
Abstract
The Ras-dependent Raf/MEK/ERK1/2 mitogen-activated protein (MAP) kinase signaling pathway is a major regulator of cell proliferation and survival. Not surprisingly, hyperactivation of this pathway is frequently observed in human malignancies as a result of aberrant activation of receptor tyrosine kinases or gain-of-function mutations in RAS or RAF genes. Components of the ERK1/2 pathway are therefore viewed as attractive candidates for the development of targeted therapies of cancer. In this article, we briefly review the basic research that has laid the groundwork for the clinical development of small molecules inhibitors of the ERK1/2 pathway. We then present the current state of clinical evaluation of MEK1/2 inhibitors in cancer and discuss challenges ahead.
Similar content being viewed by others


Introduction
Human tumorigenesis is a multistep process during which accumulation of genetic and epigenetic alterations leads to the progressive transformation of a normal cell into a malignant cancer cell. During this process, cancer cells acquire new capabilities (hallmarks) that enable them to escape from normal homeostatic regulatory defense mechanisms. These hallmarks are defined as: self-sufficiency in growth signals, insensitivity to antiproliferative signals, evasion from apoptosis, limitless replicative potential, sustained angiogenesis, and increased motility and invasiveness [1]. While the mechanisms by which cancer cells acquire these capabilities vary considerably between tumors of different types, most if not all of these physiological changes involve alteration of signal transduction pathways. Among the signaling pathways most frequently dysregulated in human cancer is the Ras-Raf-MEK-extracellular signal-regulated kinase 1 and 2 (ERK1/2) pathway.
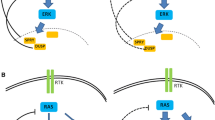
The Ras-dependent ERK1/2 mitogen-activated protein (MAP) kinase pathway is one of the best-studied signal transduction pathways (Fig. 1). Since the discovery of MAP kinases by Ray and Sturgill in 1988 [2], more than 11,000 articles have been published on this topic. ERK1/2 MAP kinases are activated by virtually all growth factors and cytokines acting through receptor tyrosine kinases, cytokine receptors or G protein-coupled receptors. Typically, ligand binding to receptor tyrosine kinases induces dimerization of the receptor and auto-phosphorylation of specific tyrosine residues in the C-terminal region. This generates binding sites for adaptor proteins, such as growth factor receptor-bound protein 2 (GRB2), which recruit the guanine nucleotide exchange factor Sos at the plasma membrane. Sos activates the membrane-bound Ras by catalyzing the replacement of GDP with GTP. In its GTP-bound form, Ras recruits Raf kinases (ARAF, BRAF and CRAF) to the plasma membrane, where they become activated by a complex interplay of phosphorylation events and protein-protein interactions. Raf acts as a MAP kinase kinase kinase (MAPKKK) and activates the MAP kinase kinases (MAPKKs) MEK1 and MEK2, which, in turn, catalyze the activation of the effector MAP kinases ERK1 and ERK2 [3]. Once activated, ERK1/ERK2 phosphorylate a panoply of nuclear and cytoplasmic substrates involved in diverse cellular responses, such as cell proliferation, survival, differentiation, motility, and angiogenesis [4].

Schematic representation of the Ras-Raf-MEK-ERK1/2 MAP kinase pathway. The figure shows the cascade of activation of the MAP kinases ERK1/ERK2 mediated by growth factor binding to receptor tyrosine kinases. See text for details. GF, growth factor; RTK, receptor tyrosine kinase.
MEK1/MEK2 and the family of MAP kinase kinases
MEK1 and MEK2 belong to the family of MAPKKs (also known as MEKs or MKKs), which are dual specificity enzymes that phosphorylate threonine and tyrosine residues within the activation loop of their MAP kinase substrates [5]. The human genome encodes seven MAPKK enzymes that regulate the activity of four distinct MAP kinase pathways (Fig. 2A). Aside from MEK1/MEK2, the MAPKKs MKK4 and MKK7 phosphorylate and activate the c-Jun N-terminal kinase (JNK) isoforms, MKK3 and MKK6 phosphorylate and activate the p38 isoforms, and MEK5 selectively activates ERK5. Depending on the cellular context, MKK4 may also contribute to the activation of the p38 pathway [6, 7].

