
Abstract
Background
PIF/Harbinger is the most recently discovered DNA transposon superfamily and is now known to populate genomes from fungi to plants to animals. Mobilization of superfamily members requires two separate element-encoded proteins (ORF1 and TPase). Members of this superfamily also mobilize Tourist-like miniature inverted repeat transposable elements (MITEs), which are the most abundant transposable elements associated with the genes of plants, especially the cereal grasses. The phylogenetic analysis of many plant genomes indicates that MITEs can amplify rapidly from one or a few elements to hundreds or thousands.
The most active DNA transposon identified to date in plants or animals is mPing, a rice Tourist-like MITE that is a deletion derivative of the autonomous Ping element. Ping and the closely related Pong are the only known naturally active PIF/Harbinger elements. Some rice strains accumulate ~40 new mPing insertions per plant per generation. In this study we report the development of a yeast transposition assay as a first step in deciphering the mechanism underlying the amplification of Tourist-MITEs.
Results
The ORF1 and TPase proteins encoded by Ping and Pong have been shown to mobilize mPing in rice and in transgenic Arabidopsis. Initial tests of the native proteins in a yeast assay resulted in very low transposition. Significantly higher activities were obtained by mutation of a putative nuclear export signal (NES) in the TPase that increased the amount of TPase in the nucleus. When introduced into Arabidopsis, the NES mutant protein also catalyzed higher frequencies of mPing excision from the gfp reporter gene. Our yeast assay retains key features of excision and insertion of mPing including precise excision, extended insertion sequence preference, and a requirement for two proteins that can come from either Ping or Pong or both elements.
Conclusions
The yeast transposition assay provides a robust platform for analysis of the mechanism underlying transposition catalyzed by the two proteins of PIF/Harbinger elements. It recapitulates all of the features of excision and reinsertion of mPing as seen in plant systems. Furthermore, a mutation of a putative NES in the TPase increased transposition both in yeast and plants.
Similar content being viewed by others

Background
Class 2 DNA transposons were discovered in maize over 60 years ago with the genetic characterization of the Ac/Ds family of autonomous and nonautonomous elements by McClintock [1]. Since then, DNA transposons have been found in all kingdoms of life and have been characterized into at least 10 superfamilies, based on the sequence of the element-encoded transposase protein [2]. The newest superfamily is PIF/Harbinger, whose existence only came to light in the last decade. PIF/Harbinger derives its name from the two founding elements: Harbinger from Arabidopsis thaliana and PIF from Zea mays, discovered by computational and genetic analyses, respectively [3, 4].
Several features of transposition distinguish PIF/Harbinger from the other superfamilies. First, virtually all coding elements characterized to date contain two genes, ORF1 and TPase [5, 6]. Unlike CACTA elements where alternative splicing produces multiple proteins [7, 8], the two genes of PIF/Harbinger elements appear to be independent [5, 6]. Both the ORF1 and TPase proteins are required for transposition [9, 10]. Second, where analysed, excision is usually perfect as both the element and one copy of the 3 bp target site duplication (TSD) generated upon insertion is excised from the donor site [9, 10]. This differs from all previously characterized plant transposable elements where the majority of excision events leave a footprint or deletion at the excision site [11]. Third, PIF/Harbinger elements display an extended target sequence preference: 9 bp in plants [4, 9, 12] and 15 bp in vertebrates [6, 10].
Another distinguishing feature of this superfamily is that PIF/Harbinger elements are responsible for the generation and amplification of Tourist-like miniature inverted repeat transposable elements (MITEs), one of the two predominant MITE families (the other being Stowaway). MITEs are small (100-500 bp) non-coding elements with the ability to amplify rapidly from one or a few near-identical elements to hundreds or thousands of copies [13]. MITEs comprise ~5% of the rice genome [14] and are abundant in the genomes of some animals including mosquitoes [15], zebrafish [16] and humans [17, 18]. Where MITEs have been analysed on a genome-wide basis, they appear to play a significant role in gene evolution as they are abundant and insert preferentially into or near genes [19]. In order to understand the success of Tourist-MITEs we need to first understand the transposition mechanism of PIF/Harbinger elements. With this goal in mind, we focus in this study on the rice mPing element, which is the only known active MITE.
Computational analysis of the sequenced genome of the rice (Oryza sativa, japonica) cultivar Nipponbare led to the identification of mPing [20]. mPing was independently discovered to be actively transposing in the rice strain Gimbozu/EG4 [21] and in rice anther culture [22]. Further analysis revealed that this 431 bp Tourist-like MITE is a perfect deletion derivative of the 4.5 kb Ping element, which is present as a single copy in the Nipponbare genome and is a member of the PIF/Harbinger superfamily [5]. Thus, it came as a surprise to find that mPing was actively transposing in an indica rice cell culture line that lacked the Ping element [20]. The most likely source of TPase was determined to be the closely related Pong element, which is present in multiple copies in all tested strains of Oryza sativa. Subsequent studies with transgenic Arabidopsis confirmed that either Ping or Pong proteins were able to mobilize mPing and that transposition required functional copies of ORF1 and TPase [9].
