
Abstract
Background
Oxidative stress may lead to an increased level of unrepaired cellular DNA damage, which is discussed as one risk for tumor initiation. Mismatch repair (MMR) enzymes act as proofreading complexes that maintain the genomic integrity and MMR-deficient cells show an increased mutation rate. One important gene in the MMR complex is the MutL homolog 1 (MLH1) gene. Since a diet rich in antioxidants has the potential to counteract harmful effects by reactive oxygen species (ROS), we investigated the impact of an antioxidant, folate, and vitamin rich diet on the epigenetic pattern of MLH1. These effects were analyzed in individuals with non-insulin depended diabetes mellitus type 2 (NIDDM2) and impaired fasting glucose (IFG).
Methods
In this post-hoc analysis of a randomized trial we analyzed DNA methylation of MLH1, MSH2, and MGMT at baseline and after 8 weeks of intervention, consisting of 300 g vegetables and 25 ml plant oil rich in polyunsaturated fatty acids per day. DNA methylation was quantified using combined bisulfite restriction enzyme analysis (COBRA) and pyrosequencing. MLH1 and DNMT1 mRNA expression were investigated by qRT-PCR. DNA damage was assessed by COMET assay. Student’s two-tailed paired t test and one-way ANOVA with Scheffé corrected Post hoc test was used to determine significant methylation and expression differences. Two-tailed Pearson test was used to determine correlations between methylation level, gene expression, and DNA strand break amount.
Results
The intervention resulted in significantly higher CpG methylation in two particular MLH1 promoter regions and the MGMT promoter. DNA strand breaks and methylation levels correlated significantly. The expression of MLH1, DNMT1, and the promoter methylation of MSH2 remained stable. CpG methylation levels and gene expression did not correlate.
Conclusion
This vitamin and antioxidant rich diet affected the CpG methylation of MLH1. The higher methylation might be a result of the ROS scavenging antioxidant rich diet, leading to lower activity of DNA demethylating enzymes. Our results suggest the hypothesis of CpG demethylation via DNA repair enzymes under these circumstances. NIDDM2 and IFG patients benefit from this simple dietary intervention involving epigenetic and DNA repair mechanisms.
Similar content being viewed by others


Background
Diabetes mellitus type 2 (T2DM) is a multifactorial disease characterized by hyperlipidemia, visceral obesity, hypercoagulability, microalbuminuria and hypertension based on genetic predisposition and environmental factors resulting in insulin resistance and hyperglycemia. Diabetic patients have often been described as being under enhanced oxidative stress [1, 2]. Long-term dietary patterns and status have a large impact on the risk developing non-communicable diseases like T2DM. Chronic exposure to elevated amounts in particular of free fatty acids and palmitate (C16 saturated fatty acid) leads to a higher reactive oxygen species (ROS) burden, while long chain polyunsaturated fatty acids such as docosahexaenoic acid (DHA) has the opposite effect [3, 4]. The phenotype of chronic hyperglycemia leads to increased production of ROS also originating from the substrate overwhelmed electron transport system in the mitochondria and the plasma membrane nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [5]. Further, the NADPH oxidase-dependent ROS production is directly proportional to accumulated body fat, although the mechanisms behind this are still not entirely clear [6, 7].
In healthy cells, ROS does not implicate harmful effects per se; when consistently regulated, ROS has an intracellular signaling role [8]. This vital balance can be disrupted by an excess of ROS and/or lack of antioxidants (AO) leading to cytotoxic and genotoxic oxidative stress, resulting in DNA strand breaks [8]. The ROS-induced spontaneous deamination of cytosine to uracil (unmethylated cytosine) and 5-hydroxyuracil (methylated cytosine) results in a G:C to A:T transition mutation, since they both preferentially pair with adenosine during DNA replication [9]. Likewise, guanine can be oxidized to 8-oxo-7,8 dihydroguanine (8-oxoG) and leads to a G:C to T:A transition mutation, due to its mispairing with adenosine [9].
In order to ensure DNA integrity, especially in diabetic patients, DNA repair enzymes are crucial. The mismatch repair (MMR) enzyme complex (MLH1 and MSH2 appear to play a key role) acts as a proofreading system during DNA synthesis and repairs 8-oxoG lesions [10, 11]. MGMT removes alkyl adducts from the DNA especially O6-methylguanine, which is read as an adenine by the DNA polymerase. Evidence shows that AO are able to increase DNA repair enzyme activities [12, 13]. MLH1 and MGMT promoter are inactivated by hypermethylation, and a promoter methylation-dependent downregulation of the corresponding gene expression in some cancer tissues has been found [14].
Environmental factors have a significant impact upon the epigenetic program of gene expression. Dietary factors have been found to alter DNA methylation both globally, and locus-specifically, leading to a changed expression of genes [15–17]. These modifications can be epigenetically inherited through DNA methylation to the next generation [18].
Numerous epidemiological studies have shown that nutritional folate, by providing one-carbon units, and AO play an important role for both DNA methylation and nucleotide biosynthesis reactions [19, 20], and as a consequence, for DNA repair. Folic acid deficiency causes epigenetic changes by diminishing remethylation of s-adenosyl-homocysteine (SAH) to s-adenosylmethionine (SAM) in the methionine cycle, which causes cytosine demethylation, global DNA hypomethylation, and chromosomal instability [21]. In addition, inadequate dietary folate increases uracil misincorporation, rate of DNA strand breaks, and chromosome breaks. Furthermore, folic acid deficiency affects DNA repair by the inhibition of thymidine and purine biosynthesis [21].
