patterns of nucleotide diversity within Brazil, we computed means of d, d S , and d N for all pairwise comparisons within and between the six Brazilian states from which we obtained sequences. Similarly, in order to examine patterns nucleotide diversity between regions throughout the world, we computed means of d, d S , and d N for all pairwise comparisons between the following geographic regions: Africa (5 sequences); Agusan del Sur, Philippines (abbreviation: ADS; 21 sequences), Brazil (93 sequences); China (8 sequences); India (15 sequences); Indonesia excluding Irian Jaya (5 sequences); Irian Jaya (1 sequence); Morong, Philippines (111 sequences); Myanmar (1 sequence); Palawan, Philippines (17 sequences); Papua New Guinea (abbreviation: PNG; 22 sequences); Solomon Islands (5 sequences); South Korea (4 sequences); Sri Lanka (3 sequences); Thailand (7 sequences); Vanuatu (2 sequences). Finally, in order to analyze nucleotide diversity within geographic regions other than Brazil, we computed means of d, d S , and d N for all pairwise comparisons within each of the above regions that was represented by at least two sequences. Following general usage, means of d, d S , and d N within populations were designated respectively π, π S , and π N .
Pairwise comparisons of d, d S , and d N are not statistically independent. Therefore, we tested hypotheses about the means of these variables using randomization (Monte Carlo) tests. Given N comparisons categorized (e.g., as within-region or between-region) by a classificatory variable X, in order to conduct simultaneous pairwise comparisons between categories with respect to the median of some continuous scalar variable Y measured on each of the N units (e.g., d, d S , or d N ), we created 1000 pseudo data sets of N units each by randomly sampling (with replacement) independently from the vector of X values and from the vector of Y values. For a two-tailed test, the level of significance of the difference between two group medians was obtained by comparing the observed absolute difference with the distribution of absolute differences obtained for the corresponding groups in the 1000 pseudo data sets.
We used a similar randomization procedure to test the significance of correlation coefficients between pairwise measures of F ST and geographical distance. We created 1000 pseudo data sets by sampling at random from replacement in order to generate a null distribution against which observed values were compared. We used a similar procedure to test the equality of mean F ST in the Brazilian data with those from Asian and Oceanian populations of comparable geographic distance.
References
Cui L, Escalante AA, Imwong M, Snounou G: The genetic diversity of Plasmodium vivax populations. Trends Parasitol. 2003, 19: 220-226. 10.1016/S1471-4922(03)00085-0.
Hughes AL, Verra F: Very large long-term effective population size in the virulent human malaria parasite Plasmodium falciparum. Proc Biol Sci. 2001, 268: 1855-1860. 10.1098/rspb.2001.1759.
Hughes MK, Hughes AL: Natural selection on Plasmodium surface proteins. Mol Biochem Parasitol. 1995, 71: 99-113. 10.1016/0166-6851(95)00037-2.
Verra F, Hughes AL: Evidence for ancient polymorphism at the Apical Membrane Antigen-1 (AMA-1) locus of Plasmodium falciparum. Mol Biochem Parasitol. 2000, 105: 149-153. 10.1016/S0166-6851(99)00162-0.
Takahata N, Nei M: Allelic genealogy under overdominant and frequency-dependent selection and polymorphism of major histocompatibility complex loci. Genetics. 1990, 124: 967-978.
Li W-H: Maintenance of genetic variability under the joint effect of mutation, selection, and random drift. Genetics. 1978, 90: 349-382.
Maruyama T, Nei M: Genetic variability maintained by mutation and overdominant selection in finite populations. Genetics. 1981, 98: 441-459.
World Healthy Organization (WHO): 2007, [http://www.who.org]
Brazilian Ministry of Health: 2007, [http://portal.saude.gov.br/saude/]
Mendis K, Sina BJ, Marchesini P, Carter R: The neglected burden of Plasmodium vivax malaria. Am J Trop Med Hyg. 2001, 64: 97-106.