Heterologous assay systems in plants and human cell culture have provided clues to the function of ORF1 protein and the reason why two proteins are required for transposition [9, 10]. For most class 2 elements, the TPase contains a conserved catalytic domain (DDE) and a DNA binding domain that recognizes and binds to the terminal inverted repeat (TIR) and/or subterminal regions [23]. In contrast, the TPase of characterized members of the PIF/Harbinger superfamily lacks an obvious binding domain. Instead, a conserved Myb-like domain in ORF1 protein was hypothesized to be involved in DNA binding [5, 6]. This model was supported by studies with Harbinger3N_DR, an artificial element whose reconstruction was guided by building consensus sequences from the zebrafish genome [10]. Harbinger3N_DR was mobilized in human cells only when both the reconstructed Harbinger3_DR TPase and ORF1 proteins were co-expressed [10]. Furthermore, ORF1 protein was shown to bind the Harbinger3N_DR TIRs and interact with TPase. Finally, this study found that interaction with ORF1 protein was required for nuclear localization of TPase [10]. These results suggest that ORF1 protein plays a critical role by positioning the TPase both in the nucleus and at the TIR where excision occurs.
We were motivated to develop a more facile assay system as a first step in the understanding of the amplification of MITEs and to dissect the complex transposition mechanism catalyzed by the PIF/Harbinger proteins. Here we report a yeast assay that recapitulates all of the features of excision and reinsertion of mPing as seen in plant systems. Furthermore, we demonstrate the validity of the yeast model by showing that a mutation of a putative nuclear export signal (NES) in the TPase increased transposition both in yeast and plants. These results provide a platform for further analysis of the regulation of other PIF/Harbinger elements and their relationships to Tourist-like MITEs.
Results
mPing transposition in yeast
In order to establish a transposition assay for mPing in yeast, we modified successful assays previously used for the maize Ac/Ds elements (the hAT superfamily) [24, 25] and the rice Osmar elements (the Mariner superfamily) [9]. The most significant modification involved the construction of separate expression vectors containing fusions of the inducible yeast GAL 1 promoter to either the Ping ORF1 or the Ping TPase coding regions (Figure 1). The third plasmid vector in this assay was a selectable marker composed of the yeast ADE2 gene with its coding region interrupted by mPing [including the TSD (TAA)] (Figure 1). Growth on medium lacking adenine requires the excision of mPing and the in-frame repair of ADE2. Expression of the Ping proteins (induced by galactose) and selection for excision was performed concurrently on solid media lacking adenine. In this assay, each colony represents a unique excision event, thus eliminating an artifactually high frequency due to founder effects from early transposition events. This strict selection is sensitive to slight changes in the health of the culture, due to factors such as age, density and growth phase. In recognition of this source of variation, we pooled the excision frequencies from two or more separate experiments, each with multiple replicates (see Additional File 1).

Constructs used in this study and steps in the yeast assay. Diagram showing the relationship of the native Ping and mPing elements to the constructs used for the yeast transposition assay (A). ORF1 (green) and TPase (orange) coding regions were placed in galactose inducible constructs, while mPing (red) was cloned into the ADE2 coding region (yellow). The transposition assay protocol (B) involves growing transformed yeast in non-inducing liquid media and then plating on inducing (galactose) media under selection for adenine autotrophy. mPing excision and repair of ADE2 results in colony formation.
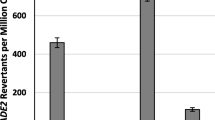
Co-expression of Ping TPase and ORF1 proteins in a single yeast cell resulted in a very low excision frequency of mPing from ADE2 (0.03 ± 0.02) (Figure 2, Additional File 1). We hypothesized that the low frequency could be due to the activity of a potential NES located in the C-terminal region of the TPase (Figure 2, Additional File 2) [26]. In order to determine the impact of this putative NES on activity, we mutated two leucine residues that are conserved in most predicted NESs (L384A, L386A)[26]. Alteration of this sequence resulted in significantly higher excision frequencies (5.9 ± 3) (Figure 2A, Additional File 1), suggesting that the NES was inhibiting TPase function in yeast. No ADE2 revertant colonies were observed when Ping ORF1 or TPase -NES proteins were expressed in isolation (data not shown).

Excision frequency in yeast catalyzed by wild type and mutant Ping and Pong proteins. Histograms showing average excision frequencies for mPing (A) and mPong (B) in yeast using different combinations of Ping and Pong proteins. TPase - nuclear export signal (NES) mutants contain alanine substitutions at the two conserved leucines in the predicted NES (Ping 384+386, Pong 418+420). Error bars represent the standard error. Sequence of the NES for the Ping and Pong TPase proteins are shown in the top left corner. Residues that match the consensus NES are in red.
