
Abstract
Microdeletion and microduplication copy number variations are found in patients with autism spectrum disorder and in a number of cases they include genes that are involved in the canonical Wnt signaling pathway (for example, FZD9, BCL9 or CDH8). Association studies investigating WNT2, DISC1, MET, DOCK4 or AHI1 also provide evidence that the canonical Wnt pathway might be affected in autism. Prenatal medication with sodium-valproate or antidepressant drugs increases autism risk. In animal studies, it has been found that these medications promote Wnt signaling, including among others an increase in Wnt2 gene expression. Notably, the available genetic information indicates that not only canonical Wnt pathway activation, but also inhibition seems to increase autism risk. The canonical Wnt pathway plays a role in dendrite growth and suboptimal activity negatively affects the dendritic arbor. In principle, this provides a logical explanation as to why both hypo- and hyperactivity may generate a similar set of behavioral and cognitive symptoms. However, without a validated biomarker to stratify for deviant canonical Wnt pathway activity, it is probably too dangerous to treat patients with compounds that modify pathway activity.
Similar content being viewed by others


Introduction
Autism is a developmental disorder that appears in the first three years of life. Diagnostic behavioral symptoms of autism are abnormal socialization, limited communication, unusually narrow interests and repetitive behaviors[1, 2]. Clinical presentation and intellectual abilities are, however, extremely heterogeneous and autism may be better described as autism spectrum disorders. There is a large difference in concordance rates between monozygotic and dizygotic twins, which indicates that autism spectrum disorders have a strong genetic basis[3–5]. Investigations of genome-wide single nucleotide polymorphisms and copy number variations have generated a long list of candidate genes[6–8]. These candidate genes have very diverse functions and interactions[9–12] and the process by which these modified genes contribute or cause autism remains poorly understood. One approach to shed some light on pathological processes is to arrange the identified candidate genes according to place and function within known intracellular signal transduction cascades. When the functional consequence of a given mutation is known, one can infer whether the signaling pathway is activated or suppressed, and ultimately one can try to estimate the functional consequences in terms of neuronal function, brain circuits and behavioral output. In the present report, mutations in the canonical Wnt (wingless-type MMTV integration site) pathway that are found in cases with autism are reviewed. Before going into detail, it is important to sketch this Wnt signal transduction cascade.
Description of the canonical Wnt pathway
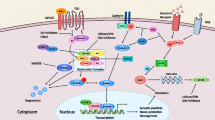
For a rapid overview of the canonical Wnt pathway, the reader may consult the cartoon in Figure1. ‘Wnts’ are lipid-modified signaling proteins that act as short range ligands to activate receptor-mediated signaling cascades. In mammals, some 19 Wnt members exist[13]. The proteins which act as cell surface receptors for Wnts are called ‘frizzled’ and, of these, 10 members have been described. Activated frizzled receptors connect to several downstream pathways[14–16]. In the so-called canonical Wnt pathway, signal transduction involves a low-density lipoprotein co-receptor (either LRP5 or LRP6) and polymerization of a protein called ‘dishevelled’ (abbreviated DVL; three isoforms)[14, 17–19]. Activation of the canonical Wnt pathway leads to dissociation of cadherin/β-catenin complexes in the cell membrane with release of β-catenin, a process involving the phosphorylation of the Tyr-142 residue of β-catenin by the hepatocyte growth factor receptor Met[20, 21].

Schematic representation of the canonical Wnt2 pathway, including all genes discussed in the current review. Wnt2 activates the 7-transmembrane-spanning Fzd9 receptor, which together with the co-receptor LRP5/6 activates dishevelled (DVL). Activated DVL inhibits the activity of the β-catenin “destruction complex” (indicated as a light blue ellipse). β-catenin is released from its complex with cadherin by the activity of the HGF receptor MET. When β-catenin is protected against destruction, it can enter the nucleus, bind the transcription factor LEF1 and co-factors to promote transcription of target genes like, for example, engrailed 2 (EN2). The functional consequence is an increase in cell growth and motility.
Once released from the membrane, the fate of β-catenin in the cytoplasm is determined by a multi-protein complex (frequently referred to as the ‘destruction complex’), consisting of two serine-threonine kinases (CK1α and GSK3), two scaffolding proteins (axin and APC) and the phosphatases PP1 and PP2A[15, 16]. Depending on the strength of the Wnt signal, this complex either promotes the catabolism of axin or that of β-catenin[17].
The factors which determine the shuttling of β-catenin between cytoplasm and nucleus are not entirely clear and its distribution seems to be determined by both cytosolic-retention factors (for example, axin, APC, cadherin) and nuclear retention factors (for example, BCL9)[20, 22, 23]. Within the nucleus, β-catenin again participates in several complexes, in this case consisting of high-mobility group (HMG) transcription factors (TCF7L 1–3; the latter is also called lymphoid enhancer factor-1; LEF1) and co-activators like for instance CREB binding protein (CREBBP)[24], PYGO[25] and BCL9[25].
In mammalian species two isoforms of GSK3 exist (α, β). Of these, the β-isoform has a higher expression level and has been studied preferentially. Activity and cellular localization of GSK3 are regulated by phosphorylation steps. Autophosphorylation at Tyr-216 of GSK3β is required for full enzymatic activity[26]. Importantly, the protein encoded by the gene “disrupted in schizophrenia-1” (DISC1) directly interacts with GSK3β and suppresses Tyr-216-autophosphorylation[27] and contributes to effective canonical Wnt signaling.
Several negative regulators of the Wnt pathway are known as well. These negative regulators act by intercepting the extracellular Wnt, by blocking the frizzled receptor or by blocking the LRP co-receptor[28]. Intracellularly, pathway activity can be reduced by phosphatases like RPTP β/ζ[29], PP1 or PP2A[15]. The full complexity of the Wnt pathway is still evolving[19].
The canonical Wnt pathway plays an important role in brain development[30–35] and synaptic function[36–38]. It is, therefore, evident that mutations in Wnt pathway genes were suspected to contribute to autism spectrum disorders and to psychiatric disorders in general[39, 40].
Overview of mutations in Wnt pathway in patients with ASD (part 1)
Multiple genome-wide screens have found evidence for linkage to autism on several chromosomes[7, 41, 42]. These loci are frequently quite large, and contain numerous potential candidate genes. A typical example is the broad linkage peak on 7q31 area that maps over 200 genes[7, 41, 43]. Among the >200 genes there are several that play a role in Wnt signal transduction, for example, the gene encoding Wnt2, the hepatocyte growth factor receptor Met (which can contribute to Wnt signaling by phosphorylating β-catenin at position Tyr-142), the phosphatase PTPRZ1 (RPTP β/ζ which reverses Tyr-142 phosphorylation), the Wnt-target gene, engrailed2 (EN2) and a gene called DOCK4 (a member of the extended β-catenin destruction complex). In the sections immediately below, the canonical Wnt2 cascade will be reviewed in detail, whereas additional genes involved in Wnt signaling will be discussed in Part 2 further down. The reader will note that the strength of the evidence for the individual genes varies considerably (actually, the evidence for individual genes is in no single case ‘compelling’; see Table1). The main purpose of the present review is, however, not to assess the validity of the individual finding, but more globally, to overview the overall pathway activity and to evaluate and estimate whether signaling is decreased or enhanced.
WNT2 (7q31.2)
Given the localization within the autism ‘hotspot’ 7q31, the WNT2 gene has been screened for non-synonymous mutations in autistic probands ascertained through the Collaborative Linkage Study of Autism by Wassink et al.[44]. The authors identified several variants that segregated with autism and severe language abnormality. Two subsequent linkage studies were not able to confirm the original findings[45, 46]; however, a more recent extended study again found an association[47]. This study involved a case–control study of 9 single-nucleotide polymorphisms (SNPs) within the WNT2 gene in 170 autism patients and 214 controls from Japan, and a follow-up of the positive results in a transmission disequilibrium test (TDT) in 98 Japanese autistic family trios. The significant associations from the initial part were replicated in the TDT part and the authors concluded that “WNT2 is a strong candidate gene for autism”[47]. The function of Wnt2 has also been investigated in laboratory experiments. In ventral midbrain cultures, administration of Wnt2 protein increased proliferation of progenitors and the number of dopamine neurons, whereas the opposite was found in WNT2 knock-out mice[48]. Thus, a too-strong Wnt2 signaling could lead to enhanced midbrain dopamine function, which eventually might relate to the repetitive behaviors seen in autism patients.
FZD9 (7q11.23)
Immuno-precipitation experiments demonstrated that Wnt2 interacts with frizzled 3 (Fzd3) and Fzd9, while an antibody for Fzd9, but not an Fzd3-antibody precipitated Wnt2[49]. This result indicates that Fzd9 is the preferred receptor for Wnt2. Evidence that Wnt2 not only binds, but also activates Fzd9 was provided by Karasawa et al.[50], who demonstrated that Wnt2 application to Fzd9 expressing HEK293 cells led to phosphorylation of dishevelled-1 (DVL1) and β-catenin-mediated gene transcription.