Jongwutiwes S, Putaporntip C, Iwasaki T, Ferreira MU, Kanbara H, Hughes AL: Mitochondrial genome sequences support ancient population expansion in Plasmodium vivax. Mol Biol Evol. 2005, 22: 1733-1739. 10.1093/molbev/msi168.
De Castro MC, Singer BH: Was malaria present in the Amazon before the European conquest? Avaiable evidence and future research agenda. J Achaeol Sci. 2005, 32: 337-340. 10.1016/j.jas.2004.10.004.
Anderson TJ, Haubold B, Williams JT, Estrada-Franco JG, Richardson L, Mollinedo R, Bockarie M, Mokili J, Mharakurwa S, French N, Whitworth J, Velez ID, Brockman AH, Nosten F, Ferreira MU, Day KP: Microsatellite markers reveal a spectrum of population structures in the malaria parasite Plasmodium falciparum. Mol Biol Evol. 2000, 17: 1467-1482.
Karunaweera ND, Ferreira MU, Munasinghe A, Barnwell JW, Collins WE, King CL, Kawamoto F, Hartl DL, Wirth DF: Extensive microsatellite diversity in the human malaria parasite Plasmodium vivax. Gene. 2008, 410: 105-112. 10.1016/j.gene.2007.11.022.
Gomez JC, McNamara DT, Bockarie MJ, Baird JK, Carlton JM, Zimmerman PA: Identification of a polymorphic Plasmodium vivax microsatellite marker. Am J Trop Med Hyg. 2003, 69: 377-379.
Leclerc MC, Durand P, Gauthier C, Patot S, Billotte N, Menegon M, Severini C, Ayala FJ, Renaud F: Meager genetic variability of the human malaria agent Plasmodium vivax. Proc Natl Acad Sci USA. 2004, 101: 14455-14560. 10.1073/pnas.0405186101.
Imwong M, Sudimack D, Pukrittayakamee S, Osorio L, Carlton JM, Day NP, White NJ, Anderson TJ: Microsatellite variation, repeat array length, and population history of Plasmodium vivax. Mol Biol Evol. 2006, 23: 1016-1018. 10.1093/molbev/msj116.
Ferreira MU, Karunaweera ND, da Silva-Nunes M, da Silva NS, Wirth DF, Hartl DL: Population structure and transmission dynamics of Plasmodium vivax in rural Amazonia. J Infect Dis. 2007, 15:195 (8): 1218-26. 10.1086/512685.
Cheng Q, Stowers A, Huang TY, Bustos D, Huang YM, Rzepczyk C, Saul A: Polymorphism in Plasmodium vivax MSA-1 gene – the result of intragenic recombinations?. Parasitol. 1993, 106: 335-345.
Figtree M, Pasay CJ, Slade R, Cheng Q, Cloonan N, Walker J, Saul A: Plasmodium vivax synonymous substitution frequencies, evolution and population structure deduced from diversity in AMA-1 and MSP-1 genes. Mol Bio Parasitol. 2000, 108: 53-66. 10.1016/S0166-6851(00)00204-8.
Lim CS, Kim SH, Kwon SI, Song JW, Song KJ, Lee KN: Analysis of Plasmodium vivax merozoite surface protein-1 gene sequences from resurgent Korean isolates. Am J Trop Med Hyg. 2000, 62: 261-265.
Gutierrez A, Vicini J, Patarroyo ME, Murillo LA, Patarroyo MA: Plasmodium vivax: polymorphism in the merozoite surface protein 1 gene from wild Colombian isolates. Exp Parasitol. 2000, 95: 215-219. 10.1006/expr.2000.4534.
Zakeri S, Dinparast Djadid N, Zeinali S: Sequence heterogeneity of the merozoite surface protein-1 gene (MSP-1) of Plasmodium vivax wild isolates in southeastern Iran. Acta Trop. 2003, 88: 91-97. 10.1016/S0001-706X(03)00192-X.