The increased activity of the NES mutant provided the experimental resolution necessary to test the impact of site-directed mutations in conserved residues of domains predicted to be necessary for transposition. To test the suspected DNA binding domain of ORF1 protein, we disrupted the Myb-like sequence with a mutation in the helix-turn-helix fold (L225P). Co-expression of ORF1 (L225P) and the TPase -NES proteins resulted in no detectible excision. Similarly, mutation of the catalytic DDE domain of the TPase protein (D295A) led to a reduction in excision frequency of ~150-fold (0.04 ± 0.02) (Additional File 1). These results are consistent with the hypothesis that the DNA binding and catalytic functions are divided between the ORF1 and TPase proteins respectively.
Cross-mobilization mediated by Ping and Pong
In rice and transgenic Arabidopsis, both Ping and Pong products can mobilize mPing even though their ORF1 and TPase proteins share only 62% and 77% amino acid identity, respectively [9, 20]. To test whether Pong proteins can catalyze mPing transposition in yeast, we replaced Ping ORF1 and TPase with their Pong counterparts and combined these in yeast strains with the mPing reporter (see Methods). The results (Figure 2, Additional File 1) indicate that: (i) Pong proteins catalyze low frequency excision of mPing; (ii) introduction of the NES mutation (L418A, L420A) into the Pong TPase significantly increases the excision frequency; and (iii) Pong ORF1 functions with Ping TPase -NES and Ping ORF1 functions with Pong TPase -NES to catalyze mPing excision.
Another way to address the question of cross-mobilization is to ask whether Ping proteins can mobilize a deletion derivative of Pong. To this end, we generated a 500 bp nonautonomous Pong (mPong) and inserted it into the exact same position in ADE2 as mPing (see Methods). As shown in Figure 2B, while mPong is mobilized by the Ping and Pong proteins, the frequency is much lower than mPing (note the different scales on the Y axes). The possible reasons underlying this difference is revisited in the discussion.
Analyses of excision and reinsertion events
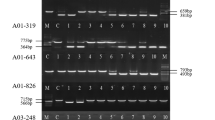
The high frequency of transposition catalyzed by the NES mutation permitted the isolation of many excision and reinsertion sites. Excision of mPing in both rice and transgenic Arabidopsis is unusual for a plant element in that it is usually precise [9, 27]. In other words, there is no alteration (footprint) at the excision site because mPing and one copy of the 3 bp TSD (usually TAA) are almost always removed. In our yeast assay, precise excision could be easily detected because it restores the Hpa I restriction site used for cloning mPing into ADE2 (see Methods). We isolated yeast ADE2 revertants [mPing mobilized by Ping ORF1 and Ping TPase (L384/386A)] and analysed the excision sites by polymerase chain reaction (PCR) and Hpa I digestion (Figure 3A). Almost all of the excision events (109/110) were precise. The one imperfect event was due to a 3 bp deletion that maintained the reading frame of ADE2 (data not shown).

Analysis of excision and insertion events. Analysis of the mPing excision sites from ADE2 revertants by polymerase chain reaction (PCR) using flanking primers and digestion with HpaI (A). mPing excision results in smaller products that can only be digested with HpaI when the ADE2 gene is repaired perfectly. Some ADE2 revertant colonies carry both the original and excised versions of the reporter plasmids, resulting in the two PCR products shown. The HpaI site is underlined, the target site duplication (TTA) is shown in bold, and mPing sequence is italicized. (Neg = mPing reporter, Pos = ADE2) The mPing insertion preference in yeast is shown as a pictogram (height of letter indicates % at that position) and the frequencies of preferred nucleotides compared to rice [12] (B).
Another unusual feature of all characterized members of the PIF/Harbinger superfamily is an insertion sequence preference that extends beyond the 3 bp TSD. This was first observed for the maize PIF element, with a 9 bp extended insertion site preference (the TSD and 3 bp on each side) [4]. mPing was similarly shown to have a 9 bp insertion preference both in rice [12] and in transgenic Arabidopsis [9]. To investigate whether a preference was manifest in the yeast assay, we used inverse PCR and transposon display to isolate mPing reinsertion sites in the yeast genome (see Methods). A consensus sequence generated from the sequences of 22 independent events revealed a 9 bp insertion site preference that was similar to the mPing insertion preference in rice and transgenic Arabidopsis (Figure 3B, Additional File 3). The insertion of mPong elements was also verified by flanking sequence analysis for four events mobilized by Ping proteins and five events catalyzed by Pong proteins (Additional File 3).
Protein localization
Our finding that the TPase NES had significant effects on activity in yeast suggested that protein localization is critical for mPing transposition. Many other TPase proteins harbor nuclear localization signals (NLS) that facilitates their access to the DNA substrate [28, 29]. Analysis of Ping proteins with the nuclear prediction software NucPred indicated that the ORF1 protein has a potential NLS and, as such, is likely to be nuclear-localized (score = 0.9) [30]. In contrast, the TPase has a much lower NucPred score (0.37), indicating it may not utilize a NLS to gain access to the DNA.