Therefore, our primary aim was to assess the impact of the antioxidant- and vitamin-rich diet on the epigenetic pattern of MLH1 in non-insulin-dependent T2DM (NIDDM2) and patients with impaired fasting glucose (IFG). In this study, we show that a changed diet rich in AO and vitamins (especially folate) has the ability to alter DNA methylation, and compensate ROS-induced epigenetic lesions.
Study design
This is a post-hoc analysis of a subgroup (n = 15) of a randomized trial (DIAPLANT) conducted at the Department of Nutritional Sciences at the University of Vienna between January and June 2010. We analyzed the DNA methylation of MLH1, MGMT, and MSH2 at baseline and after the 8 weeks ongoing intervention in patients with NIDDM2 and IFG (Table 1). To assess the natural and methodological variability of DNA methylation a lean control group (LC) consisting of 8 volunteer, healthy adults, who did not receive the intervention was analyzed separately.
Briefly, the aim of the DIAPLANT study was to investigate the positive effects of a dietary intervention with 300 g vegetables and 25 ml walnut oil per day, comprising approximately 73% polyunsaturated fatty acids (PUFA) and 18% monounsaturated fatty acids (MUFA), for 8 weeks, on the risk factors for late diabetic complications in T2DM subjects. The focus was on antioxidant and vitamins (especially folate), and rich green vegetables (broccoli, zucchini, brussels sprouts, green beans, cabbage turnip, maize, carrots, peas, cauliflower, soya beans, cos lettuce and spinach).
Subjects were recruited from a local diabetic clinic (Diabetes outpatient Clinic, Health Centre South, Vienna, Austria) during their annual health assessment. All subjects receiving the intervention had to have stable metabolic control (constant medication regarding glucose, lipid and uric acid metabolism), glycated hemoglobin (HbA1c) concentration < 9.5%, serum total cholesterol (TC) < 300 mg/dl (< 7.76 mmol/l), serum triglycerides (TG) < 500 mg/dl (< 5.7 mmol/l) and serum creatinine < 2.5 mg/dl (< 221 μmol/l). Only subjects with stable body weight, constant dietary habits and physical activity levels for at least four weeks before entry to the study were included. Subjects who intended to change dietary habits, frequency of physical activity or body weight within the study period were not allowed to participate. Exclusion criteria also included smoking, intake of fish oil capsules and other fatty acids. All medical therapies of subjects were continued unchanged throughout the study. Results of the DIAPLANT study will be reported elsewhere. The study protocol was approved by the Ethical Committee of the City of Vienna (EK09-218-VK_NZ). All participants gave their written consent.
Results
Oral folate intake
The calculated mean ± SD of folate contained in the intervention vegetables was 153.00 μg ± 82.32 μg per 300 g. Oral folate intake, assessed by 24-h recalls, was significantly increased after the intervention compared to baseline values (127.66 μg ± 195.27, P = 0.024).
Analysis of MLH1 methylation
The Bst UI restriction site (CGCG) in the MLH1 promoter analyzed by combined bisulfite restriction analysis (COBRA) was hypomethylated at all time points and possible minor methylation shifts were not detectable (see Additional file 1) [22]. DNA methylation differences between baseline (T0) and after 8 weeks of the intervention (T1) were not significantly different in the intervention groups.
Pyrosequencing of the MLH1 region1 (MLH1-1)
Quantitative analysis of the investigated area (65 bp, Figure 1 and Figure 2) on the forward strand showed significant changes over time at CpG sites numbers 1, 4 and 6 (Table 2). Site number 2 was not analyzed due to the long 5-nucleotide homopolymer T stretch (Figure 3), which has also been observed to cause problems in analysis, as in other studies [23]. The mean methylation over the seven CpGs was significantly (P = 0.001) elevated after the intervention (Figures 4 and 5). This area includes a sequence motif previously analyzed by COBRA [24] at CpG numbers 6 and 7 that has not yet been analyzed by pyrosequencing.

MLH1 Region 1 sequence and primers. MLH1 region 1: Bisulfite-converted MLH1 promoter 5′-3′sequence and primers used for pyrosequencing. Analyzed CpGs within the sequenced area are shaded orange.

Analyzed regions within the MLH1 promoter. CpG methylation assay overview within the MLH1 promoter region 5′- 3′. Approximately 730 to 240 bp upstream of the translational start site, 25 CpG sites were analyzed on forward and/or reverse strand.

MLH1 region 1 pyrogram. MLH1-1 assay showing seven analyzed sites (sample P44 T0). CpG number 2 was not considered for analysis. Three bisulfite treatment controls (16, 24, 38) confirm the complete conversion of unmethylated (non-CpG motif) cytosine residues.

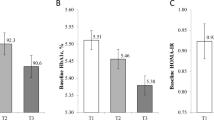
Mean DNA methylation levels. Mean DNA methylation levels in the intervention groups. Non-insulin-dependent diabetes mellitus type 2 (NIDDM2) and impaired fasting glucose (IFG) groups were pooled as they showed almost the same picture and no group-specific DNA methylation differences between baseline and T1 were found. P-values are based on the paired t-test.