The 7q11.23 area is known to be relevant for the Williams-Beuren syndrome (WBS)[51, 52]. Copy number variants of the WBS-region are responsible for a complex neurological, cognitive and behavioral syndrome with frequent involvement of multiple additional other organ systems[53]. There are interesting similarities and differences in clinical features of patients with a 7q11.23 deletion compared to those with duplications (reviewed by[51]). Developmental delay, mental retardation and Attention Deficit Hyperactivity Disorder (ADHD) are found in both groups, but whereas patients with a deletion are excessively social and verbally skilled, patients carrying a duplication display severe delays in language development and deficits in social interaction[51, 54, 55]. Male duplication patients, furthermore, show hyperactivity, self-injury and aggression[54]. The critical region is approximately 1.4 to 1.5 Mb[52, 53] and contains some 20 genes, including FZD9. The consequence of a loss of the Fzd9 receptor has been investigated in mice. Homozygous deletion of FZD9 resulted in severe deficits in visuospatial learning and memory, in apoptosis in the dentate gyrus and in a lowered seizure threshold[56]. To a lesser extent, these effects were also observed in heterozygous FZD9-KO mice (that is, the situation analogous to WBS ‘deletion’ patients)[56]. There is evidence that Fzd9 is the main Fzd-subtype expressed on neuronal progenitor cells[57]. Given this information, it is conceivable that alterations in FZD9 gene-dose contribute the behavioral phenotype of patients with 7q11.23 copy number variations.
BCL9 (1q21)
BCL9 contributes to transduction of the Wnt signal by promoting transcriptional activity and nuclear retention of β-catenin[23, 25, 58]. BCL9 is located on 1q21.1, an area for which, as for FZD9, both microduplications and microdeletions are described[59, 60]. Mefford and colleagues[59] detected a duplication of 1q21.1 in 9 out of 5,218 patients with unexplained mental retardation, autism or congenital abnormalities. From these, 50% had autism or autistic behaviors; 62% had macrocephaly and mild dysmorphic features and in seven out of eight cases there was a delay in learning or development. Also, Brunetti-Pierri et al.[60] described cases with both duplications and deletions and confirm the presence of autism, dysmorphic features and seizures in each patient group. Aggression and ADHD were seen in both groups, while patients with a 1q21.1 microdeletion had notable short statures and microcephaly. The critical minimal area was determined to be about 1.35 Mb and contains 14 genes[60]. It is of note that both research groups consider hydrocephalus-inducing homologue 2 (HYDIN2) as the most likely candidate gene and do not discuss BCL9.
Discussion (Part 1)
The data reviewed above in principle describe the canonical Wnt cascade for Wnt2: Wnt2 activates its preferred receptor (Fzd9) and the intracellular signaling ultimately leads to BCL9-assisted β-catenin-mediated gene-transcription (see Figure1). Several aspects are noteworthy. Under the assumption that duplication-CNVs increase and deletions decrease Wnt pathway activity, it seems that both activation and inhibition is associated with autism. This observation is suggestive for a bell-shaped dose–response relationship between Wnt2 pathway activity and cognitive/linguistic development. But there are also differences between the groups. Patients with deletions of the Fzd9 genomic area are described to be highly socially active and empathic. This is in evident contrast with the aggression and social inhibition encountered in patients in whom the Wnt2 pathway (presumably) is overactive (Fzd9 duplication, BCL9 duplication). The canonical Wnt pathway has an important influence on organ sizes, including the brain[35]. In agreement with this, microcephaly was noted in BCL9 deletion patients, while macrocephaly was reported BCL9 duplication patients. These results indicate a linear dose-dependency between Wnt pathway activity and skull size. In the following sections, further genes that influence canonical Wnt signaling will be reviewed.
Overview of mutations in Wnt pathway in patients with ASD (Part 2)
WIF1 (12q14.3)
A genome-wide linkage analysis found autism to be linked to the 12q14 region[61]. The most significant linkage peak reason encompasses approximately 19 genes, including the Wnt inhibitory factor (WIF1). WIF1 is mainly known for its function as tumor suppressor and disruptive mutations and/or epigenetic silencing enhanced cancer risk by activating Wnt signaling. It is, however, unknown if mutations in WIF1 are responsible for the linkage to 12q14.
MARK1 (14q41)
MARK1 (microtubule affinity regulating kinase-1, also: PAR1) is one of the kinases which phosphorylates and, thereby, activates dishevelled[62, 63]. Several SNPs in the MARK1 gene were associated with ASD and one of these SNPs was reported to affect transcription rate[64]. Consistent with this, increased mRNA levels of MARK1 have been found in post mortem frontal cortex samples from autism subjects[64]. These data suggest that the autism-related mutations in MARK1 activate the Wnt pathway. Interestingly, both overexpression and silencing of MARK1 was found to result in shortened dendrites in mouse neocortical neurons, indicating a bell-shaped dose–response curve[64].
PTPRZ1 (7q31.3)
As described above, there are some 200 genes underneath the broad linkage peak on the long arm of chromosome 7, but fortunately specific copy number variations may allow some locus refinement. Quite recently a submicroscopic deletion of 5.4 Mb size was detected in a three-year-old boy with autism spectrum disorder that encompassed just 20 genes[65]. These 20 genes included the autism-candidate genes, CADPS2 and TSPAN12, but also the receptor tyrosine phosphatase, RPTP β/ζ (PTPRZ1). Since PTPRZ1 is a negative regulator of the Wnt pathway, the expected functional consequence of a haplo-insufficiency would be an increase in Wnt pathway activity. The clinical case description does, however, not fully support Wnt pathway hyperactivity since the patient’s head circumference was 2.4 SD below control and not, as expected for Wnt-pathway activation, above average.
MET (7q31)
MET is the receptor for hepatocyte growth factor (HGF). Activated by HGF, MET phosphorylates membrane-bound β-catenin at Tyr-142, which promotes dissociation of the β-catenin/cadherin complex[21, 66]. This releases β-catenin for nuclear signaling and, furthermore, by limiting cell-cell adhesion, it promotes cellular motility[21, 67, 68]. Cellular motility is crucial for interneuron migration, dendrite extension and synapse formation, and consequently, these processes are reduced by genetic disruption of MET[69, 70]. HGF-MET signaling, therefore, contributes to neuronal differentiation, to development of cerebral cortex and cerebellum and to axon growth[66, 69–71]. MET was shown to be associated with autism spectrum disorder in four independent family cohorts (reviewed by[72]), while one of risk alleles negatively regulated gene transcription[73]. A two-fold reduction of MET expression was found in post mortem temporal cortex of patients with autism[73]. In addition, two autism patients with a deletion CNV that encompassed MET were reported by Marshall et al.[74]. Finally, MET transcription is regulated by FOXP2, a further autism risk-gene[72]. These data indicate that reduction in MET function, perhaps paralleled by a reduction in Wnt signaling, contributes to autism susceptibility.
Classical cadherins: CDH8 (16q21), CDH10 (5p14.1), CDH13 (16q23.3) and CDH15 (16q24.3)
The cadherin family is composed of more than 80 members of which about one quarter are so-called “classical” cadherins[75]. Classical cadherins form a complex with β-catenin and play a role in cell-cell adhesion[76]. Loss of function mutations in classical cadherins lead to decreased cell adhesion, an increase in cell motility, β-catenin release and an increase in canonical Wnt signaling[13]. Pagnamenta et al.[77] described two families with a rare 1.6 Mb microdeletion of the classical cadherin, CDH8, in which affected family members suffered from autism and learning disability. Also, the classical cadherin, CDH13, was found disrupted by a microdeletion, albeit thus far in a single patient only[52]. Furthermore, a genome-wide association study in 780 families with autism spectrum disorder produced a strong association signal for SNPs located between CDH9 and CDH10 on chromosome 5p14.1[78]. Both cadherins are expressed in the brain, but the functional consequence of the SNPs was not investigated[78]. A further classical cadherin that may be implicated in autism is CDH15[79]. The authors described patients with a microdeletion of 16q24.3, an area just distal to CDH15. The predicted functional consequence of haplotype-insufficiencies of these cadherins would be enhanced β-catenin release and activation of the Wnt pathway.
TSC1 (9q34) and TSC2 (16p13.3)
Although mainly known for their role in the tuberous sclerosis syndrome, the tumor suppressors, tuberin (TSC2) and hamartin (TSC1), also participate in Wnt signaling. Both TSC1 and TSC2 were found to co-immunoprecipitate with axin and β-catenin[80]. Overexpression of TSC1 or TSC2 led to reduction of Wnt-induced β-catenin signaling, whereas mutations in TSC1 or TSC2, as found in tuberous sclerosis patients, led to increased canonical Wnt signaling[80]. In the brain, TSC1 and TSC2 have been implicated in cell body size, dendritic arborization, axonal outgrowth, neuronal migration, cortical lamination and spine formation[81]. The co-occurrence of autism and tuberous sclerosis has been recognized for decades and features of autism are present in up to half of the patients with tuberous sclerosis[81, 82]. These findings support the contention that increased Wnt signaling may contribute to autism.