We used cerulean florescent protein (CFP) fusions to test the localization of the Ping proteins in yeast. ORF1 protein was found to be primarily nuclear and co-localized with the nuclear marker protein, NLS-EYFP (Figure 4) [31]. In contrast, wild type TPase showed a diffuse staining pattern. Figure 4 shows that the TPase (L384/386A) NES mutant shows increased CFP fluorescence in the nucleus, confirming that the NES plays a role in nuclear export of the TPase. Co-expression of the Ping ORF1 and TPase proteins together did not significantly alter the localization pattern of either protein (data not shown).

Protein localization. Fluorescence microscopy of yeast expressing cerulean fluorescent protein fusions of Ping ORF1, TPase or TPase (L384/386A) proteins (magenta). The nuclear localization signal-yellow fluorescent protein (green) was co-expressed to provide a nuclear marker.
Analysis of the NES mutant in transgenic Arabidopsis
Our findings indicate that the NES encoded by wild type TPase inhibits mPing transposition in yeast and that mutation of this motif increases nuclear localization and transposition activity. In order to determine if the NES also reduced transposition in plants, we modified the previously developed Arabidopsis transposition assay to test the effect of the TPase (L384/386) NES mutation. The reporter construct for this assay was mPing inserted between the CaMV 35S promoter and a gfp reporter gene (Figure 5A). We transformed the CaMV 35S:Ping cDNA with and without the TPase NES mutation (L384/386A) because the wild type construct was shown previously to express both ORF1 and TPase proteins from a single construct in Arabidopsis (see Methods) [9]. A comparison of the number of transgenic plants that showed mPing excision (determined by green fluorescent protein (GFP) visualization and PCR analysis) indicated that the alteration of the NES resulted in significantly higher excision activity (40% versus 15%, P < 0.005). In addition, the NES mutation resulted in more GFP fluorescence in both the cotyledons and mature leaves (Figure 5B). Finally, PCR analysis using primers that flank the mPing insertion site showed an increase in the amount of the ~400 bp (excision) product in seedlings containing the mutant TPase.

Arabidopsis transposition assay. Comparison of the transposition activity of wild type TPase and the TPase (L384/386A) nuclear export signal mutant in Arabidopsis containing an mPing gfp reporter construct. ORF1 and TPase proteins were expressed from a cDNA that contains both genes [TPase is presumably expressed by a native promoter within the intergenic region] (A). The relative frequency of mPing excision for each construct is shown together with a representative green fluorescent protein (GFP) expressing plant (B). GFP expression produces green sectors on a red chlorophyll background. Polymerase chain reaction analysis of representative GFP expressing plants from each treatment using primers (shown in A) that flank the excision site (C).
Discussion
In this study we report the first yeast transposition assay for the PIF/Harbinger superfamily. The assay retains many of the unusual features of this superfamily including precise excision, insertion sequence preference and the requirement for the two element-encoded proteins. In order to develop this assay we used the related rice elements Ping and Pong, the only naturally active members of this superfamily, and mPing, the only naturally active Tourist MITE. Below, we discuss some of the features of this assay and how the wealth of yeast genetics could be exploited to understand the transposition mechanism and, perhaps, the success of Tourist MITEs.
Excision and insertion
Excision of mPing in both rice and transgenic Arabidopsis is usually perfect as it involves the removal of both mPing and one copy of the TSD. This stands in marked contrast to all other plant class 2 elements where one or more nucleotides usually remain at the excision site as a so-called transposon footprint [11]. While all but one of the excision events of mPing in yeast were perfect (109/110) (Figure 3), one could argue that this result is an artifact of selection for ADE2 reversion. This would be the case if only the wild type sequence at this site was able to restore ADE2 function. However, in a parallel study of a rice Stowaway element, inserted at exactly the same site in ADE2 as mPing, all ADE2 revertants had footprints with insertions of 6, 9 or-12 bp [32].
Thus, excision site repair in yeast is influenced by currently unknown differences in the catalytic properties of the transposase source. Plant transposable element excision sites are thought to be repaired primarily by the non-homologous end-joining pathway (NHEJ), which is well characterized in yeast [33]. In a prior study, Yu et al. [25] analysed the excision of the maize Ds element by the Ac transposase (a member of the hAT superfamily) in yeast strains that were mutant for various steps in the NHEJ pathway. They found that mutations in a subset of the genes tested influenced both the quality and frequency of excision site repair [25]. In this regard, we have begun to analyse excision of mPing and the rice mariner element OsStow35 [32] in the same yeast mutant backgrounds to address why mPing but not Ds or OsStow is repaired perfectly.
Another distinguishing feature of the transposition reaction of characterized PIF/Harbinger elements is an extended insertion sequence preference of 9-15 bp [4–6]. That the target preference is conserved (to the extent that our yeast sample size can differentiate, Figure 3) indicates that one or both of the element-encoded proteins is responsible for this specificity. As such, the yeast assay should be a valuable tool that could allow the rapid analysis of mutant ORF1 and/or TPase proteins in order to determine if they contribute to insertion preference.