MLH1 region 1 mean DNA methylation levels. Detail of MLH1 region 1. Mean DNA methylation levels over seven CpGs in the intervention groups, non-insulin-dependent diabetes mellitus type 2 (NIDDM2) and impaired fasting glucose (IFG), and the lean control group (LC). P-values are based on the paired t-test.
Pyrosequencing of the MLH1 region2 (MLH1-2)
This region covers a part of the reverse strand of the MLH1-1 region (see Figures 2 and 6.). The mean methylation was significantly higher (0.564% ± 0.696, P = 0.007) after the intervention in both NIDDM2 and IFG subjects (Figure 4).

MLH1 region 2 pyrogram. The extended MLH1-2 assay pyrogram showing 13 analyzed sites (sample P27 T0).
Pyrosequencing of the MLH1 region 3 (MLH1-3)
This region is situated 300 bp upstream of the other two MLH1 pyrosequenced regions (see Figure 2.). The methylation levels were very low (0% to 2%) and remained stable over time (Figure 4). No group-specific pattern could be found.
Analysis of MGMT methylation
The MGMT mean promoter (see Additional file 2) methylation was significantly higher (0.337% ± 0.468, P = 0.023) after the intervention (Figure 4 and Table 3). The percentages of methylated cytosine were generally low (≤ 5%). No correlation of CpG methylation with the MLH1 expression could be found. DNA methylation differences between baseline and T1 were not significantly different in the intervention groups. Patient number 76 (IFG) showed a baseline methylation level of 11% and 2% after intervention at CpG number 8, leading to a distorted statistic at this position. Excluding patient 76 from pooled statistics leads to + 0.733% overall methylation (SE = 0.146, P < 0.001). A European Molecular Biology Open Software Suite (EMBOSS) transcription factor prediction within shared motifs (> 5 bp; 5′-AGCCCG-3′ and 5′-GGACAGC-3′) with MLH1 region 1 was negative.
Quantitative analysis of MSH2 promoter (see Additional file 3) methylation did not reveal any aberrant methylation in the investigated samples. The percentages of methylated cytosine were generally low (≤ 5%). The methylation level remained stable over time. Neither a group-specific pattern nor a correlation of CpG methylation with the MLH1 expression could be found.
MLH1 and DNMT1 m-RNA expression
Changes in MLH1 and DNMT1 gene expression in response to the intervention were quantified by quantitative real-time reverse-transcriptase (qRT)-PCR. Melting curve analysis showed specific product peaks at 87.26°C, 89.34°C, and 83.67°C for GAPDH, DNMT1, and MLH1, respectively. Neither a significant expression shift between the two time points, nor a correlation with methylation levels of CpGs or DNA damage could be found. MLH1 and DNMT1 expressions were not affected by the intervention (P = 0.306 and P = 0.932, respectively).
DNA damage
Baseline levels of DNA strand breaks were comparable between the intervention groups. After the intervention (T1), more DNA strand breaks were measured in the IFG group by the H2O2-induced oxidative DNA damage test (P = 0.006) compared to the NIDDM2 group. We found significant correlations between the occurrence of DNA strand breaks and the methylation level of CpG number 1 (P < 0.01; r = −0.471; see Figure 7), number 3 (P < 0.05; r = −0.370), and number 4 (P < 0.01; r = −0.486) as well as the mean methylation (P = 0.05; r = −0.361) within the MLH1-1 assay in the NIDDM2 and IFG group at both time points (Figure 4, and Additional files 4 and 5).

Correlation between DNA strand breaks and CpG number 4 methylation. Significant correlation over all time points between the occurrence of DNA strand breaks and the DNA methylation level at CpG number 4 within the MLH1 region 1 (P < 0.01; r = −0.486).
Discussion
While several studies have found that higher ROS levels induce aberrant DNA hypomethylation [25, 26], this study is among the first to report that a changed diet can reverse this effect. Our DNA methylation and gene expression studies reveal a link between specific MLH1 promoter CpG methylations and an optimized diet in IFG and NIDDM2 patients, but no group-specific DNA methylation differences between baseline and T1 could be found. A novel finding is the identification of three variable CpG methylation positions situated in the MLH1 promoter, which to our knowledge has not been published before. Due to the AO- and folate-rich dietary intervention, the methylation levels of these CpGs were higher compared to the baseline. Our analysis was not intended to show differences in methylation variability between NIDDM2/IFG patients and lean control subjects. Nevertheless, there is clear evidence of a negative correlation between methylation levels and age in our study population.
Chang et al. demonstrated that non-cytotoxic levels of H2O2 significantly reduced the activity of the MMR system in repairing single-base and insertion/deletion loop mismatches in a dose-dependent manner. The different MMR activity was not a result of altered expression levels, but based on posttranslational enzyme degradations by H2O2. They further propose that ROS-induced stress reduces MMR function and may play a role in the low frequency of microsatellite instability (MSI) detected in inflamed tissues [27]. Therefore, our gene expression data may not reflect MMR activity. Nevertheless, we showed that the variable MLH1 promoter methylation levels are not affecting its expression, hence not affecting MMR activity.