DISC1 (1q42)
The ‘disrupted in schizophrenia 1’ (DISC1) gene is disrupted by a balanced chromosomal translocation (1; 11) (q42; q14.3) in a Scottish family with a high incidence of bipolar disorder, major depression and schizophrenia[83]. DISC1 can be considered an endogenous GSK3β inhibitor and in line with that activity, it promotes canonical Wnt to β-catenin signaling[27]. Expression in mice of the truncated DISC1-form identified in the Scottish family led to an attenuated neurite outgrowth of primary cortical neurons and behavioral hyperactivity[84]. To date, several linkage and association studies have confirmed the role of DISC1 in neuropsychiatric disorders[85, 86], including one study on autism and Asperger syndrome[87]. This study found an association between autism and a DISC1 intragenic microsatellite marker and, furthermore, an intragenic three-SNP haplotype and Asperger syndrome[87]. About 3% of patients tested by Kilpinen and colleagues[87] had a double diagnosis of autism plus either schizophrenia or bipolar disorder. In fact, the same haplotype was found to be associated with schizophrenia and bipolar disorder[88]. Analysis of the promoter region of DISC1 showed that FOXP2 suppresses DISC1 gene expression and protein levels[89]. Interestingly, autism-related mutations in FOXP2 diminished the suppressive effect on DISC1 transcription[89]. So it could be that both diminished and enhanced DISC1 function could contribute to autism spectrum disorder.
APC (5q21-q22)
Adenomatous polyposis coli (APC) is a negative regulator of the canonical Wnt pathway and functionally disruptive mutations are known to predispose for colorectal cancer. Barber et al.[90] describe a patient who originally was referred for autism and who was subsequently found to carry an APC deletion and had developed rectal cancer. Zhou and colleagues[91] reported a two part study. In a retrospective study in 75 autism spectrum disorder patients and 476 controls an association was found between a SNP in the 3’ untranslated region of the APC gene and autism. In the second part, the authors performed a prospective study in a new set of 75 ASD patients and 280 new controls on 4 SNPs spanning the entire 100 kB gene. While the individual SNPs were not significantly associated, one of the possible haplotypes (TGAG) was[91]. Unfortunately, the functional consequence of the TGAG haplotype regarding Wnt pathway activity remains unknown.
DOCK4 (7q31.1)
Also, the DOCK4 gene is located under the broad linkage peak on 7q31. Single nucleotide polymorphisms within DOCK4 were associated with autism risk in different populations[41]. Further evidence for a role of DOCK4 in autism was provided by the finding of a microdeletion CNV in an autistic sib-pair[41] and a deletion in a family with dyslexia[92]. DOCK4 is a component of the β-catenin destruction complex and decreasing its expression by siRNA reduced Wnt-induced TCF transcriptional activity[93]. DOCK4 is also involved in Wnt-induced activation of the GTPase, Rac, which is required for cell migration and synaptic function[93]. A study in rats showed that DOCK4 is highly expressed in the hippocampus and DOCK4 expression levels increase during periods of dendrite growth[94]. The data thus suggest that diminished DOCK4 level (and presumably, function), as found in autism, suppresses Wnt signaling and dendrite growth.
AHI1 (6q23.3)
Joubert syndrome is characterized by ataxia, abnormal breathing patterns, sleep apnea, abnormal eye and tongue movements and hypotonia, as well as distinct malformations of cerebellum and brain stem. A high percentage of patients with Joubert syndrome have been diagnosed with autism spectrum disorder[95]. One of the first genes identified to be involved in the pathogenesis of this syndrome was ‘Abelson’s Helper Integration 1’ (AHI1)[96]. The AHI1 gene was found to bear several mutations that give rise to non-functional variants of the encoded protein, Jouberin[96]. Dysfunction of Jouberin may thus lead to autism. In mice, AHI1 was distributed throughout cytoplasm, dendrites and axons of neurons and was expressed from embryonal Day 10.5 onwards[97]. In particular, AHI1 mRNA was expressed in cell bodies of midline-crossing neurons, providing an explanation for axonal abnormalities found in Joubert-syndrome[98]. Jouberin participates in the Wnt pathway by facilitating the nuclear accumulation of β-catenin[99], but it is currently not known if this is causally involved in the autism symptoms of Joubert syndrome patients.
EN2 (7q36)
EN2 (engrailed-2) is involved in regionalization, patterning and neuronal differentiation of the mid- and hindbrain and is strongly expressed in these areas during embryonic development. A low level of expression is maintained in adulthood; for instance, in the hippocampus and cerebral cortex[100]. The transcription of EN2 is enhanced by stimulation of the canonical Wnt pathway[101]. Several studies have shown an association between autism and SNPs in EN2 (for review, see[102]). Two intronic SNPs were over-transmitted to affected individuals both singly and as haplotype in separate data sets from North American origin[102]. The risk haplotype (A-C) led to higher transcription of EN2 than the opposite haplotype[102]. Remarkably, a study in Han Chinese autism cases confirmed EN2 as a susceptibility gene, but found the A-C haplotype to be protective[103]. It is conceivable that both deficits and overexpression of EN2 are disruptive for normal brain development. Knock-out mice, which lack both copies of EN2, display subtle cerebellar neuropathology and a behavior that could be interpreted as autism-like, for example, decreased play, reduced social sniffing and grooming, and reduced aggression[104]. For a better interpretation, one should also study EN2-overexpressing mice, but this has, to my knowledge, not been done. EN1 and, to a lesser degree, EN2 are expressed in dopamine neurons in the substantia nigra and ventral tegmental area[105].
CREBBP (16p13.3)
CREB binding protein (CREBBP; 16p13.3) and its close analogue EP300 (22q13.2) are transcriptional co-activators of β-catenin[24]. Mutations and deletions of the EP300 or CREBBP genes give rise to the Rubinstein-Taybi syndrome (characterized by broad thumbs and toes, short stature, distinctive facial features, impairments in cognitive and motor skills and micro- or macrocephaly; for review see[106]). Patients with deletions of CREBBP show cognitive impairment, autistic features and seizures[107]. Patients with a duplication of the 16p13.3 region, invariably encompassing the CREBBP gene, have also been described[108, 109]. The behavioral phenotype of these patients is relatively mild, but can include autism spectrum disorder[109], speech deficits and moderate mental retardation[108]. Experiments in rats have shown that CREB activation is required for hepatocyte growth factor-induced dendritic arborization during brain development[71].
Discussion (Part 2)
The literature reviewed above supports the contention that modification of genes affecting the activity of the canonical Wnt pathway can contribute to individual forms of autism spectrum disorder. Functionally active polymorphisms (SNPs) and copy number variations (CNVs) suggest both increases in Wnt signaling (SNPs in MARK1 and EN2 that increase gene transcription, PTPRZ1 deletion, cadherin deletion-CNVs, CREB duplication-CNV), as well as decreases in Wnt signaling (SNPs and CNVs in MET that reduce transcription, deletion-CNVs in DOCK4 and CREB, as well as disruptive SNPs in DISC1 and AHI1). While reviewing the functions of the Wnt-pathway genes, the reader may notice a recurrent theme: their effect on cellular motility (neurite growth, spine and synapse formation). Thus, hepatocyte growth factor activation of MET, disruption of cadherin /β-catenin complexes and downstream activation of CREB are involved in neurite extension and development of the dendritic arbor. TSC1 and TSC2 play a role in these processes, too. DOCK4 is expressed during periods of dendrite growth, while AHI1 is important for midline-crossing axons. A crucial observation is that both overexpression and genetic silencing of MARK1 resulted in too short dendrites. This indicates that both too much and too little Wnt pathway activity is deleterious for dendrite growth. Consequently, both hyperactivation and hypoactivity of the Wnt pathway will negatively affect cognitive function and, since language development is a cognitive skill, it is conceivable that linguistic capabilities are reduced too.