The MAP kinase kinases family. (A) MAP kinases and their upstream MAPKKs. (B) Schematic representation of human MAPKKs. MAPKKs are composed of a kinase catalytic domain (in blue) flanked by N- and C-terminus extensions of varying lengths. The percentage of identity of the kinase domain with MEK1 is indicated. An NES, only present in MEK1 and MEK2, is indicated in yellow.
Structurally, MAPKKs are proteins of ~45-50 kDa that share 37-44% amino acid identity with MEK1/MEK2 in the kinase domain (Fig. 2B). MEK1 and MEK2 are themselves 86% identical in the catalytic domain. In addition to their kinase domain, MEK1 and MEK2 contain a strong leucine-rich nuclear export signal (NES) at their N-terminal extremity [8], a feature not found in other MAPKK family members. Contrary to MAP kinases, MAPKKs have very narrow substrate specificity. It is assumed, from lack of evidence to the contrary, that the MAP kinases ERK1/ERK2 are the only substrates of MEK1 and MEK2. However, the possibility that MEK1/MEK2 have other non-catalytic effectors cannot be excluded. For example, a recent study showed that MEK1 interacts with peroxisome proliferator-activated receptor γ (PPARγ) to induce its nuclear export and attenuate its transcriptional activity [9].
The high sequence identity between MEK1 and MEK2, and their significant similarity with MEK5 have important pharmacological implications. First, this explains why small molecule MEK1/2 inhibitors developed so far are non-selective with regard to MEK1 and MEK2 isoforms.
Although it is commonly believed that the two MAPKK isoforms are functionally equivalent, there is evidence, however, that they are regulated differentially and may not be interchangeable in all cellular contexts [10–13]. Intriguingly, it has been reported that activated MEK1 but not MEK2 induces epidermal hyperplasia in transgenic mice [14]. RNA interference and gene invalidation studies have also suggested that MEK1 and MEK2 may contribute differentially to tumorigenesis [15, 16]. The physiopathological relevance of these observations to human cancer remains unclear. Second, it helps understand why the first-generation MEK1/2 inhibitors PD98059, U0126 and PD184352 were also found to inhibit MEK5 and the ERK5 MAP kinase pathway at higher concentrations [17, 18]. Elucidation of the crystal structures of MEK1 and MEK2 has revealed that MEK5 share 83% amino acid identity with MEK1 in the PD184352-like inhibitor-binding pocket [19]. These MEK1/2 inhibitors have been used in thousands of papers and have proven extremely useful tools to investigate the biological functions of the ERK1/2 MAP kinase pathway. However, their inhibitory activity towards MEK5, albeit weaker, indicates that we should be cautious in the interpretation of data obtained at high concentrations of inhibitor.
The ERK1/2 MAP kinase pathway is a key regulator of cell proliferation and survival
Multiple lines of evidence have implicated the ERK1/2 MAP kinase pathway in the control of cell proliferation [20]. First, ERK1 and ERK2 are activated in response to virtually all mitogenic factors. Second, several studies have reported that the mitogenic response to growth factors is correlated with their ability to induce sustained ERK1/2 activity [21–23]. Third, expression of kinase-dead mutants of ERK1 or anti-sense ERK1 RNA inhibited the activation of ERK1/ERK2 and exerted a dominant-negative effect on cell proliferation [24]. These early findings were confirmed by subsequent RNA interference-based studies showing that silencing of ERK1/ERK2 expression inhibits the proliferation of various cell types [25–27]. Fourth, treatment with small molecule inhibitors of MEK1/MEK2 was reported to inhibit the proliferation of a variety of cell types [28–30]. Reciprocally, expression of constitutively-active forms of MEK1 was sufficient to stimulate cell proliferation and relax growth factor dependency [31–33]. Further demonstration of the essential role of ERK1/2 signaling in cell proliferation was provided by gene invalidation studies in mice showing that loss of Erk1 or Erk2 gene function results in impaired proliferation of specific cell types [34–37].