The detected MLH1 methylation levels were consistently low (0% to 12%), independently from the time point. Contrary to findings in cancer studies [28], the expression of MLH1 was not affected by the methylation levels. This suggests that the expression is not regulated by the methylation levels of the analyzed sites in non-cancer patients. Consistent with other findings [29, 30], increases in methylation are not explained by higher DNMT1 expressions. Although the oral intake of folate had increased significantly after the intervention, it had no effect on the DNMT1 expression. This may indicate that no folate- or methyldonor-deficiency was present before the intervention [31].
Possible histone modification involved in the regulation of MLH1 expression and its effect on DNA methylation [32–34] do not offer explanations for our results. Histone modifications such as H3K9me3, H3K27me2, and H3K27me3, apparently do not directly affect MLH1 expression but may serve to index the promoter region for additional epigenetic control [35]. We cannot rule out the involvement of histone modifications leading to higher methylation levels, but in stark contrast we hypothesize that the intervention did not directly induce higher methylation levels, but suppresses the evident loss of methylation before the intervention. CpG-relevant ROS-induced DNA damages are 8-oxoguanine and the conversion of 5-methyl-cytosine (5mC) to 5-hydroxymethylcytosine (5hmC) and 5-hydroxymethyluracil (5hmU), predominantly repaired through the MMR, base excision repair (BER), and nucleotide excision repair (NER) pathway, respectively [36, 37]. So far it remains unclear how exactly CpG methylation marks are removed from the DNA, even though active mechanisms have been discussed [38]. Evidence is provided for both direct and indirect demethylation [39, 40]. Barreto et al. propose that active and direct DNA demethylation is accomplished through a GADD45-dependent process of DNA repair (NER and BER) that involves nucleotide exchange, replacing 5-methyl-cytosine with unmodified cytosine, and it is possible that this is the physiological mechanism that operates typically in vivo. In vitro, GADD45 knockdown resulted in similar methylation differences to our results (see Barreto et al. Figure 3c-d) [39]. Different enzymes (for example TET, AID/APOBEC) have been found to modify the methylated cytosine (by hydroxylation, deamination, oxidation, or a combination of these modifications), leading to its replacement by DNA repair. They act indirectly to mediate DNA demethylation. TET1, 2, and 3 catalyze the conversion of 5mC to 5hmC, which is then replaced with an unmethylated cytosine by the BER enzymes via DNA repair [41].
Conclusions
Based on these findings we propose that the low initial methylation levels on MLH1 and MGMT are the result of increased oxidative stress, which causes DNA lesions resulting in increased DNA repair activity. The latter leads to a loss of methylation marks by direct and/or indirect demethylation. The nutritional intervention might have led to a lower ROS burden and diminished the demethylating effects, resulting in higher overall methylation levels, and helped to maintain crucial and tissue-specific methylation marks with possible regulatory function. This finding need to be further investigated to reveal the exact underlying mechanisms behind the ROS-induced demethylation in relation to MSI and cancer-relevant promoter demethylation of oncogene, leading to initiation of tumors.
Methods
Sample collection
Blood was sampled before (T0) and after 8 weeks of dietary intervention (T1) using PAXgene Blood DNA tubes and the PAXgene Blood RNA tubes (Qiagen, Hilden, Germany). The samples were stored until analyses at −20°C.
Folate quantification
On the day before blood sampling, 24-h recalls of food intake were obtained from the subjects. The dietary composition of the vegetables and 24-h recalls were evaluated using the nutritional software NUT.S (BLS II.3.1., Karlsruhe, Germany).
DNA isolation and bisulfite conversion
DNA was extracted using the PAXgene Blood DNA Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. For methylation analysis, all samples were bisulfite-converted with the EpiTectBisulfite Kit (Qiagen, Hilden, Germany), resulting in the deamination of unmethylated cytosine to uracil, whereas methylated cytosine remain unchanged. The EpiTect Whole Bisulfitome Kit (Qiagen, Hilden, Germany) was used to amplify 2 μl converted DNA. The concentration of DNA was measured on a Pico100 (Picodrop Limited, Hinxton, UK). All reactions were carried out according to manufacturer’s protocols. Samples were stored at −20°C.
RNA isolation and reverse transcription
Total RNA was extracted from blood samples using the PAXgene Blood RNA Kit (Qiagen, Hilden, Germany) following the producer’s handbook. The random primers (hexamers) of the Phusion RT-PCR Kit (Finnzymes, Vantaa, Finland) were used to reverse-transcribe 5 μl of total RNA into single-stranded DNA. The concentration of cDNA was measured on a Pico100 (Picodrop Limited, Hinxton, UK). The samples were stored at −20°C.