Medications that influence the canonical Wnt pathway
A study by Rasalam et al.[110] noted that 1 out of 10 children born from mothers taking antiepileptic medication had social, behavioral and language difficulties. Valproate was the drug that was most commonly associated with autistic disorder[110, 111]. This is supported by data from animal studies. When rats were prenatally (Day 12.5) exposed to a single dose of sodium-valproate, after birth they exhibited a lower sensitivity to pain, but a higher sensitivity to non-painful stimuli[112]. Furthermore, the animals displayed hyper-locomotion and stereotypy and lower exploratory activity, a decreased number of social behaviors and longer latency to social behavior. All behaviors appeared prior to puberty. Prenatal valproate use in rats is considered an animal model of autism[112]. Interestingly, prenatal exposure to valproate led to increases in mRNA and protein levels of WNT1 and WNT2 in the prefrontal cortex and hippocampus, and genes under transcriptional control of the Wnt pathway (for example, EN1, cyclin D1) were up-regulated[113]. Other medications which are suspected of contributing to autism prevalence are antidepressants, in particular the serotonin re-uptake inhibitors (SSRIs)[114]. A prospective population-based study by Croen et al.[115], in which prenatal SSRI exposure of autistic children was compared to SSRI exposure in control children, found a doubling of the risk of autism when the mother took an SSRI during the year before delivery, while the most pronounced risk was seen when exposure occurred during the first trimester. The authors concluded that exposure to SSRIs during the first trimester of pregnancy modestly increases the risk for autism spectrum disorder. Rats that were exposed to the SSRI, citalopram, during postnatal Days 8 to 21 displayed altered branching characteristic in hippocampal and neocortical neurons, had reduced myelination of callosal axons and, furthermore, showed impaired social behavior and response to novelty[116]. This result shows that alterations in central serotonin levels may interfere with normal brain development. Subchronic treatment of rats with the antidepressants citalopram, fluoxetine, venlafaxine and atamoxetine increased the expression of several Wnt-pathway genes; the effect shared by all antidepressants was an increase in WNT2- expression, involving both mRNA and protein levels[117]. It is evident that antidepressant-induced Wnt signaling has the propensity to influence brain development, and reviewed data provide further support for the contention that altered Wnt pathway activity is a risk factor for autism spectrum disorder.
Discussion (Part 3)
When particular medications like anticonvulsants or antidepressants can increase risk for autism, it is conceivable that appropriate medications can reduce autism risk. The information reviewed above suggests that alterations in the activity of the canonical Wnt pathway could contribute to autism risk and, consequently, pharmacotherapeutic correction of the aberrant pathway activity might help to improve symptoms. It seems that both hyperactivity and hypoactivity can generate symptoms, implying that patients have to be stratified according to their Wnt pathway activity status before pharmacotherapy can be initiated. How should this stratification be done? One possibility would be to stratify according to skull size. Unfortunately, the neuro-developmental mechanisms that regulate brain and skull growth are multiple, and involve not only the canonical Wnt pathway, but also growth-factor pathways like the ERK-mitogen-activated protein-kinase pathway, the PI3K-PKB-mTOR pathway, the Sonic hedgehog pathway (and so on) and, also, include nuclear receptor activators like retinoic acid, thyroid hormone, corticosteroids and gonadal steroids (for review see[118]). A more direct way to determine canonical Wnt-pathway activity would be to screen for proteins regulated by Wnt (for example, c-Myc, cyclin-D1 or ADAM10)[119–123]. Blood plasma levels of such proteins might become a biomarker for pathway activity.
Consequences for the treatment of autism spectrum disorders
Given the importance of the canonical Wnt pathway for the development of the brain and other organs, modifying its activity, in particular in young children, is a quite hazardous enterprise. Furthermore, since autism is a neurodevelopmental disorder, it cannot be excluded that drug treatment will be only effective during a narrow period, while treatment outside this critical period is inactive[124], and thus dangerous. Therefore, pharmaco-therapeutic treatment would only be justifiable if a valid surrogate marker for canonical Wnt pathway activity would be available. Under such circumstances, one could then consider treatment with ‘mild’ pathway-modifying drugs. Lithium is such a drug: it activates the canonical Wnt pathway[125] without at the same time raising cancer risk[126, 127]. Patients with Williams-Beuren syndrome, MET mutations, DOCK4 microdeletions or Joubert syndrome might indeed benefit from lithium treatment; however, it would probably be contra-indicated in patients with a cadherin haplo-insufficiency, tuberous sclerosis or MARK1 mutations. ‘Soft’ treatments of autism disorder related to Wnt pathway-hyperactivity can in principle be found among anti-cancer drugs. The non-steroidal, anti-inflammatory compound sulindac could be an option. This compound inhibits polymerization of dishevelled[128] and consequently inhibits β-catenin signaling[128–130]. Interestingly, the activity of sulindac is not related to COX-inhibition since sulindac-sulphone, a metabolite devoid of COX-inhibition, is equally effective as a Dvl-inhibitor as sulindac itself[128]. However, at present it is clearly premature to propose sulindac as treatment for autism (respectively, autism spectrum disorders).
Conclusion
Taken as a whole it seems safe to conclude that the activity of the canonical Wnt pathway is altered at least in a subset of patients with autism spectrum disorder. Whether correction of the deviant pathway activity leads to symptomatic improvement remains unknown. It is important to realize deviations from the optimum in both directions seem to increase the risk for autism spectrum disorder. This implies that patients, depending on their Wnt pathway activity, will have to be treated differentially.
Author’s contribution
HOK searched, read and summarized the literature, wrote the article and created the figure.
Author’s information
HOK worked for 29 years in the nervous system department of Novartis and led several research programs (including GSK3-inhibitors and AMPA-receptor antagonists). He is now retired.
Abbreviations
- ADAM:
-
A desintegrin and metalloproteinase
- ADHD:
-
Attention deficit hyperactivity disorder
- AHI:
-
Abelson’s helper integration
- APC:
-
Adenomatous polyposis coli
- BCL9:
-
B-cell lymphoma-9)
- CBP:
-
CREB binding protein
- CDH:
-
Cadherin
- CK:
-
Casein-kinase
- CNV:
-
Copy number variation
- DISC1:
-
Disrupted in schizophrenia-1
- DOCK4:
-
Dedicator of cytokinesis-4
- EN:
-
Engrailed
- FOXP:
-
Forkhead box P
- FZD:
-
Frizzled
- GSK3:
-
Glycogen-synthase kinase-3
- HEK293:
-
Human embryonic kidney 293
- HGF:
-
Hepatocyte growth factor
- LRP:
-
Low-density lipoprotein receptor-related protein
- MARK:
-
Microtubule affinity regulating kinase
- PP:
-
Protein phosphatase
- PTPR:
-
Protein tyrosine phosphatase receptor-type
- SNP:
-
Single nucleotide polymorphism
- SSRI:
-
Selective serotonin re-uptake inhibitor
- TCF:
-
T-cell factor
- TDT:
-
Transmission disequilibrium test
- TSC:
-
Tuberosclerosis
- WBS:
-
Williams-Beuren syndrome
- Wnt:
-
Wingless-type mouse mammary tumor virus integration site
- WIF:
-
Wnt inhibitory factor.
References
Beglinger LJ, Smith TH: A review of subtyping in autism and proposed dimensional classification model. J Autism Dev Disord. 2001, 31: 411-422. 10.1023/A:1010616719877.
Volkmar FR, State M, Klin A: Autism and autism spectrum disorders: diagnostic issues for the coming decade. J Child Psychol Psychiatry. 2009, 50: 108-115. 10.1111/j.1469-7610.2008.02010.x.
Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff A, Yuzda E, Rutter M: Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995, 25: 63-77. 10.1017/S0033291700028099.
Rosenberg RE, Law JK, Yenokyan G, McGready J, Kaufmann WE, Law PA: Characteristics and concordance of autism spectrum disorders among 277 twin pairs. Arch Pediatr Adolesc Med. 2009, 163: 907-914. 10.1001/archpediatrics.2009.98.
Gillis RF, Rouleau GA: The ongoing dissection of the genetic architecture of autistic spectrum disorder. Mol Autism. 2011, 2: 12-10.1186/2040-2392-2-12.
Polleux F, Lauder JM: Toward a developmental neurobiology of autism. Ment Retard Dev Disabil Res Rev. 2004, 10: 303-317. 10.1002/mrdd.20044.
Freitag CM: The genetics of autistic disorders and its clinical relevance: a review of the literature. Mol Psychiatry. 2007, 12: 2-22. 10.1038/sj.mp.4001896.
Toro R, Konyukh M, Delorme R, Leblond C, Chaste P, Fauchereau F, Coleman M, Leboyer M, Gillberg C, Bourgeron T: Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends Genet. 2010, 26: 363-372. 10.1016/j.tig.2010.05.007.
Anney RJ, Kenny EM, O’Dushlaine C, Yaspan BL, Parkhomenka E, Buxbaum JD, Sutcliffe J, Gill M, Gallagher L, Autism Genome Project: Gene-ontology enrichment in two independent family-based samples highlights biologically plausible processes for autism spectrum disorders. Eur J Hum Genet. 2011, 19: 1082-1089. 10.1038/ejhg.2011.75.
Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, Vitkup D: Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron. 2011, 70: 898-907. 10.1016/j.neuron.2011.05.021.
Sakai Y, Shaw CA, Dawson BC, Dugas DV, Al-Mohtaseb Z, Hill DE, Zoghbi HY: Protein interactome reveals converging molecular pathways among autism disorders. Sci Transl Med. 2011, 3: 86ra49-10.1126/scitranslmed.3002166.