Leclerc MC, Menegon M, Cligny A, Noyer JL, Mammadov S, Aliyev N, Gasimov E, Majori G, Severini C: Genetic diversity of Plasmodium vivax isolates from Azerbaijan. Mal J. 2004, 3: 40-10.1186/1475-2875-3-40.
Leclerc MC, Gauthier C, Villegas L, Urdaneta L: Genetic diversity of merozoite surface protein-1 gene of Plasmodium vivax isolates in mining villages of Venezuela (Bolivar State). Acta Trop. 2005, 95: 26-32. 10.1016/j.actatropica.2005.03.007.
Zakeri S, Mehrizi AA, Mamaghani S, Noorizadeh S, Snounou G, Djadid ND: Population structure analysis of Plasmodium vivax in areas of iran with different malaria endemicity. Am J Trop Med Hyg. 2006, 74: 394-400.
Kim JR, Imwong M, Nandy A, Chotivanich K, Nontprasert A, Tonomsing N, Maji A, Addy M, Day NP, White NJ, Pukrittayakamee S: Genetic diversity of Plasmodium vivax in Kolkata, India. Mal J. 2006, 5: 71-10.1186/1475-2875-5-71.
Bonilla JA, Validum L, Cummings R, Palmer CJ: Genetic diversity of Plasmodium vivax Pvcsp and Pvmsp1 in Guyana, South America. Am J Trop Med Hyg. 2006, 75: 830-835.
Kain KC, Brown AE, Webster HK, Wirtz RA, Keystone JS, Rodriguez MH, Kinahan J, Rowland M, Lanar DE: Circumsporozoite genotyping of global isolates of Plasmodium vivax from dried blood specimens. J Clin Microbiol. 1992, 30: 1863-1866.
Santos-Ciminera PD, Alecrim MG, Roberts DR, Quinnan GV: Molecular epidemiology of Plasmodium vivax in the State of Amazonas, Brazil. Acta Trop. 2007, 102: 38-46. 10.1016/j.actatropica.2007.02.013.
Cheng Q, Saul A: Sequence analysis of the apical membrane antigen I (AMA-1) of Plasmodium vivax. Mol Biochem Parasitol. 1994, 65: 183-187. 10.1016/0166-6851(94)90127-9.
Han ET, Park JH, Shin EH, Choi MH, Oh MD, Chai JY: Apical membrane antigen-1 (AMA-1) gene sequences of re-emerging Plasmodium vivax in South Korea. Kor J Parasitol. 2002, 40: 157-162.
Chung JY, Chun EH, Chun JH, Kho WG: Analysis of the Plasmodium vivax apical membrane antigen 1 gene from re-emerging Korean isolates. Parasitol Res. 2003, 90: 325-329. 10.1007/s00436-002-0777-2.
Rodrigues MH, Rodrigues KM, Oliveira TR, Comodo AN, Rodrigues MM, Kocken CH, Thomas AW, Soares IS: Antibody response of naturally infected individuals to recombinant Plasmodium vivax apical membrane antigen-1. Int J Parasitol. 2005, 35: 185-192. 10.1016/j.ijpara.2004.11.003.
Gunasekera AM, Wickramarachchi T, Neafsey DE, Ganguli I, Perera L, Premaratne PH, Hartl D, Handunnetti SM, Udagama-Randeniya PV, Wirth DF: Genetic Diversity and Selection at the Plasmodium vivax Apical Membrane Antigen-1 (PvAMA-1) locus in a Sri Lankan population. Mol Biol Evol. 2007, 24: 939-947. 10.1093/molbev/msm013.
Rajesh V, Elamaran M, Vidya S, Gowrishankar M, Kochar D, Das A: Plasmodium vivax: Genetic diversity of the apical membrane antigen-1 (AMA-1) in isolates from India. Exp Parasitol. 2007, 116: 252-256. 10.1016/j.exppara.2007.01.006.