Requirement for 2 element-encoded proteins
Perhaps the most unusual feature of the PIF/Harbinger superfamily is that transposition requires two element-encoded proteins, ORF1 and TPase [9, 10]. Prior analysis of the reconstructed Harbinger3N_DR element from zebrafish suggested that ORF1 protein binds to the TIR and is required in human cells for nuclear localization of TPase (which lacks a nuclear localization sequence). In this study, the use of the wild type ORF1 and TPase proteins from either Ping or Pong resulted in negligible excision of mPing (Figure 2). This result was unexpected as these proteins catalyzed robust excision of mPing from the gfp reporter in transgenic Arabidopsis [9]. Mutagenesis of a predicted NES in the C-terminal region of the TPase from both Ping and Pong (Additional File 2) led to significant increases in the excision frequency of mPing (Figure 2A).
Increased nuclear localization of the mutant versus the wild type Ping TPase (Figure 4) suggests that the low excision frequency catalyzed by wild type TPase is a direct result of its export from the nucleus. However, if this is the case, why does wild type Ping and Pong TPase catalyze mPing excision in both rice and Arabidopsis [9, 20]? One reason may be that yeast, like other simple eukaryotes, undergoes a 'closed' mitosis where the nuclear envelope does not break down [34]. Unlike the situation in plants, TPase exported to the cytoplasm in yeast cannot get back into the nucleus during mitosis. Thus, mutation of the NES prevents or slows nuclear export of TPase and keeps it in the nucleus.
An alternative model to explain the difference in activity of the wild type TPase in yeast and plants is that the NES may not function in rice and Arabidopsis. To address this question, we generated transgenic Arabidopsis plants with wild type and mutant TPase (Figure 5A). Although excision frequency is difficult to quantify precisely, we found that both the percentage of plants showing excision events and PCR products resulting from mPing excision were significantly increased in plants harbouring the TPase with the mutant NES (Figure 5). The identification of a hyperactive mutation in yeast, and its subsequent validation in plants, indicates the potential value of the yeast assay as a rapid and facile screen for ORF1 and TPase mutations that reduce or increase the frequency of transposition or alter the quality of excision or insertion events.
Using the yeast assay to dissect the success of mPing
In addition to its potential utility at dissecting the catalytic features of the PIF/Harbinger superfamily, the yeast assay may be of value in determining the reason why mPing, and not other nonautonomous elements, become MITEs. As is true for many relatively recent discoveries, the definition of just what is a MITE is continuously being refined as an increasing number of genome sequences become available. Stated simply, a MITE is a class 2 nonautonomous element that can attain very high copy numbers (hundreds or thousands) through the rapid amplification of one or a few identical or nearly identical founder elements [35, 36]. MITEs are usually short (~100-500 bp) and are either of unknown origin or, like mPing, are deletion derivatives of a larger autonomous element.
One of the outstanding questions about MITEs is whether their ability to attain high copy numbers is an inherent feature of the element or a fortuitous circumstance. In other words, is there something special about the structure of MITEs that, for example, makes them better substrates for transposase? Alternatively, are MITEs just average nonautonomous elements that happen to be in the genome when, for example, an unrelated autonomous element is producing transposase? The results of this study favour the former model where MITEs, such as mPing, have distinctive structural features (compared to other low copy nonautonomous elements) that facilitate their rapid amplification and spread in the genome. Although mPing and mPong are both deletion derivatives of larger autonomous elements, the excision frequency of the former is much higher than the latter (Figure 2A, B). In fact, the frequency of mPing excision is ~40- to 50-fold higher than that of mPong when either Ping or Pong proteins catalyze transposition. The relative success of mPing in yeast mirrors what occurs in rice; there are less than five deletion derivatives of mPong in all of the sequenced rice genomes, while most characterized strains of japonica rice have 25-50 mPing copies and a few have over 1000 [12]. As such, the yeast assay can be used to determine the excision frequencies of a variety of constructs, including additional deletion derivatives of Pong and Ping, chimeric elements where regions of mPing and mPong are swapped and site-directed mutations throughout mPing. In this way we should be able to determine the cis-features that distinguish MITEs from low copy number nonautonomous elements.
Conclusions
This is the first report of the transposition of members of the PIF/Harbinger superfamily in yeast. This breakthrough facilitated the characterization of the NES encoded by the Ping and Pong TPase proteins and confirms a role for the control of nuclear access in regulating transposition. The yeast assay also provides a platform for the analysis of the unique characteristics of the transposition reaction including precise excision and insertion sequence preference. Our finding that Ping and Pong proteins can function in a cooperative manner suggests that a new layer of cross-mobilization may have to be considered in future studies of how transposable element families evolve. Finally, the transposition of mPing in yeast will facilitate investigations into the features underlying the success of Tourist-like MITEs.