Hussman JP, Chung RH, Griswold AJ, Jaworski JM, Salyakina D, Ma D, Konidari I, Whitehead PL, Vance JM, Martin ER, Cuccaro ML, Gilbert JR, Haines JL, Pericak-Vance MA: A noise-reduction GWAS analysis implicates altered regulation of neurite outgrowth and guidance in autism. Mol Autism. 2011, 2: 1-10.1186/2040-2392-2-1.
Nelson WJ, Nusse R: Convergence of Wnt, β-catenin, and cadherin pathways. Science. 2004, 303: 1483-1487. 10.1126/science.1094291.
Gao C, Chen YG: Dishevelled: the hub of Wnt signaling. Cell Signal. 2010, 22: 717-727. 10.1016/j.cellsig.2009.11.021.
Buechling T, Boutros M: Wnt signaling: signaling at and above the receptor level. Curr Top Dev Biol. 2011, 97: 21-53.
Kikuchi A, Yamamoto H, Sato A, Matsumoto S: New insights into the mechanism of Wnt signaling pathway activation. Int Rev Cell Mol Biol. 2011, 291: 21-71.
Hay E, Fauchau C, Suc-Royer I, Touitou R, Stiot V, Vayssiere B, Baron R, Roman-Roman S, Rawadi G: Interaction between LRP5 and Frat1 mediates the activation of the Wnt canonical pathway. J Biol Chem. 2005, 280: 13616-13623. 10.1074/jbc.M411999200.
Zeng X, Huang H, Tamai K, Zhang X, Harada Y, Yokota C, Almeida K, Wang J, Doble B, Woodgett J, Wynshaw-Boris A, Hsieh JC, He X: Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development. 2008, 135: 367-375.
Metcalfe C, Bienz M: Inhibition of GSK3 by Wnt signalling – two contrasting models. J Cell Sci. 2011, 124: 3537-3544. 10.1242/jcs.091991.
Brembeck FH, Rosario M, Birchmeier W: Balancing cell adhesion and Wnt signaling, the key role of β-catenin. Curr Opin Genet Dev. 2006, 16: 51-59. 10.1016/j.gde.2005.12.007.
Heuberger J, Birchmeier W: Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb Perspect Biol. 2010, 2: a002915-10.1101/cshperspect.a002915.
Henderson BR, Fagotto F: The ins and outs of APC and β-catenin nuclear transport. EMBO Rep. 2002, 3: 834-839. 10.1093/embo-reports/kvf181.
Städeli R, Hoffmans R, Basler K: Transcriptional control of nuclear Arm/β-catenin. Curr Biol. 2006, 16: R378-R385. 10.1016/j.cub.2006.04.019.
Hecht A, Vleminckx K, Stemmler MP, van Roy F, Kemler R: The p300/CBP acetyltransferases function as transcriptional coactivators of β-catenin in vertebrates. EMBO J. 2000, 19: 1839-1850. 10.1093/emboj/19.8.1839.
Kramps T, Peter O, Brunner E, Nellen D, Froesch B, Chatterjee S, Murone M, Züllig S, Basler K: Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear β-catenin – TCF complex. Cell. 2002, 109: 47-60. 10.1016/S0092-8674(02)00679-7.
Lochhead PA, Kinstrie R, Sibbet G, Rawjee T, Morrice N, Cleghon V: A chaperone-dependent GSK3β transitional intermediate mediates activation-loop autophosphorylation. Mol Cell. 2006, 24: 627-633. 10.1016/j.molcel.2006.10.009.
Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, Tassa C, Berry EM, Soda T, Singh KK, Biechele T, Petryshen TL, Moon RT, Haggarty SJ, Tsai LH: Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3β/β-catenin signaling. Cell. 2009, 136: 1017-1031. 10.1016/j.cell.2008.12.044.
Lustig B, Behrens J: The Wnt signaling pathway and its role in tumor development. J Cancer Res Clin Oncol. 2003, 129: 199-221.
Meng K, Rodriguez-Pena A, Dimitrov T, Chen W, Yamin M, Noda M, Deuel TF: Pleiotrophin signals increased tyrosine phosphorylation of β-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase β/ζ. Proc Nat Acad Sci U S A. 2000, 97: 2603-2608. 10.1073/pnas.020487997.
Galceran J, Miyashita-Lin EM, Devaney E, Rubinstein JL, Grosschedl R: Hippocampus development and generation of dentate gyrus granule cells is regulated by LEF1. Development. 2000, 127: 469-482.
Lee SM, Tole S, Grove E, McMahon AP: A local Wnt-3a signal is required for development of the mammalian hippocampus. Development. 2000, 127: 457-467.
Zhou CJ, Zhao C, Pleasure SJ: Wnt signaling mutants have decreased dentate granule cell production and radial glial scaffolding abnormalities. J Neurosci. 2004, 24: 121-126. 10.1523/JNEUROSCI.4071-03.2004.
Yu X, Malenka RC: β-catenin is critical for dendritic morphogenesis. Nat Neurosci. 2003, 6: 1169-1177. 10.1038/nn1132.
Lie DC, Colamarino SA, Song HJ, Desire L, Mira H, Consiglio A, Lein ES, Jessberger S, Lansford H, Dearie AR, Gage FH: Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005, 437: 1370-1375. 10.1038/nature04108.
Rosso SB, Sussman D, Wynshaw-Boris A, Salinas PC: Wnt signaling through dishevelled, Rac and JNK regulates dendritic development. Nat Neurosci. 2005, 8: 34-42. 10.1038/nn1374.
Krylova O, Herreros J, Cleverley KE, Ehler E, Henriquez JP, Hughes SM, Salinas PC: WNT-3, expressed by motoneurons, regulates terminal arborization of neurotrophin-3-responsive spinal sensory neurons. Neuron. 2002, 35: 1043-1056. 10.1016/S0896-6273(02)00860-7.
Packard M, Koo ES, Gorczyca M, Sharpe J, Cumberledge S, Budnik V: The drosophila Wnt, Wingless, provides an essential signal for pre- and postsynaptic differentiation. Cell. 2002, 111: 319-330. 10.1016/S0092-8674(02)01047-4.
Chen J, Park CS, Tang SJ: Activity-dependent synaptic Wnt release regulates hippocampal long term potentiation. J Biol Chem. 2006, 281: 11910-11916. 10.1074/jbc.M511920200.
De Ferrari GV, Moon RT: The ups and downs of Wnt signaling in prevalent neurological disorders. Oncogene. 2006, 25: 7545-7553. 10.1038/sj.onc.1210064.
Okerlund ND, Cheyette BN: Synaptic Wnt signaling – a contributor to major psychiatric disorders. J Neurodev Disord. 2011, 3: 162-174. 10.1007/s11689-011-9083-6.
Maestrini E, Pagnamenta AT, Lamb JA, Bacchelli E, Sykes NH, Sousa I, Toma C, Barnby G, Butler H, Winchester L, Scerri TS, Minopoli F, Reichert J, Cai G, Buxbaum JD, Korvatska O, Schellenberg GD, Dawson G, de Bildt A, Minderaa RB, Mulder EJ, Morris AP, Bailey AJ, Monaco AP, IMGSAC: High density SNP association study and copy number variation analysis of the AUTS1 and AUTS5 loci implicate the IMMP2L-DOCK4 gene region in autism susceptibility. Mol Psychiatry. 2010, 15: 954-968. 10.1038/mp.2009.34.
Yang MS, Gill M: A review of gene linkage, association and expression studies in autism and an assessment of convergent evidence. Int J Dev Neurosci. 2007, 25: 69-85. 10.1016/j.ijdevneu.2006.12.002.
Folstein SE, Mankoski RE: Chromosome 7q: where autism meets language disorder?. Am J Hum Genet. 2000, 67: 278-281. 10.1086/303034.
Wassink TH, Piven J, Vieland VJ, Huan J, Swiderski RE, Pietila J, Braun T, Beck G, Folstein SE, Haines JL, Sheffield VC: Evidence supporting WNT2 as an autism susceptibility gene. Am J Med Genet. 2001, 105: 406-413. 10.1002/ajmg.1401.
McCoy PA, Shao Y, Wolpert CM, Donnely SL, Ashley-Koch A, Abel HL, Ravan SA, Abramson RK, Wright HH, DeLong GR, Cuccaro ML, Gilbert JR, Pericak-Vance MA: No association between the WNT2 gene and autistic disorder. Am J Med Genet. 2002, 114: 106-109. 10.1002/ajmg.10182.
Li J, Nguyen L, Gleason C, Lotspeich L, Spiker D, Risch N, Myers RM: Lack of evidence for an association between WNT2 and RELN polymorphisms and autism. Am J Med Genet. 2004, 126B: 51-57. 10.1002/ajmg.b.20122.
Marui T, Funatogawa I, Kishi S, Yamamoto K, Matsumoto H, Hashimoto O, Jinde S, Nishida H, Sugiyama T, Kasia K, Watanabe K, Kano Y, Kato N: Association between autism and variants in the wingless-type MMTV integration site family member 2 (WNT2) gene. Int J Neuropsychopharmacol. 2010, 13: 443-449. 10.1017/S1461145709990903.