Peterson MG, Marshall VM, Smythe JA, Crewther PE, Lew A, Silva A, Andres RF, Kemp DJ: Integral membrane protein located in the apical complex of Plasmodium falciparum. Mol Cell Biol. 1989, 9: 3151-3154.
Narum DL, Thomas AW: Differential localization of full-length and processed forms of PF83/AMA-1: an apical membrane antigen of Plasmodium falciparum merozoites. Mol Biochem Parasitol. 1994, 67: 59-68. 10.1016/0166-6851(94)90096-5.
Galinski MR, Barnwell JW: Plasmodium vivax: merozoítos, invasion of reticulocytes and considerations for malaria vaccine development. Parasitol Today. 1996, 12: 20-29. 10.1016/0169-4758(96)80641-7.
Riley EM, Wagner GE, Ofori MF, Wheeler JG, Akanmori BD, Tetteh K, McGuinness D, Bennett S, Nkrumah FK, Anders RF, Koram KA: Lack of association between maternal antibody and protection of African infants from malaria infection. Infect Immun. 2000, 68: 5856-5863. 10.1128/IAI.68.10.5856-5863.2000.
Hodder AN, Crewther PE, Mattew MLSM, Reid GE, Moritz RL, Simpson RJ, Anders RF: The disulphide bond structure of Plasmodium apical membrane antigen-1. J Biol Chem. 1996, 271: 29446-29452. 10.1074/jbc.271.46.29446.
Crewther PE, Culvenor JG, Silva A, Cooper JA, Anders RF: Plasmodium falciparum: two antigens of similar size are located in different compartiments of the rhoptry. Exp Parasitol. 1990, 70: 193-206. 10.1016/0014-4894(90)90100-Q.
Mitchell GH, Thomas AW, Margos G, Dluzewski AR, Bannister LH: Apical Membrane Antigen 1, a Major Malaria Vaccine Candidate, Mediates the Close Attachment of Invasive Merozoites to Host Blood Cells. Infect Immun. 2004, 72: 154-158. 10.1128/IAI.72.1.154-158.2004.
Hodder AN, Crewther PE, Anders RF: Specificity of the protective antibody response to apical membrane antigen 1. Infect Immun. 2001, 69: 3286-3294. 10.1128/IAI.69.5.3286-3294.2001.
Healer J, Murphy V, Hodder AN, Masciantonio R, Gemmil AW, Anders RF, Cowman AF, Batchelor A: Allelic polymorphism in apical membrane antigen-1 are responsible for evasion of antibody-mediated inhibition in Plasmodium falciparum. Mol Microbiol. 2004, 52: 159-168. 10.1111/j.1365-2958.2003.03974.x.
Castro MC, Monte-Mór RL, Sawyer DO, Singer BH: Malaria risk on the Amazon frontier. Proc Natl Acad Sci USA. 2006, 103: 2452-2457. 10.1073/pnas.0510576103.
Castro MC, Sawyer DO, Singer BH: Spatial patterns of malaria in the Amazon: implications for surveillance and targeted interventions. Health and Place. 2007, 13: 368-380. 10.1016/j.healthplace.2006.03.006.
Gil LH, Tada MS, Katsuragawa TH, Ribolla PE, Pereira da Silva LH: Urban and suburban malaria in Rondônia (Brazilian Western Amazon) II. Perennial transmissions with high anopheline densities are associated with human environmental changes. Mem Inst Oswaldo Cruz. 2007, 102: 271-276.
Tada MS, Marques RP, Mesquita E, Dalla Martha RC, Rodrigues JA, Costa JD, Pepelascov RP, Katsuragawa TH, Pereira-da-Silva LH: Urban malaria in the Brazilian Western Amazon Region I. High prevalence of asymptotic carriers in an urban riverside district is associated with a high level of clinical malaria. Mem Inst Oswaldo Cruz. 2007, 102: 263-269. 10.1590/S0074-02762007005000012.