PCR and restriction enzymatic digestion - COBRA
All bisulfite-treated samples were investigated by PCR and endonuclease digestion. The primers of Deng et al. were used for PCR [24]. The two methylation-specific primers MLH1-C-FW and MLH1-C-RE (Table 4) were chosen for the touchdown PCR amplification. The PCR was always carried out in 50 μl reaction mixtures; these contained 25 μl 2× SensiMix Probe polymerase mastermix (Bioline, London, UK), 625 nM of each primer, and 20 ng bisulfite-converted DNA. PCR conditions were 95°C for 10 minutes, 15 cycles of 95°C for 10 s, 55°C to 51°C for 1 minute, -0.2°C per cycle, 72°C for 1 minute followed by 25 cycles of 95°C for 40 s, 51°C for 40 s, 72°C for 40 s and a final elongation at 72°C for 10 minutes. After amplification, the PCR products were purified by isopropanol-precipitation. For enzymatic digestion, 10 μl PCR product was digested with 1U of BstUI (New England Biolabs, Frankfurt am Main, Germany) and NEB4 Buffer in 25 μl total volume at 60°C for 3 h. The digested fragments were separated by 2% agarose gel electrophoresis. Both CpG sites within the BstUI need to be methylated to be digested by BstUI. If the recognition site (CGCG) is methylated, its ssPCR product (294 bp) is digested into a 206 bp and an 88 bp fragment. If the CGCG motif is not methylated, the amplicon is not digested. The digested DNA was separated on 3.5% agarose gels in 1× tris-acetate ethylenediaminetetraacetic acid (TAE) and stained with GelRed (Biotium, Hayward, CA). Bands were analyzed using ImageJ 1.44p and quantified relative to the synthetic fully methylated /unmethylated control-DNA (Qiagen, Hilden, Germany). The proportion of methylated versus unmethylated DNA was determined from the relative intensities of cut and uncut PCR product.
Quantitative promoter methylation analyses by DNA pyrosequencing
We performed quantitative methylation analyses of the MLH1, MGMT, and MSH2 promoter by pyrosequencing of sodium bisulfite-modified DNA using the PyroMark Q24 (Qiagen, Hilden, Germany). DNA methylation was determined in three different regions inside the MLH1 promoter (Figure 2). PCR were performed in 25 μl reaction mixtures with 12.5 μl PyroMark 2× PCR master mix, 280 nM of each primer (Table 4), 2.5 ml CoralLoad Concentrate 10× (Qiagen, Hilden, Germany), and 25 ng bisulfite converted DNA for MGMT and 25 ng of EpiTect Whole Bisulfitome (Qiagen, Hilden, Germany)-amplified DNA for all other analyses. PCR conditions were 95°C for 15 minutes; 45 cycles of 94°C for 30 s, a.) for MGMT 47.5°C for 45 s, b.), for all other genes, 56°C for 30 s; 72°C for 30 s, and a final elongation at 72°C for 10 minutes. One of the PCR primers was biotinylated to purify the final PCR product using streptavidin-coated Sepharose® beads (GE Healthcare, Vienna, Austria). Additionally three PyroMark CpG assay kits (Qiagen, Hilden, Germany) were used following the manufacturer’s instructions (Table 5). The MLH1-2 assay was manually extended to analyze thirteen CpGs instead of six. PCR products were checked by 3.5% agarose gel electrophoresis. A total of 12.5 μl of the PCR product was used for subsequent pyrosequencing using a PyroMark Q24 System (Qiagen, Hilden, Germany). Briefly, the sepharose beads containing the immobilized PCR product were washed and denatured using 0.2 M NaOH solution and rewashed using the pyrosequencing Vacuum Prep Tool (Qiagen, Hilden, Germany). The purified single-stranded PCR product was released to the annealing buffer (Qiagen, Hilden, Germany) containing the corresponding pyrosequencing primer (300 nM). Subsequent quantification of CpG methylation levels was performed using the PyroMark Q24 software (Qiagen, Hilden, Germany). Each pyrosequencing assay was performed on duplicates. For quality control, each experiment included non-CpG cytosines as internal controls to verify efficient sodium bisulfite DNA conversion. We also included synthetic fully methylated /unmethylated control-DNA (Qiagen, Hilden, Germany).
qRT-PCR
The mRNA level of MLH1, DNMT1, and GAPDH as an endogenous control gene for normalization was analyzed in triplicates on a StepOne Plus real-time PCR system (Applied Biosystems, Vienna, Austria) using the SYBR Green approach. Each reaction mix consisted of 5 μl SensiFAST SYBRgreen (Bioline GmbH, London, UK), 300nM primers (Table 4), and 100 ng cDNA to a total volume of 10 μl. PCR conditions were 95°C for 2 minutes, 40 cycles of 95°C for 30 s, 60°C for 10 s, and 72°C for 5 s, followed by a melt curve analysis. The efficiency for DNMT1, MLH1, and GAPDH, was 103.5%, 106.7%, and 109.4%, respectively. Calculations were performed using the comparative Ct method.
Comet assay
A Comet assay was performed based on the protocol published by Azqueta [46]. For the evaluation of DNA damage, Komet 5.5 image analysis software (Kinetic Imaging Limited, Nottingham, UK) was used, which was linked to a fluorescent microscope. For every sample two gels with 50 cells each were randomly scored. The percentage of DNA in the tail (% tail DNA) was determined and the mean was calculated.
Statistical analyses
Methylation levels and expression data of 23 paired samples were analyzed with SPSS 20 (IBM, Armonk, NY). The Kolmogorov-Smirnov test was used to test for normality of the distributions. The Student’s two-tailed paired t-test and one-way analysis of variance (ANOVA) with the Scheffé post hoc correction test were used to determine significant differences. The two-tailed Pearson test was used to determine correlations between methylation level, gene expression, and amount of DNA strand break. All statistics were tested for possible associations with age and sex; no significant associations were found. A P-value < 0.05 was considered statistically significant. All data shown are mean ± SD unless otherwise indicated.