ERK1/2 signaling is required for the progression of cells from the G0/G1 to S phase [20, 38]. Activation of the ERK1/2 pathway is associated with induction of the positive cell cycle regulators cyclin D1 [39] and c-Myc [40], and with down-regulation of anti-proliferative proteins such as Tob1 [23], Foxo3a [41] and p21 [42]. In addition to its direct role in the cell division cycle, the ERK1/2 MAP kinase pathway also regulates cell growth by stimulating protein and nucleotide biosynthesis [20, 43]. One mechanism by which the ERK1/2 pathway increases global protein translation is through phosphorylation and inactivation of tuberin (also known as TSC2), a negative regulator of the master growth regulator mammalian target of rapamycin (mTOR), resulting in increased mTOR signaling [44, 45].
Studies in several experimental systems have highlighted the important role of the Raf-MEK-ERK1/2 MAP kinase pathway in the control of cell survival [46, 47]. Early studies have shown that activation of the ERK1/2 pathway prevents apoptosis induced by growth factor withdrawal, loss of matrix attachment or cytoskeletal disruption in cultured cells [48–51]. These findings were reinforced by genetic studies showing that loss of ERK1/ERK2 or MEK1/MEK2 induces cell death in various mouse tissues [37, 52, 53]. ERK1/2 signaling promotes cell survival by repressing the expression or activity of pro-apoptotic Bcl-2 family proteins, such as Bim and Bad, and by inducing the expression of pro-survival members like Bcl-2 and Mcl-1 [47].
Hyperactivation of the ERK1/2 MAP kinase pathway in cancer
Given the central role of the Raf-MEK-ERK1/2 signaling pathway in cell proliferation and survival signaling, it is therefore not surprising that alterations in this pathway are highly prevalent in human cancer. Multiple genetic changes can lead to hyperactivation of the ERK1/2 pathway in cancer (Fig. 3). Aberrant activation of receptor tyrosine kinases such as the epidermal growth factor (EGF) receptor, as a result of gene amplification or gain-of-function mutations, is frequently observed in carcinomas and brain tumors [54, 55]. Activating mutations in RAS genes, most often in KRAS, are found in ~30% of cancers and are generally acquired early in the tumorigenic process [56]. More recently, large-scale resequencing studies have revealed that BRAF is mutated in ~20% of all cancers and in more than 40% of melanomas [57]. The majority of these mutations are clustered in the kinase domain of B-Raf and lead to the stimulation of ERK1/2 activity in cells [58]. It is noteworthy that RAS and BRAF mutations are generally mutually exclusive in tumors, suggesting an epistatic relationship. Also, activating mutations in MEK1 gene are found at low prevalence in lung carcinomas, melanomas and colon carcinomas [59, 60]. However, no mutation in the ERK1 or ERK2 gene has been reported to date in tumors. Consistent with these observations, numerous studies using clinical specimens have documented the hyperactivation of MEK1/MEK2 and ERK1/ERK2 in solid tumor and hematological malignancies [61, 62].

Genetic alterations of the Ras-dependent ERK1/2 pathway in cancer.
Studies in cultured cells have revealed that expression of activated alleles of MEK1 or MEK2 is sufficient to deregulate the proliferation and trigger transformation of immortalized fibroblast and epithelial cell lines [15, 31, 32, 63, 64]. Orthotopic transplantation of mammary or intestinal epithelial cells expressing activated MEK1/MEK2 into mice induces the formation of aggressive tumors that progress up to the metastatic stage [15, 64]. Similarly, expression of activated Raf mutants in various cell lines, including melanocytes, stimulates MEK1/2 and ERK1/2 signaling, and induces the formation of tumors in nude mice [65]. The oncogenic activity of the Raf-MEK-ERK1/2 pathway was further tested in transgenic mouse models. Transgenic expression of activated MEK1 in mouse skin induces hyperproliferative and inflammatory lesions and inhibits epidermal differentiation, mimicking features of squamous cell carcinomas [14, 66, 67]. In the same way, targeted expression of activated forms of C-Raf or B-Raf in various tissues of transgenic mice was shown to drive lung, skin, thyroid, and prostate tumorigenesis [65, 68, 69]. Importantly, deinduction of activated B-Raf expression in a conditional lung cancer mouse model leads to dramatic tumor regression concomitant to inactivation of ERK1/2 signaling, suggesting a dependency of B-Raf-induced lung tumors on the ERK1/2 pathway [70].