Methods
Yeast strains and construct construction
Saccharomyces cerevisiae strain DG2523 and the pWL89a vector were described previously [24, 37]. The following primers were used to prepare the elements for ligation into the HpaI site of ADE2: mPing F 5'-AAGGCCAGTCACAATGGGG-3', mPing R 5'-AGGCCAGTCACAATGGCTAG-3'; mPong F 5'-AAGGCCAGTCACAATGGGG-3'; and mPong R 5'-AGGCCATTCACAATGCAGT-3'. The mPong element was constructed in order to contain 251 bp from the 5' end of Pong and 245 bp from the 3' end of Pong (sequence available upon request). Genes from cDNA clones [GenBank: AK068363.1, GenBank: AK068654.1] or genomic DNA were cloned into the Gateway cloning vectors, pENTR or pDONR Zeo, and then transferred to destination vectors with LR Clonase reactions (Invitrogen, California, USA). Sequence alterations were made using the Quikchange II Mutagenesis Kit (Stratagene, California, USA).
Yeast transposition assay
Ping and Pong ORFs (wild type and mutant) were cloned into pAG413GAL-ccdb (ORF1) and pAG415GAL-ccdb (TPase) vectors [38]. Transformed yeast were grown to saturation (36-48 h) in 5 ml of CSM -his-leu-ura with dextrose, washed with 5 ml sterile water, resuspended in 0.5 ml water and plated onto CSM -ade -his -leu -ura with galactose as the sole carbon source. Colonies were counted after incubation at 30°C for 15 days. Viable counts were made by plating 50 μl or 100 μl of a 4 × 104 dilution on yeast extract peptone dextrose plates.
Excision and insertion site analysis
ADE2 revertant yeast was grown in selective media and genomic DNA isolations were performed using an E.Z.N.A. Yeast DNA Kit (Omega Bio-tek, Georgia, USA). PCR [PCR Master Mix (Promega, Wisconsin, USA)] using primers flanking the mPing excision site were digested with HpaI (New England Biolabs, Massachusetts, USA) and then analysed by gel electrophoresis. PCR products were cloned for sequencing using Topo-pCR2.1 (Invitrogen). Inverse PCR and transposon display was performed as described [9]. The insertion site analysis figure was made using the program Pictogram http://genes.mit.edu/pictogram.html.
Microscopy
The yeast nuclear marker vector NLS-EYFP (pPS1888) was described previously [31]. N-terminal cerulean fusion protein constructs were constructed using pAG415GAL-cerulean-ccdb [38]. Yeast were grown on selective media with galactose, visualized with a Zeiss Axio Imager M1 microscope equipped with a 63× oil objective and SlideBook 4.0 software (Intelligent Imaging Innovations, Denver, USA). GFP expression was detected in seedlings with a Zeiss Discovery V12 fluorescence stereoscope.
Arabidopsis transposition assay
The coding regions of the Ping cDNA [GenBank: AK068363.1] were cloned into the pENTR vector for mutagenesis and sequencing before transfer to the pEarleyGate 204 vector [39]. Wild type and NES mutant versions of TPase were transformed into Arabidopsis thaliana (Columbia ecotype) containing a previously transformed single copy of the mPing reporter [9] using a simple Agrobacterium tumefaciens (GV3103) floral dip method [40]. Seedlings were germinated on 0.2% phytagel plates (0.5 Murashige and Skoog, 1% sucrose) containing the appropriate selection reagents (7.5 μg/ml PPT, 50 μg/ml Kan, 150 μg/ml Timentin) in a Percival growth chamber (12 hrs light, 21°C) and analysed by fluorescent microscopy. DNA from 2-week-old seedlings was extracted by the CTAB method with PCR performed as described to confirm transformation and detect mPing excision [9].
Abbreviations
- CFP:
-
cerulean fluorescent protein
- GFP:
-
green florescent protein
- MITE:
-
miniature inverted repeat transposable element
- NES:
-
nuclear export signal
- NHEJ:
-
non-homologous end joining
- NLS:
-
nuclear localization signal
- PCR:
-
polymerase chain reaction
- TIR:
-
terminal inverted repeat
- TSD:
-
target site duplication
- YFP:
-
yellow fluorescent protein.
References
McClintock B: The origin and behavior of mutable loci in maize. Proceedings of the National Academy of Sciences of the United States of America. 1950, 36: 344-355. 10.1073/pnas.36.6.344.
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O: A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007, 8: 973-982. 10.1038/nrg2165.
Kapitonov VV, Jurka J: Molecular paleontology of transposable elements from Arabidopsis thaliana. Genetica. 1999, 107: 27-37. 10.1023/A:1004030922447.
Zhang XY, Feschotte C, Zhang Q, Jiang N, Eggleston WB, Wessler SR: P instability factor: An active maize transposon system associated with the amplification of Tourist-like MITEs and a new superfamily of transposases. Proceedings of the National Academy of Sciences of the United States of America. 2001, 98: 12572-12577. 10.1073/pnas.211442198.
Zhang XY, Jiang N, Feschotte C, Wessler SR: PIF- and Pong-like transposable elements: distribution, evolution and relationship with Tourist-like miniature inverted-repeat transposable elements. Genetics. 2004, 166: 971-986. 10.1534/genetics.166.2.971.