Abbreviations
- ANOVA:
-
one-way analysis of variance
- AO:
-
antioxidants
- BER:
-
base excision repair
- COBRA:
-
combined bisulfite restriction analysis
- DHA:
-
docosahexaenoic acid
- HbA1c:
-
glycated hemoglobin
- IFG:
-
impaired fasting glucose
- MMR:
-
mismatch repair
- MSI:
-
microsatellite instability
- MUFA:
-
monounsaturated fatty acids
- NADPH:
-
nicotinamide adenine dinucleotide phosphate
- NER:
-
nucleotide excision repair
- 8-oxoG:
-
8-oxo-7,8 dihydroguanine
- PSQ:
-
pyrosequencing
- PUFA:
-
polyunsaturated fatty acids
- qRT-PCR:
-
quantitative real-time reverse-transcriptase polymerase chain reaction
- ROS:
-
reactive oxygen species
- SAH:
-
s-adenosyl-homocysteine
- SAM:
-
s-adenosylmethionine
- TAE:
-
tris-acetate ethylenediaminetetraacetic acid
- TC:
-
total cholesterol
- T2DM:
-
diabetes mellitus type 2
- TG:
-
serum triglycerides.
References
Tatsch E, Bochi GV, Piva SJ, de Carvalho JA, Kober H, Torbitz VD, Duarte T, Signor C, Coelho AC, Duarte MM, Montagner GF, Da Cruz IB, Moresco RN: Association between DNA strand breakage and oxidative, inflammatory and endothelial biomarkers in type 2 diabetes. Mutat Res. 2012, 732: 1-5. 10.1016/j.mrfmmm.2012.02.003.
Newsholme P, Haber EP, Hirabara SM, Rebelato EL, Procopio J, Morgan D, Oliveira-Emilio HC, Carpinelli AR, Curi R: Diabetes associated cell stress and dysfunction: role of mitochondrial and non-mitochondrial ROS production and activity. J Physiol. 2007, 583: 1-16. 10.1113/jphysiol.2007.139923.
Nakamura S, Takamura T, Matsuzawa-Nagata N, Takayama H, Misu H, Noda H, Nabemoto S, Kurita S, Ota T, Ando H, Miyamoto K, Kaneko S: Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J Biol Chem. 2009, 284: 14809-14818. 10.1074/jbc.M901488200.
Han CY, Umemoto T, Omer M, den Hartigh LJ, Chiba T, LeBoeuf R, Buller CL, Sweet IR, Pennathur S, Abel ED, Chait A: NADPH oxidase-derived reactive oxygen species increases expression of monocyte chemotactic factor genes in cultured adipocytes. J Biol Chem. 2012, 287: 10379-10393. 10.1074/jbc.M111.304998.
Brownlee M: Biochemistry and molecular cell biology of diabetic complications. Nature. 2001, 414: 813-820. 10.1038/414813a.
Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I: Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004, 114: 1-10.
Styskal J, van Remmen H, Richardson A, Salmon AB: Oxidative stress and diabetes: what can we learn about insulin resistance from antioxidant mutant mouse models?. Free Radic Biol Med. 2012, 52: 46-58. 10.1016/j.freeradbiomed.2011.10.441.
Waris G, Ahsan H: Reactive oxygen species: role in the development of cancer and various chronic conditions. Journal of carcinogenesis. 2006, 5: 1-8. 10.1186/1477-3163-5-1.
Hegde ML, Hazra TK, Mitra S: Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008, 18: 1-21. 10.1038/cr.2008.9.
Boiteux S, Gellon L, Guibourt N: Repair of 8-oxoguanine in Saccharomyces cerevisiae: interplay of DNA repair and replication mechanisms. Free Radic Biol Med. 2002, 32: 1244-1253. 10.1016/S0891-5849(02)00822-5.
Haracska L, Yu SL, Johnson RE, Prakash L, Prakash S: Efficient and accurate replication in the presence of 7,8-dihydro-8-oxoguanine by DNA polymerase eta. Nat Genet. 2000, 25: 1-4.
Collins AR, Azqueta A, Langie SA: Effects of micronutrients on DNA repair. Eur J Nutr. 2012, 51: 261-279. 10.1007/s00394-012-0318-4.
Niture SK, Velu CS, Smith QR, Bhat GJ, Srivenugopal KS: Increased expression of the MGMT repair protein mediated by cysteine prodrugs and chemopreventative natural products in human lymphocytes and tumor cell lines. Carcinogenesis. 2007, 28: 378-389.
Esteller M: CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002, 21: 1-14. 10.1038/sj.onc.1205020.
Ingrosso D, Cimmino A, Perna AF, Masella L, de Santo NG, de Bonis ML, Vacca M, D’Esposito M, D’Urso M, Galletti P, Zappia V: Folate treatment and unbalanced methylation and changes of allelic expression induced by hyperhomocysteinaemia in patients with uraemia. Lancet. 2003, 361: 1-7. 10.1016/S0140-6736(03)12101-0.