Pre-clinical pharmacological studies have demonstrated that blockade of the ERK1/2 pathway with small-molecule MEK1/2 inhibitors markedly restrains the proliferation of various carcinoma and leukemic cell lines by inducing cell cycle arrest and apoptosis [28, 30, 71, 72]. In vivo studies further established that administration of orally available MEK1/2 inhibitors elicits significant tumor regression in mouse xenograft models [30, 72–74]. The strategic position of MEK1 and MEK2 in the Ras-dependent ERK1/2 pathway in conjunction with a promising pre-clinical profile have provided strong rationale for the development of small-molecule inhibitors of MEK1/2 for chemotherapeutic intervention in cancer [62].
Clinical development of MEK1/2 inhibitors
PD98059 was the first small-molecule inhibitor of MEK1/2 to be disclosed in 1995 [28]. Biochemical studies indicated that PD98059 inhibits the activity of both MEK1 and MEK2 isoforms, but fails to inhibit a panel of other Ser/Thr kinases [75, 76]. Two other potent inhibitors of MEK1/2, U0126 [77] and Ro 09-2210 [78], were subsequently identified in cell-based assays. None of these compounds was moved to clinical evaluation because of their pharmaceutical limitations. However, PD98059 and U0126 have proven to be invaluable academic research tools to investigate the role of the ERK1/2 MAP kinase pathway in normal cell physiology and disease.
To date, eleven MEK1/2 inhibitors have been tested clinically or are currently undergoing clinical trial evaluation (Table 1). The chemical structures of some of these inhibitors are given in Fig. 4.

Chemical structures of small molecule MEK1/2 inhibitors.
CI-1040 (PD184352)
The benzhydroxamate derivative CI-1040 (Pfizer) was the first MEK1/2 inhibitor to enter clinical trials [79]. CI-1040 is a potent (IC50 of 17 nM on purified MEK1) and highly selective inhibitor of MEK1 and MEK2 that was identified by screening a library compound with an in vitro ERK1 reactivation assay [30]. Similar to PD98059 and U0126, CI-1040 and its analogs inhibit MEK1/2 in a non-ATP and non-ERK1/2 competitive manner. Structural studies have revealed that CI-1040-related analogs bind into a hydrophobic pocket adjacent to but not overlapping with the Mg-ATP binding site of MEK1 and MEK2 [19]. Binding of the inhibitor induces a conformational change in unphosphorylated MEK1/2 that locks the kinase into a close catalytically inactive form. This binding pocket is located in a region with low sequence homology to other kinases (except for MEK5), which explains the high selectivity of these compounds and their noncompetitive kinetics of inhibition. In pre-clinical studies, CI-1040 was shown to inhibit the growth of colon carcinomas by as much as 80% in mouse xenograft models [30]. Importantly, antitumor activity was achieved at well-tolerated doses and correlated with a reduction in the levels of phosphorylated ERK1/2 in excised tumors.
A phase I study of orally administered CI-1040 was undertaken in 77 patients with advanced cancers [79]. Results of this study indicated that the compound was well tolerated at doses resulting in a median 73% inhibition of phospho-ERK1/2 expression in tumor biopsies. About 60% of patients experienced adverse effects, mostly grade 1 or 2, with no patient having drug-related grade 4 events. The most common toxicities included diarrhea, asthenia, rash, nausea, and vomiting. Interestingly, one patient with pancreatic cancer achieved a partial response with significant symptomatic improvement that lasted 12 months, and 19 additional patients suffering from a variety of cancers had disease stabilization lasting 4 to 17 months. This encouraging study provided the first demonstration that MEK1/2 can be inhibited in vivo in humans, and the first evidence of clinical activity for this class of agents. On this basis, a phase II study was initiated in 67 patients with advanced breast, pancreatic, colon and non-small cell lung cancers [80]. Unfortunately, results of this trial were disappointing. No patient achieved a complete or partial response, and stabilization of disease (median of 4.4 months) was observed in only 8 patients. The insufficient antitumor activity, poor solubility and low bioavailability of CI-1040 precluded further clinical development of this compound.