Sousa KM, Villaescusa JC, Cajanek L, Ondr JK, Castelo-Branco G, Hofstra W, Bryja V, Palmberg C, Bergman T, Wainwright B, Lang RA, Arenas E: Wnt2 regulates progenitor proliferation in the developing ventral midbrain. J Biol Chem. 2010, 285: 7246-7253. 10.1074/jbc.M109.079822.
Wang HX, Tekpetey FR, Kidder GM: Identification of Wnt/β-catenin signaling pathway components in human cumulus cells. Mol Hum Reprod. 2009, 15: 11-17. 10.1093/molehr/gan070.
Karasawa T, Yokokura H, Kitajewski J, Lombroso PJ: Frizzled-9 is activated byWnt-2 and functions in Wnt/ β-catenin signaling. J Biol Chem. 2002, 277: 37479-37486. 10.1074/jbc.M205658200.
Merla G, Brunetti-Pierri N, Micale L, Fusco C: Copy number variants at Williams-Beuren syndrome 7q11.23 region. Hum Genet. 2010, 128: 3-26. 10.1007/s00439-010-0827-2.
Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, Mason CE, Bilguvar K, Celestino-Soper PB, Choi M, Crawford EL, Davis L, Wright NR, Dhodapkar RM, DiCola M, DiLullo NM, Fernandez TV, Fielding-Singh V, Fishman DO, Frahm S, Garagaloyan R, Goh GS, Kammela S, Klei L, Lowe JK, Lund SC: Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011, 70: 863-885. 10.1016/j.neuron.2011.05.002.
Van der Aa N, Rooms L, Vandeweyer G, van de Ende J, Reyniers E, Fichera M, Romano C, Della Chiaie B, Mortier G, Menten B, Destrée A, Maystadt I, Männik K, Kurg A, Reimand T, McMullan D, Oley C, Brueton L, Bongers EM, van Bon BW, Pfund R, Jacquemont S, Ferrarini A, Martinet D, Schrander-Stumpel C, Stegmann AP, Frints SG, de Vries BB, Ceulemans B, Kooy RF: Fourteen new cases contribute to the characterization of the 7q11.23 microduplication syndrome. Eur J Med Genet. 2009, 52: 94-100. 10.1016/j.ejmg.2009.02.006.
Berg JS, Brunetti-Pierri N, Peters SU, Kang SH, Fong CT, Salamone J, Freedenberg D, Hannig VL, Prock LA, Miller DT, Raffalli P, Harris DJ, Erickson RP, Cunniff C, Clark GD, Blazo MA, Peiffer DA, Gunderson KL, Sahoo T, Patel A, Lupski JR, Beaudet AL, Cheung SW: Speech delay and autism spectrum behaviors are frequently associated with duplications of the 7q11.23 Williams-Beuren syndrome region. Genet Med. 2007, 9: 427-441. 10.1097/GIM.0b013e3180986192.
Morris CA: The behavioral phenotype of Williams syndrome: a recognizable patter of neurodevelopment. Am J Med Genet. 2010, 154C: 427-431. 10.1002/ajmg.c.30286.
Zhao C, Aviles C, Abel RA, Almli CR, McQuillen P, Pleasure SJ: Hippocampal and visuospatial learning defects in mice with a deletion of frizzled 9, a gene in the Williams syndrome deletion interval. Development. 2005, 132: 2917-2927. 10.1242/dev.01871.
Rawal N, Castelo-Branco G, Sousa KM, Kele J, Kobayashi H, Arenas E: Dynamic temporal and cell type-specific expression of Wnt signaling components in the developing midbrain. Exp Cell Res. 2006, 312: 1626-1636. 10.1016/j.yexcr.2006.01.032.
Adachi S, Jigami T, Yasui T, Nakano T, Ohwada S, Omori Y, Sugano S, Ohkwara B, Shibuya H, Nakamura T, Akiyama T: Role of BCL9-related β-catenin binding protein, B9L, in tumorigenesis induced by aberrant activation of Wnt signaling. Cancer Res. 2004, 64: 8496-8501. 10.1158/0008-5472.CAN-04-2254.
Mefford H, Sharp A, Baker C, Itsara A, Jiang Z, Buysse K, Huang S, Maloney V, Crolla J, Barella D, Collins A, Mercer C, Norga K, de Ravel T, Devriendt K, Bongers EM, de Leeuw N, Reardon W, Gimelli S, Bena F, Hennekam RC, Male A, Gaunt L, Clayton-Smith J, Simonic I, Park SM, Mehta SG, Nik-Zainal S, Woods CG, Firth HV: Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med. 2008, 359: 1685-1699. 10.1056/NEJMoa0805384.
Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA, Sahoo T, Lalani SR, Graham B, Lee B, Shinawi M, Shen J, Kang SH, Pursley A, Lotze T, Kennedy G, Lansky-Shafer S, Weaver C, Roeder ER, Grebe TA, Arnold GL, Hutchison T, Reimschisel T, Amato S, Geragthy MT, Innis JW, Obersztyn E, Nowakowska B, Rosengren SS, Bader PI, Grange DK: Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet. 2008, 40: 1466-1471. 10.1038/ng.279.
Ma DQ, Cuccaro ML, Jaworski JM, Haynes CS, Stephan DA, Parod J, Abramson RK, Wright HH, Gilbert JR, Haines JL, Pericak-Vance MA: Dissecting the locus heterogeneity of autism: significant linkage to chromosome 12q14. Mol Psychiatry. 2007, 12: 376-384. 10.1038/sj.mp.4001927.
Sun TQ, Lu B, Feng JJ, Reinhard C, Jan YN, Fantl WJ, Williams LT: PAR-1 is a dishevelled-associated kinase and positive regulator of Wnt signalling. Nat Cell Biol. 2001, 3: 628-636. 10.1038/35083016.
Bernatik O, Ganji RS, Dijksterhuis JP, Konik P, Cervenka I, Polonio T, Krejci P, Schulte G, Bryja V: Sequential activation and inactivation of dishevelled in the Wnt/β-catenin pathway by casein kinases. J Biol Chem. 2011, 286: 10396-10410. 10.1074/jbc.M110.169870.
Maussion G, Carayol J, Lepagnol-Bestel AM, Tores F, Loe-Mie Y, Milbreta U, Rousseau F, Fontaine K, Renaud J, Moalic JM, Philippi A, Chedotal A, Gorwood P, Ramoz N, Hager J, Simonneau M: Convergent evidence identifying MAP/microtubule affinity-regulating kinase 1 (MARK1) as a susceptibility gene for autism. Hum Mol Genet. 2008, 17: 2541-2551. 10.1093/hmg/ddn154.
Okamoto N, Hatsukawa Y, Shimojima K, Yamamoto T: Submicroscopic deletion in 7q31 ecompassing CADPS2 and TSPAN12 in a child with autism spectrum disorder. Am J Med Genet. 2011, 155A: 1568-1573.
David MD, Yeramian A, Dunach M, Llovera M, Canti C, de Herreros AG, Comella JX, Herreros J: Signalling by neurotrophins and hepatocyte growth factor regulates axon morphogenesis by differential β-catenin phosphorylation. J Cell Sci. 2008, 121: 2718-2730. 10.1242/jcs.029660.
Papkoff J, Aikawa M: WNT-1 and HGF regulate GSK3β activity and β-catenin signaling in mammary epithelial cells. Biochem Biophys Res Commun. 1998, 247: 851-858. 10.1006/bbrc.1998.8888.
Hiscox S, Jiang WG: Association of the HGF/SF receptor, c-met, with the cell-surface adhesion molecule, E-cadherin, and catenins in human tumor cells. Biochem Biophys Res Commun. 1999, 261: 406-411. 10.1006/bbrc.1999.1002.
Powell EM, Campbell DB, Stanwood GD, Davis C, Noebels JL, Levitt P: Genetic disruption of cortical interneuron development causes region- and GABA cell type-specific deficits, epilepsy, and behavioral dysfunction. J Neurosci. 2003, 23: 622-631.
Judson MC, Bergman MY, Campbell DB, Eagleson KL, Levitt P: Dynamic gene and protein expression patterns of the autism-associated Met receptor tyrosine kinase in the developing mouse forebrain. J Comp Neurol. 2009, 513: 511-531. 10.1002/cne.21969.
Finsterwald C, Martin JL: Cellular mechanisms underlying the regulation of dendritic development by hepatocyte growth factor. Eur J Neurosci. 2011, 34: 1053-1061. 10.1111/j.1460-9568.2011.07839.x.
Mukamel Z, Konopka G, Wezler E, Osborn G, Dong H, Bergman MY, Levitt P, Geschwind DH: Regulation of MET by FOXP2, genes implcated in higher cognitive dysfunction and autism risk. J Neurosci. 2011, 31: 11437-11442. 10.1523/JNEUROSCI.0181-11.2011.