Wasson GR, McGlynn AP, McNulty H, O’Reilly SL, McKelvey-Martin VJ, McKerr G, Strain JJ, Scott J, Downes CS: Global DNA and p53 region-specific hypomethylation in human colonic cells is induced by folate depletion and reversed by folate supplementation. J Nutr. 2006, 136: 1-6.
Milagro FI, Campión J, García-Díaz DF, Goyenechea E, Paternain L, Martínez JA: High fat diet-induced obesity modifies the methylation pattern of leptin promoter in rats. J Physiol Biochem. 2009, 65: 1-9. 10.1007/BF03165964.
Vickers MH, Cupido C, Gluckman PD: Developmental programming of obesity and type 2 diabetes. Fetal and Maternal Medicine Review. 2007, 18: 1-23. 10.1017/S096553950700188X.
Ahmed F: The role of oxidative stress in environmental carcinogenesis. J Environ Sci Health C. 1999, 17: 111-142.
Wei Q, Shen H, Wang L, Duphorne CM, Pillow PC, Guo Z, Qiao Y, Spitz MR: Association between low dietary folate intake and suboptimal cellular DNA repair capacity. Cancer Epidemiol Biomarkers Prev. 2003, 12: 963-969.
Duthie SJ: Folate and cancer: how DNA damage, repair and methylation impact on colon carcinogenesis. J Inherit Metab Dis. 2011, 34: 101-109. 10.1007/s10545-010-9128-0.
Goedecke S, Schlosser S, Mühlisch J, Hempel G, Frühwald MC, Wünsch B: Determination of DNA methylation by COBRA: a comparative study of CGE with LIF detection and conventional gel electrophoresis. Electrophoresis. 2009, 30: 3063-3070. 10.1002/elps.200900204.
Rahim A, Coutelle C, Harbottle R: High-throughput Pyrosequencing of a phage display library for the identification of enriched target-specific peptides. Biotechniques. 2003, 35: 322-324.
Deng D, Deng G, Zhou J, Xin H: Detection of CPG methylations in human mismatch repair gene hMLH1 promoter by denaturing high-performance liquid chromatography (DHPLC). Chinese Journal of Cancer Research. 2000, 12: 171-191. 10.1007/BF02983460.
Tunc O, Tremellen K: Oxidative DNA damage impairs global sperm DNA methylation in infertile men. J Assist Reprod Genet. 2009, 26: 537-544. 10.1007/s10815-009-9346-2.
Bhusari SS, Dobosy JR, Fu V, Almassi N, Oberley T, Jarrard DF: Superoxide dismutase 1 knockdown induces oxidative stress and DNA methylation loss in the prostate. Epigenetics. 2010, 5: 18-
Chang CL, Marra G, Chauhan DP, Ha HT, Chang DK, Ricciardiello L, Randolph A, Carethers JM, Boland CR: Oxidative stress inactivates the human DNA mismatch repair system. Am J Physiol Cell Physiol. 2002, 283: 1-8.
Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R: Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997, 57: 808-811.
Yauk CL, Polyzos A, Rowan-Carroll A, Kortubash I, Williams A, Kovalchuk O: Tandem repeat mutation, global DNA methylation, and regulation of DNA methyltransferases in cultured mouse embryonic fibroblast cells chronically exposed to chemicals with different modes of action. Environ Mol Mutagen. 2008, 49: 26-35. 10.1002/em.20359.
Trasler J, Deng L, Melnyk S, Pogribny I, Hiou-Tim F, Sibani S, Oakes C, Li E, James SJ, Rozen R: Impact of Dnmt1 deficiency, with and without low folate diets, on tumor numbers and DNA methylation in Min mice. Carcinogenesis. 2003, 24: 1-7. 10.1093/carcin/24.1.1.
Ghoshal K, Li X, Datta J, Bai S, Pogribny I, Pogribny M, Huang Y, Young D, Jacob ST: A folate- and methyl-deficient diet alters the expression of DNA methyltransferases and methyl CpG binding proteins involved in epigenetic gene silencing in livers of F344 rats. J Nutr. 2006, 136: 1522-1527.
Sun H, Zhou X, Chen H, Li Q, Costa M: Modulation of histone methylation and MLH1 gene silencing by hexavalent chromium. Toxicol Appl Pharmacol. 2009, 237: 258-266. 10.1016/j.taap.2009.04.008.
Fahrner JA, Eguchi S, Herman JG, Baylin SB: Dependence of histone modifications and gene expression on DNA hypermethylation in cancer. Cancer Res. 2002, 62: 7213-7218.
Lin JC, Jeong S, Liang G, Takai D, Fatemi M, Tsai YC, Egger G, Gal-Yam EN, Jones PA: Role of nucleosomal occupancy in the epigenetic silencing of the MLH1 CpG island. Cancer Cell. 2007, 12: 432-444. 10.1016/j.ccr.2007.10.014.
McGarvey KM, Fahrner JA, Greene E, Martens J, Jenuwein T, Baylin SB: Silenced tumor suppressor genes reactivated by DNA demethylation do not return to a fully euchromatic chromatin state. Cancer Res. 2006, 66: 3541-3549. 10.1158/0008-5472.CAN-05-2481.
Kow YW: Repair of deaminated bases in DNA. Free Radic Biol Med. 2002, 33: 1-8. 10.1016/S0891-5849(02)00827-4.