Kapitonov VV, Jurka J: Harbinger transposons and an ancient HARBI1 gene derived from a transposase. DNA and Cell Biology. 2004, 23: 311-324. 10.1089/104454904323090949.
Pereira A, Cuypers H, Gierl A, Schwarzsommer Z, Saedler H: Molecular analysis of the En/Spm transposable element system of Zea mays. Embo Journal. 1986, 5: 835-841.
Masson P, Rutherford G, Banks JA, Fedoroff N: Essential large transcripts of the maize Spm transposable element are generated by alternative splicing. Cell. 1989, 58: 755-765. 10.1016/0092-8674(89)90109-8.
Yang GJ, Zhang F, Hancock CN, Wessler SR: Transposition of the rice miniature inverted repeat transposable element mPing in Arabidopsis thaliana. Proceedings of the National Academy of Sciences of the United States of America. 2007, 104: 10962-10967. 10.1073/pnas.0702080104.
Sinzelle L, Kapitonov VV, Grzela DP, Jursch T, Jurka J, Izsvak Z, Ivics Z: Transposition of a reconstructed Harbinger element in human cells and functional homology with two transposon-derived cellular genes. Proceedings of the National Academy of Sciences of the United States of America. 2008, 105: 4715-4720. 10.1073/pnas.0707746105.
Sutton WD, Gerlach WL, Schwartz D, Peacock WJ: Molecular analysis of Ds controlling element mutations at the Adhl locus of maize. Science. 1983, 223: 1265-1268. 10.1126/science.223.4642.1265.
Naito K, Cho E, Yang GJ, Campbell MA, Yano K, Okumoto Y, Tanisaka T, Wessler SR: Dramatic amplification of a rice transposable element during recent domestication. Proceedings of the National Academy of Sciences of the United States of America. 2006, 103: 17620-17625. 10.1073/pnas.0605421103.
Wessler SR, Bureau TE, White SE: LTR-retrotransposons and MITEs - Important players in the evolution of plant genomes. Current Opinion in Genetics & Development. 1995, 5: 814-821.
Jiang N, Wessler SR: Insertion preference of maize and rice miniature inverted repeat transposable elements as revealed by the analysis of nested elements. Plant Cell. 2001, 13: 2553-2564. 10.1105/tpc.13.11.2553.
Tu ZJ: Three novel families of miniature inverted-repeat transposable elements are associated with genes of the yellow fever mosquito, Aedes aegypti. Proceedings of the National Academy of Sciences of the United States of America. 1997, 94: 7475-7480. 10.1073/pnas.94.14.7475.
Izsvak Z, Ivics Z, Shimoda N, Mohn D, Okamoto H, Hackett PB: Short inverted-repeat transposable elements in teleost fish and implications for a mechanism of their amplification. Journal of Molecular Evolution. 1999, 48: 13-21. 10.1007/PL00006440.
Pace JK, Feschotte C: The evolutionary history of human DNA transposons: evidence for intense activity in the primate lineage. Genome Research. 2007, 17: 422-432. 10.1101/gr.5826307.
Smit AFA, Riggs AD: Tiggers and other DNA transposon fossils in the human genome. Proceedings of the National Academy of Sciences of the United States of America. 1996, 93: 1443-1448. 10.1073/pnas.93.4.1443.
Kidwell MG, Lisch D: Transposable elements as sources of variation in animals and plants. Proceedings of the National Academy of Sciences of the United States of America. 1997, 94: 7704-7711. 10.1073/pnas.94.15.7704.
Jiang N, Bao ZR, Zhang XY, Hirochika H, Eddy SR, McCouch SR, Wessler SR: An active DNA transposon family in rice. Nature. 2003, 421: 163-167. 10.1038/nature01214.
Nakazaki T, Okumoto Y, Horibata A, Yamahira S, Teraishi M, Nishida H, Inoue H, Tanisaka T: Mobilization of a transposon in the rice genome. Nature. 2003, 421: 170-172. 10.1038/nature01219.
Kikuchi K, Terauchi K, Wada M, Hirano HY: The plant MITE mPing is mobilized in anther culture. Nature. 2003, 421: 167-170. 10.1038/nature01218.
Haren L, Ton-Hoang B, Chandler M: Integrating DNA: transposases and retroviral integrases. Annual Review of Microbiology. 1999, 53: 245-281. 10.1146/annurev.micro.53.1.245.
Weil CF, Kunze R: Transposition of maize Ac/Ds transposable elements in the yeast Saccharomyces cerevisiae. Nature Genetics. 2000, 26: 187-190. 10.1038/82827.
Yu JH, Marshall K, Yamaguchi M, Haber JE, Weil CF: Microhomology-dependent end joining and repair of transposon-induced DNA hairpins by host factors in Saccharomyces cerevisiae. Molecular and Cellular Biology. 2004, 24: 1351-1364. 10.1128/MCB.24.3.1351-1364.2004.
la Cour T, Kiemer L, Molgaard A, Gupta R, Skriver K, Brunak S: Analysis and prediction of leucine-rich nuclear export signals. Protein Engineering Design & Selection. 2004, 17: 527-536.