Campbell DB, D’Oronzio R, Garbett K, Ebert PJ, Mirnics K, Levitt P, Persico AM: Disruption of cerebral cortex MET signaling in autism spectrum disorder. Ann Neurol. 2007, 62: 243-250. 10.1002/ana.21180.
Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, Thiruvahindrapduram B, Fiebig A, Schreiber S, Friedman J, Ketelaars CE, Vos YJ, Ficicioglu C, Kirkpatrick S, Nicolson R, Sloman L, Summers A, Gibbons CA, Teebi A, Chitayat D, Weksberg R, Thompson A, Vardy C, Crosbie V, Luscombe S, Baatjes R: Structural variations of chromosomes in autism disorder. Am J Hum Genet. 2008, 82: 477-488. 10.1016/j.ajhg.2007.12.009.
Yagi T, Takeichi M: Cadherin superfamily genes: functions, genomic organization, and neurologic diversity. Genes Dev. 2000, 14: 1169-1180.
Arikkath J, Reichardt LF: Cadherins and catenins at synapses: roles in synaptogenesis and synaptic plasticity. Trends Neurosci. 2008, 31: 487-494. 10.1016/j.tins.2008.07.001.
Pagnamenta AT, Khan H, Walker S, Gerrelli D, Wing K, Bonaglia MC, Giorda R, Berney T, Mani E, Molteni M, Pinto D, Le Couteur A, Hallmayer J, Sutcliffe JS, Szatmari P, Paterson AD, Scherer SW, Vieland VJ, Monaco AP: Rare familial 16q21 microdeletions under a linkage peak indicate cadherin 8 (CDH8) in susceptibility to autism and learning disability. J Med Genet. 2011, 48: 48-54. 10.1136/jmg.2010.079426.
Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, Salyakina D, Imielinski M, Bradfield JP, Sleiman PM, Kim CE, Hou C, Frackelton E, Chiavacci R, Takahashi N, Sakurai T, Rappaport E, Lajonchere CM, Munson J, Estes A, Korvatska O, Piven J, Sonnenblick LI, Alvarez Retuerto AI, Herman EI, Dong H, Hutman T, Sigman M, Ozonoff S, Klin A: Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009, 459: 528-533. 10.1038/nature07999.
Willemsen MH, Fernandez BA, Bacino CA, Gerkes E, de Brouwer APM, Pfundt R, Sikkema-Raddatz B, Scherer SW, Marshall CR, Potocki L, van Bokhoven H, Kleefstra T: Identification of ANKRD11 and ZNF778 as candidate genes for autism and variable cognitive impairment in the novel 16q24.3 microdeletion syndrome. Eur J Hum Genet. 2010, 18: 429-435. 10.1038/ejhg.2009.192.
Mak BC, Takemaru KI, Kenerson HL, Moon RT, Yeung RS: The tuberin-hamartin complex negatively regulates β-catenin signaling activity. J Biol Chem. 2003, 278: 5947-5951. 10.1074/jbc.C200473200.
Orlova KA, Crino PB: The tuberous sclerosis complex. Ann N Y Acad Sci. 2010, 1184: 87-105. 10.1111/j.1749-6632.2009.05117.x.
Wiznitzer M: Autism and tuberous sclerosis. J Child Neurol. 2004, 19: 675-679.
St Clair D, Blackwood D, Muir W, Carothers A, Walker M, Spowart G, Gosden C, Evans HJ: Association within a family of a balanced autosomal translocation with major mental illness. Lancet. 1990, 336: 13-16. 10.1016/0140-6736(90)91520-K.
Pletnikov MV, Ayhan Y, Nikolskaia O, Xu Y, Ovanesov MV, Huang H, Mori S, Moran TH, Ross CA: Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol Psychiatry. 2008, 13: 173-186. 10.1038/sj.mp.4002079.
Brandon NJ, Sawa A: Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat Rev Neurosci. 2011, 12: 707-722. 10.1038/nrn3120.
Porteous DJ, Millar JK, Brandon NJ, Sawa A: DISC1 at 10: connecting psychiatric genetics and neuroscience. Trends Mol Med. 2011, 17: 699-706. 10.1016/j.molmed.2011.09.002.
Kilpinen H, Ylisaukko-oja T, Hennah W, Palo OM, Varilo T, Vanhala R, Nieminen-von Wendt T, von Wendt L, Paunio T, Peltonen L: Association of DISC1 with autism and Asperger syndrome. Mol Psychiatry. 2008, 13: 187-196. 10.1038/sj.mp.4002031.
Hennah W, Thomson P, Peltonen L, Porteous D: Genes and schizophrenia: the role of DISC1 in major mental illness. Schizophr Bull. 2006, 32: 409-416.
Walker RM, Hill AE, Newman AC, Hamilton G, Torrance HS, Anderson SM, Ogawa F, Derizioti P, Nicod J, Vernes SC, Fisher SE, Thomson PA, Porteous DJ, Evans KL: The DISC1 promoter: characterization and regulation by FOXP2. Hum Mol Genet. 2012, 21: 2862-2872. 10.1093/hmg/dds111.
Barber JC, Ellis KH, Bowles LV, Delhanty JD, Ede RF, Male BM, Eccles DM: Adenomatous polyposis coli and a cytogenic deletion of chromosome 5 resulting from a maternal intrachromosomal insertion. J Med Genet. 1994, 31: 312-316. 10.1136/jmg.31.4.312.
Zhou XL, Giacobini M, Anderlid BM, Anckarsäter H, Omrani D, Gillberg C, Nordenskjöld M, Lindblom A: Association of adenomatous polyposis coli (APC) gene polymorphisms with autism spectrum disorder (ASD). Am J Med Genet. 2007, 144B: 351-354. 10.1002/ajmg.b.30415.
Pagnamenta AT, Bacchelli E, de Jonge MV, Mirza G, Scerri TS, Minopoli F, Chiocchetti A, Ludwig KU, Hoffmann P, Paracchini S, Lowy E, Harold DH, Chapman JA, Klauck SM, Poustka F, Houben RH, Staal WG, Ophoff RA, O'Donovan MC, Williams J, Nöthen MM, Schulte-Körne G, Deloukas P, Ragoussis J, Bailey AJ, Maestrini E, Monaco AP, International Molecular Genetic Study Of Autism Consortium: Characterization of a family with rare deletions in CNTNAP5 and DOCK4 suggests novel risk loci for autism and dyslexia. Biol Psychiatry. 2010, 68: 320-328. 10.1016/j.biopsych.2010.02.002.
Upadhyay G, Goessling W, North TE, Xavier R, Zon LI, Yajnik V: Molecular association between β-catenin degradation complex and Rac guanine exchange factor DOCK4 is essential for Wnt/β-catenin signaling. Oncogene. 2008, 27: 5845-5855. 10.1038/onc.2008.202.
Ueda S, Fujimoto S, Hiramoto K, Negishi M, Katoh H: Dock4 regulates dendritic development in hippocampal neurons. J Neurosci Res. 2008, 86: 3052-3061. 10.1002/jnr.21763.
Alvarez Retuerto AI, Cantor RM, Gleeson JG, Ustaszewska A, Schackwitz WS, Pennacchio LA, Geschwind DH: Association of common variants in the Joubert syndrome gene (AHI1) with autism. Hum Mol Genet. 2008, 17: 3887-3896. 10.1093/hmg/ddn291.
Parisi MA, Doherty D, Chance PF, Glass IA: Joubert syndrome (and related disorders) (OMIM 213300). Eur J Hum Genet. 2007, 15: 511-521. 10.1038/sj.ejhg.5201648.
Doering JE, Kane K, Hsiao YC, Yao C, Shi B, Slowik AD, Dhagat B, Scott DD, Ault JG, Page-McCaw PS, Ferland RJ: Species differences in the expression of Ahi1, a protein implicated in the neurodevelopmental disorder Joubert syndrome, with preferential accumulation to stigmoid bodies. J Comp Neurol. 2008, 511: 238-256. 10.1002/cne.21824.
Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, Al-Nouri D, Al-Rumayyan A, Topcu M, Gascon G, Bodell A, Shugart YY, Ruvolo M, Walsh CA: Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet. 2004, 36: 1008-1013. 10.1038/ng1419.
Lancaster MA, Louie CM, Silhavy JL, Sintasath L, DeCambre M, Nigam SK, Willert K, Gleeson JG: Impaired Wnt-β-catenin signaling disrupts adult renal homeostasis and leads to cystic kidney ciliopathy. Nat Med. 2009, 15: 1046-1054. 10.1038/nm.2010.
Tripathi PP, Sgado P, Scali M, Viaggi C, Casarosa S, Simon HH, Vaglini F, Corsini GU, Bozzi Y: Increased susceptibility to kainic acid-induced seizures in Engrailed-2 knockout mice. Neuroscience. 2009, 159: 842-849. 10.1016/j.neuroscience.2009.01.007.