Cao K, Moormann AM, Lyke KE, Masaberg C, Sumba OP, Doumbo OK, Koech D, Lancaster A, Nelson M, Meyer D, Single R, Hartzman RJ, Plowe CV, Kazura J, Mann DL, Sztein MB, Thomson G, Fernández-Viña : Differentiation between African populations is evidenced by the diversity of alleles and haplotypes of HLA class I loci. Tissue Antigens. 2004, 63: 293-325. 10.1111/j.0001-2815.2004.00192.x.
Hughes AL, Nei M: Pattern of nucleotide substitution at MHC class I loci reveals overdominant selection. Nature. 1988, 335: 167-170. 10.1038/335167a0.
Hughes AL, Hughes MK: Natural selection on the peptide-binding regions of major histocompatibility complex molecules. Immunogenetics. 1995, 42: 233-243. 10.1007/BF00176440.
Hughes AL, Verra F: Extensive polymorphism and ancient origin of Plasmodium falciparum. Trends Parasitol. 2002, 18: 348-351. 10.1016/S1471-4922(02)02290-0.
Thompson JD, Higgings DG, Gibson TJ: CLUSTALW: improving the sensitivity of progressive multiple sequence alignment thought sequence weighting, position specific gap penalties and weight matrix choice. Nucleics Acids Res. 1994, 22: 4673-4680. 10.1093/nar/22.22.4673.
Kumar S, Tamura K, Nei M: MEGA3.1. Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform. 2004, 5: 150-163. 10.1093/bib/5.2.150.
Saitou N, Nei M: The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987, 4: 406-425.
Jukes TH, Cantor CR: Evolution of protein molecules. Mammalian Protein Metabolism. Edited by: Munro HN. 1969, New York: Academic Press, 21-132.
Felsenstein J: Confidence limits on phylogenies: An approach using the bootstrap. Evolution. 1985, 39: 783-791. 10.2307/2408678.
Maynard Smith J: Analyzing the mosaic structure of genes. J Mol Evol. 34: 126-129.
Martin DR, Williamson C, Posada D: RDP2: recombination detection and analysis from sequence alignments. Bioinformatics. 2005, 21: 260-262. 10.1093/bioinformatics/bth490.
Nei M, Gojobori T: Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986, 3: 418-426.
Acknowledgements
This work has been supported by Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG Grant # CBB53/03 and CBB625/06) and by grant GM43940 from the U.S. National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
PG carried out the molecular analyses, participated in statistical analyses, and drafted the manuscript. CJFF was responsible for acquisition of the data. ALH participated in design of the study and performed statistical analyses. EMB conceived the study and participated in its design and coordination.
Electronic supplementary material
12862_2007_692_MOESM1_ESM.doc
Additional file 1: List of the accession numbers of all sequences used in this study. The table lists the accession number of the sequences and the analyses in which each was used. (DOC 92 KB)
12862_2007_692_MOESM2_ESM.doc
Additional file 2: PvAMA-1 domain I polymorphic sites in Brazilian isolates compared with previously reported sequences. The table displays the polymorphisms (amino acid and nucleotide position) of pvama-1 domain I found in Brazilian isolates and compares them with previously reported sequences. (DOC 62 KB)
12862_2007_692_MOESM3_ESM.doc
Additional file 3: The nucleotide part represents the exclusive or particular features of Brazilian samples; the amino acid part represents the Brazilian haplotypes. This table illustrates the 27 haplotypes found in Brazilian samples, with amino acids residues and nucleotides positions. (DOC 181 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Grynberg, P., Fontes, C.J.F., Hughes, A.L. et al. Polymorphism at the apical membrane antigen 1 locus reflects the world population history of Plasmodium vivax. BMC Evol Biol 8, 123 (2008). https://doi.org/10.1186/1471-2148-8-123
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-8-123