Lin X, Long L, Shan X, Zhang S, Shen S, Liu B: In planta mobilization of mPing and its putative autonomous element Pong in rice by hydrostatic pressurization. Journal of Experimental Botany. 2006, 57: 2313-2323. 10.1093/jxb/erj203.
Liu D, Crawford NM: Characterization of the putative transposase mRNA of Tag1, which is ubiquitously expressed in Arabidopsis and can be induced by Agrobacterium-mediated transformation with dTag1 DNA. Genetics. 1998, 149: 693-701.
Ivics Z, Izsvak Z, Minter A, Hackett PB: Identification of functional domains and evolution of Tc1-like transposable elements. Proceedings of the National Academy of Sciences of the United States of America. 1996, 93: 5008-5013. 10.1073/pnas.93.10.5008.
Brameier M, Krings A, MacCallum RM: NucPred - predicting nuclear localization of proteins. Bioinformatics. 2007, 23: 1159-1160. 10.1093/bioinformatics/btm066.
Damelin M, Silver PA: Mapping interactions between nuclear transport factors in living cells reveals pathways through the nuclear pore complex. Molecular Cell. 2000, 5: 133-140. 10.1016/S1097-2765(00)80409-8.
Yang GJ, Holligan-Nagel D, Feschotte C, Hancock CN, Wessler SR: Tuned for transposition: molecular determinants underlying the hyperactivity of a Stowaway MITE. Science. 2009, 325: 1391-1394. 10.1126/science.1175688.
Daley JM, Palmbos PL, Wu D, Wilson TE: Nonhomologous end joining in yeast. Annu Rev Genet. 2005, 39: 431-451. 10.1146/annurev.genet.39.073003.113340.
Gant TM, Wilson KL: Nuclear assembly. Annu Rev Cell Dev Biol. 1997, 13: 669-695. 10.1146/annurev.cellbio.13.1.669.
Feschotte C, Jiang N, Wessler SR: Plant transposable elements: where genetics meets genomics. Nature Reviews Genetics. 2002, 3: 329-341. 10.1038/nrg793.
Feschotte C, Zhang XY, Wessler SR: Miniature Inverted-repeat Transposable Elements (MITEs) and their relationship with established DNA transposons. Mobile DNA II. Edited by: Craig NL, Craige R, Gellert M, Lambowitz A. 2002, Washington D.C.: American Society of Microbiology Press
Yang GJ, Weil CF, Wessler SR: A rice TC1/mariner-like element transposes in yeast. Plant Cell. 2006, 18: 2469-2478. 10.1105/tpc.106.045906.
Alberti S, Gitler AD, Lindquist S: A suite of gateway (R) cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast. 2007, 24: 913-919. 10.1002/yea.1502.
Earley KW, Haag JR, Pontes O, Opper K, Juehne T, Song KM, Pikaard CS: Gateway-compatible vectors for plant functional genomics and proteomics. Plant Journal. 2006, 45: 616-629. 10.1111/j.1365-313X.2005.02617.x.
Clough SJ, Bent AF: Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant Journal. 1998, 16: 735-743. 10.1046/j.1365-313x.1998.00343.x.
Acknowledgements
We thank Guojun Yang, Jim Burnette and Eunyoung Cho for their technical assistance and advice. Our thanks are also due to the many undergraduates, including Lee B Sharpe, Lucy Fu, Catherine Bridges and the Spring 09 HHMI Dynamic Genome class, who helped with aspects of the project. We are grateful to Clay Isom and Cody Diehl for their critical review of the manuscript. This research funded by grants from the National Institute of Health, and Howard Hughes Medical Institute Professor award to SRW.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
CNH designed and carried out the Arabidopsis transposition assays, microscopy and coordinated/participated in all of the yeast transposition assays and the associated sequence analysis. FZ participated in the design of the study, produced some yeast constructs and performed a portion of the insertion site analysis. SRW conceived the study, participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
13100_2009_5_MOESM1_ESM.XLSX
Additional file 1: Table of yeast excision frequency data. Excel file containing the data set used to produce Figure 2. Frequency = ADE2 revertants/total cells plated. (XLSX 20 KB)
13100_2009_5_MOESM2_ESM.PDF
Additional file 2: NetNES output. PDF file containing the prediction of the nuclear export signal (NES) signal in the C-terminal region of the Ping TPase protein. NN indicates the neural network score, HMM indicates the hidden Markov model score. Scores above the threshold indicate potential NES signals. (PDF 8 KB)
13100_2009_5_MOESM3_ESM.XLS
Additional file 3: Insertion site sequences. Excel file containing a table of the mPing and mPong insertions in yeast. (XLS 26 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hancock, C.N., Zhang, F. & Wessler, S.R. Transposition of the Tourist-MITE mPing in yeast: an assay that retains key features of catalysis by the class 2 PIF/Harbinger superfamily. Mobile DNA 1, 5 (2010). https://doi.org/10.1186/1759-8753-1-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1759-8753-1-5