McGrew LL, Takemaru KI, Bates R, Moon RT: Direct regulation of Xenopus engrailed-2 promoter by the Wnt signaling pathway, and a molecular screen for Wnt-responsive genes, confirm a role for Wnt signaling during neural patterning in Xenopus. Mech Dev. 1999, 87: 21-32. 10.1016/S0925-4773(99)00136-7.
Benayed R, Choi J, Matteson PG, Gharani N, Kamdar S, Brzustowicz LM, Millonig JH: Autism-associated haplotype affects the regulation of the homeobox gene, ENGRAILED 2. Biol Psychiatry. 2009, 66: 911-917. 10.1016/j.biopsych.2009.05.027.
Yang P, Shu BC, Hallmayer JF, Lung FW: Intronic single nucleotide polymorphisms of Engrailed Homeobox 2 modulate the disease vulnerability of autism in a Han Chinese population. Neuropsychobiology. 2010, 62: 104-115. 10.1159/000315441.
Cheh MA, Millonig JH, Roselli LM, Ming X, Jacobsen E, Kamdar S, Wagner GC: En2 knockout mice display neurobehavioral and neurochemical alterations relevant to autism spectrum disorder. Brain Res. 2006, 1116: 166-176. 10.1016/j.brainres.2006.07.086.
Di Nardo AA, Nedelec S, Trembleau A, Volovitch M, Prochiantz A, Montesinos ML: Dendritic localization and activity-dependent translation of Engrailed1 transcription factor. Mol Cell Neurosci. 2007, 35: 230-236. 10.1016/j.mcn.2007.02.015.
Roelfsema JH, Peters DJ: Rubinstein-Taybi syndrome: clinical and molecular overview. Exp Rev Mol Med. 2007, 9: 1-16.
Schorry EK, Keddache M, Lanphear N, Rubinstein JH, Srodulski S, Fletcher D, Blough-Pfau RI, Grabowski GA: Genotype-phenotype correlations in Rubinstein-Taybi syndrome. Am J Med Genet. 2008, 146A: 2512-2519. 10.1002/ajmg.a.32424.
Marangi G, Leuzzi V, Orteschi D, Grimaldi ME, Lecce R, Neri G, Zollino M: Duplication of the Rubinstein-Taybi region on 16p13.3 is associated with a distinctive phenotype. Am J Med Genet. 2008, 146A: 2313-2317. 10.1002/ajmg.a.32460.
Thienpont B, Bena F, Breckpot J, Philip N, Menten B, Van Esch H, Scalais E, Salamone JM, Fong CT, Kussmann JL, Grange DK, Gorski JL, Zahir F, Yong SL, Morris MM, Gimelli S, Fryns JP, Mortier G, Friedman JM, Villard L, Bottani A, Vermeesch JR, Cheung SW, Devriendt K: Duplications of the critical Rubinstein-Taybi deletion region on chromosome 16p13.3 cause a novel recognisable syndrome. J Med Genet. 2010, 47: 155-161. 10.1136/jmg.2009.070573.
Rasalam AD, Hailey H, Williams JHG, Moore SJ, Turnpenny PD, Lloyd DJ, Dean JCS: Characteristics of fetal anticonvulsant syndrome associated autistic disorder. Dev Med Child Neurol. 2005, 47: 551-555. 10.1017/S0012162205001076.
Moore SJ, Turnpenny P, Quinn A, Glover S, Lloyd DJ, Montgomery T, Dean JC: A clinical study of 57 children with fetal anticonvulsant syndromes. J Med Genet. 2000, 37: 489-497. 10.1136/jmg.37.7.489.
Schneider T, Przewlocki R: Behavoral alterations in rats prenatally exposed to valproic acid: animal model of autism. Neuropsychopharmacology. 2005, 30: 80-89. 10.1038/sj.npp.1300518.
Wang Z, Xu L, Zhu X, Cui W, Sun Y, Nisjijo H, Peng Y, Li R: Demethylation of specific Wnt/β-catenin pathway genes and its upregulation in rat brain induced by prenatal valproate exposure. Anat Rec. 2010, 293: 1947-1953. 10.1002/ar.21232.
Levitt P: Serotonin and the autisms; a red flag or a red herring?. Arch Gen Psychiatry. 2011, 68: 1093-1094. 10.1001/archgenpsychiatry.2011.98.
Croen LA, Grether JK, Yoshida CK, Odouli R, Hendrick V: Antidepressant use during pregnancy and childhood autism spectrum disorders. Arch Gen Psychiatry. 2011, 68: 1104-1112. 10.1001/archgenpsychiatry.2011.73.
Simpson KL, Weaver KJ, de Villers-Sidani E, Lu JY, Cai Z, Pang Y, Rodriguez-Porcel F, Paul IA, Merzenich M, Lin RC: Perinatal antidepressant exposure alters cortical network function in rodents. Proc Nat Acad Sci U S A. 2011, 108: 18465-18470. 10.1073/pnas.1109353108.
Okamoto H, Voleti B, Banasr M, Sarhan M, Duric V, Girgenti MJ, DiLeone RJ, Newton SS, Duman RS: Wnt2 expression and signaling is increased by different classes of antidepressant treatments. Biol Psychiatry. 2010, 68: 521-527. 10.1016/j.biopsych.2010.04.023.
McCaffery P, Deutsch CK: Macrocephaly and the control of brain growth in autistic disorders. Prog Neurobiol. 2005, 77: 38-56. 10.1016/j.pneurobio.2005.10.005.
Haegele L, Ingold B, Naumann H, Tabatabai G, Ledermann B, Brandner S: Wnt signalling inhibits neural differentiation of embryonic stem cells by controlling bone morphogenic protein expression. Mol Cell Neurosci. 2003, 24: 696-708. 10.1016/S1044-7431(03)00232-X.
Vlad A, Röhrs S, Klein-Hitpass L, Müller O: The first five years of the Wnt targetome. Cell Signal. 2008, 20: 795-802. 10.1016/j.cellsig.2007.10.031.
Hooper C, Killick R, Fernandes C, Sugden D, Lovestone S: Transcriptomic profiles of Wnt3a and insulin in primary cultured rat cortical neurones. J Neurochem. 2011, 118: 512-520. 10.1111/j.1471-4159.2011.07349.x.
Wan XZ, Li B, Li YC, Yang XL, Zhang W, Zhong L, Tang SJ: Activation of NMDA receptors upregulates a disintegrin and metalloproteinase 10 via a Wnt/MAPK signaling pathway. J Neurosci. 2012, 32: 3910-3916. 10.1523/JNEUROSCI.3916-11.2012.
Gavert N, Sheffer M, Raveh S, Spaderna S, Shtutman M, Brabletz T, Barany F, Paty P, Notterman D, Domany E, Ben-Ze ev A: Expression of L1-CAM and ADAM10 in human colon cancer cells induces metastasis. Cancer Res. 2007, 67: 7703-7712. 10.1158/0008-5472.CAN-07-0991.
LeBlanc JJ, Fagiolini M: Autism: a “critical period” disorder?. Neural Plast. 2011, 201 (1): 921680-
Wexler EM, Geschwind DH, Palmer TD: Lithium regulates adult hippocampal progenitor development through canonical Wnt pathway activation. Mol Psychiatry. 2008, 13: 285-292. 10.1038/sj.mp.4002093.
Gould TD, Gray NA, Manji HK: Effects of a glycogen synthase kinase-3 inhibitor, lithium, in adonomatous polyposis coli mutant mice. Pharmacol Res. 2003, 48: 49-53.
Cohen Y, Chetrit A, Cohen Y, Sirota P, Modan B: Cancer morbidity in psychiatric patients: influence of lithium carbonate treatment. Med Oncol. 1998, 15: 32-36. 10.1007/BF02787342.
Lee HJ, Wang NX, Shi DL, Zheng JJ: Sulindac inhibits canonical Wnt signaling by blocking the PDZ domain of the protein dishevelled. Angew Chem Int Ed Engl. 2009, 48: 6448-6452. 10.1002/anie.200902981.
Han A, Song Z, Tong C, Hu D, Bi X, Augenlicht LH, Yang W: Sulindac suppresses β-catenin expression in human cancer cells. Eur J Pharmacol. 2008, 583: 26-31. 10.1016/j.ejphar.2007.12.034.
Wilkinson MB, Dias C, Magida J, Mazei-Robinson M, Lobo MK, Kennedy P, Dietz D, Covington H, Russo S, Neve R, Ghose S, Tamminga C, Nestler EJ: A novel role of the WNT-dishevelled-GSK3β signaling cascade in the mouse nucleus accumbens in a social defeat model of depression. J Neurosci. 2011, 31: 9084-9092. 10.1523/JNEUROSCI.0039-11.2011.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
At the time of writing, the author’s salary was paid by Novartis. The author owns Novartis stock.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kalkman, H.O. A review of the evidence for the canonical Wnt pathway in autism spectrum disorders. Molecular Autism 3, 10 (2012). https://doi.org/10.1186/2040-2392-3-10
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2040-2392-3-10