Colussi C, Parlanti E, Degan P, Aquilina G, Barnes D, Macpherson P, Karran P, Crescenzi M, Dogliotti E, Bignami M: The mammalian mismatch repair pathway removes DNA 8-oxodGMP incorporated from the oxidized dNTP pool. Curr Biol. 2002, 12: 912-918. 10.1016/S0960-9822(02)00863-1.
Paroush Z, Keshet I, Yisraeli J, Cedar H: Dynamics of demethylation and activation of the alpha-actin gene in myoblasts. Cell. 1990, 63: 1229-1237. 10.1016/0092-8674(90)90418-E.
Barreto G, Schäfer A, Marhold J, Stach D, Swaminathan SK, Handa V, Döderlein G, Maltry N, Wu W, Lyko F, Niehrs C: Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007, 445: 1-5.
Cedar H, Bergman Y: Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009, 10: 1-10.
Bhutani N, Burns DM, Blau HM: DNA demethylation dynamics. Cell. 2011, 146: 866-872. 10.1016/j.cell.2011.08.042.
Mikeska T, Bock C, El-Maarri O, Hübner A, Ehrentraut D, Schramm J, Felsberg J, Kahl P, Büttner R, Pietsch T, Waha A: Optimization of quantitative MGMT promoter methylation analysis using pyrosequencing and combined bisulfite restriction analysis. The Journal of molecular diagnostics: JMD. 2007, 9: 368-381. 10.2353/jmoldx.2007.060167.
Chang DK, Ricciardiello L, Goel A, Chang CL, Boland CR: Steady-state regulation of the human DNA mismatch repair system. J Biol Chem. 2000, 275: 18424-18431. 10.1074/jbc.M001140200.
Mizuno S, Chijiwa T, Okamura T, Akashi K, Fukumaki Y, Niho Y, Sasaki H: Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood. 2001, 97: 1172-1179. 10.1182/blood.V97.5.1172.
Adachi M, Liu Y, Fujii K, Calderwood SK, Nakai A, Imai K, Shinomura Y: Oxidative stress impairs the heat stress response and delays unfolded protein recovery. PLoS One. 2009, 4: e7719-10.1371/journal.pone.0007719.
Azqueta A, Shaposhnikov S, Collins AR: DNA oxidation: investigating its key role in environmental mutagenesis with the comet assay. Mutat Res. 2009, 674: 1-8. 10.1016/j.mrgentox.2009.01.005.
Acknowledgement
This work was funded by the EU (grant N° N00039), through the cross-border cooperation programme Slovakia-Austria 2007–2013 (http://www.sk-eu.at) and the Austrian Ministry of Health.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
OJS initiated the study, performed the methylation and expression experiments, analyzed the data, and wrote the manuscript. EM participated in the DIAPLANT study design, performed the COMET assay experiments, and analyzed the data. HB participated in designing the DIAPLANT study, recruited the study subjects and was responsible for their medical treatment. EA participated to the elaboration of the study and experiment design. K-HW participated in designing the DIAPLANT study. AGH conceived and designed the study. All authors read and approved the final manuscript.
Electronic supplementary material
13148_2012_34_MOESM1_ESM.png
{kind=link}
Additional file 1:Figure S1. COBRA gelelectorphoresis. Figure showing gelelectorphoresis of combined bisufite restriction analysis (COBRA) before (10 μl) and after (10 μl and 15 μl) BST UI digestion. 100 bp DNA ladder. (PNG 332 kb) (PNG 333 KB)
13148_2012_34_MOESM2_ESM.png
{kind=link}
Additional file 2:Figure S2. MGMT pyrosequencing location. Figure showing methylation assay overview within the MGMT promoter region 5′- 3′. Ten CpG sites were analyzed by reverse-sequencing the upper strand. CpG island concentration is shown in the lower green. (PNG 56 kb) (PNG 56 KB)
13148_2012_34_MOESM3_ESM.png
{kind=link}
Additional file 3:Figure S3. MSH2 pyrosequencing location. Figure showing methylation assay overview within the MSH2 promoter region 5′- 3′. Approximately 260 to 230 bp upstream of the translational start site, seven CpG sites were analyzed on the forward strand. CpG island concentration is shown in the lower green. (PNG 32 kb) (PNG 32 KB)
13148_2012_34_MOESM4_ESM.png
{kind=link}
Additional file 4:Figure S4. Correlation between DNA strand breaks and CpG number 1 methylation. Figure showing significant correlation over all time points between the occurrence of DNA strand breaks and the DNA methylation level at CpG number 1 within the MLH1 region 1 (P <0.01; r = −0.471). (PNG 33 kb) (PNG 34 KB)
13148_2012_34_MOESM5_ESM.png
{kind=link}
Additional file 5:Figure S5. Correlation between DNA strand breaks and mean methylation. Figure showing correlation over all time points between the occurrence of DNA strand breaks and the mean DNA methylation level of the MLH1 region 1 (P = 0.05; r = −0.361). (PNG 31 kb) (PNG 31 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Switzeny, O.J., Müllner, E., Wagner, KH. et al. Vitamin and antioxidant rich diet increases MLH1 promoter DNA methylation in DMT2 subjects. Clin Epigenet 4, 19 (2012). https://doi.org/10.1186/1868-7083-4-19
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1868-7083-4-19