PD0325901
The CI-1040 structural analogue PD0325901 (Pfizer) is a second-generation MEK1/2 inhibitor with significantly improved pharmaceutical properties [81]. Optimization of the diphenylamine core and modification of the hydroxamate side chain imparted PD0325901 with increases in potency, solubility and bioavailability. PD0325901 has an IC50 value of 1 nM against purified MEK1/MEK2, and inhibits the proliferation of various tumor cell lines at subnanomolar concentrations (100-fold more potent than CI-1040) [62, 72]. In vivo studies have demonstrated that PD0325901 potently inhibits the growth of human tumor xenografts bearing activating mutations of B-Raf, concomitant with suppression of ERK1/2 phosphorylation [72]. The growth of Ras mutant tumors was also inhibited partially.
The clinical activity of PD0325901 was first evaluated in a phase I-II study of 35 patients with advanced solid tumors employing a dose-escalating design [82, 83]. Doses ≥ 2 mg BID efficiently suppressed ERK1/2 phosphorylation (average of 84%) and Ki67 expression (average of 60%) in tumor biopsies. Anticancer activity of PD0325901 was evaluated from 27 assessable patients. Two partial responses were observed in melanoma patients, while 8 patients achieved stable disease lasting 3-7 months [84]. The phase I study was extended and clinical activity was documented by 3 partial responses in melanoma patients and 24 cases of disease stabilization (22 melanoma and 2 non-small cell lung cancer) in 66 patients [85]. However, PD0325901 was associated with more severe toxicity than CI-1040, including blurred vision as well as acute neurotoxicity in patients receiving more than 15 mg BID of the drug. The clinical development of this drug has been discontinued in 2008.
AZD6244 (ARRY-142886)
The benzimidazole derivative AZD6244 (Array BioPharma/AstraZeneca) is another second-generation potent inhibitor of MEK1/MEK2 [86]. AZD6244 selectively inhibits purified active MEK1 and MEK2 with an IC50 of 14 nM by a mechanism not competitive with ATP. In cellular assays, the compound inhibits basal and growth factor-stimulated phosphorylation of ERK1/2 with IC50 concentrations < 40 nM, and exerts antiproliferative effects on tumor cell lines harboring BRAF or RAS mutations [86–88]. AZD6244 has demonstrated potent dose-dependent antitumor activity against a panel of mouse xenograft models of colorectal, pancreatic, liver, skin, and lung cancer [86–89]. Inhibition of tumor growth was found tocorrelate with the reduction of phospho-ERK1/2 levels in tumors. Based on promising pre-clinical activity, AZD6244 was advanced into clinical development.
A phase I clinical trial was undertaken to assess the safety, pharmacokinetics and pharmacodynamics of AZD6244 in 57 patients with advanced cancer [90]. Results of this study showed that the 50% maximal tolerated dose (100 mg BID) was well tolerated with skin rash being the most frequent and dose-limiting toxicity. Most other adverse events were of grade 1 or 2. Notably, 7 patients developed transient and reversible blurred vision, an adverse effect also observed with PD0325901. A strong reduction in ERK1/2 phosphorylation (mean inhibition of 79%) was observed in tumor biopsies. Nine patients showed disease stabilization lasting for at least 5 months.
Preliminary results from four randomized phase II clinical trials of AZD6244 have been recently reported. In a first study, AZD6244 was compared to the alkylating agent temozolomide in advanced melanoma patients. Antitumor activity of AZD6244 was observed, but there was no significant difference in progression-free survival between the two treatment arms [91]. A second study compared the efficacy of AZD6244 with the antimetabolite pemetrexed as second- or third-line treatment of patients with non-small cell lung cancer. Again, the study showed evidence of single agent antitumor activity, but failed to demonstrate a difference for the primary disease progression endpoint [92]. In a third study, AZD6244 was compared to capecitabine in patients with metastatic colorectal cancer who had failed prior irinotecan and/or oxaliplatin regimens. Similarly, no difference was observed between the two treatments in the number of patients with disease progression [93]. Finally, the results of a phase II study of AZD6244 in patients with advanced or metastatic hepatocellular carcinoma were recently reported. The study was stopped prematurely due to the lack of radiographic response [94]. Other phase II trials are currently ongoing in a variety of tumor types.
GDC-0973 (XL518)
GDC-0973 (Exelixis/Genentech) is a potent, selective, orally active inhibitor of MEK1/2 with an IC50 of <1 nM in vitro [95]. In cellular studies, the compound inhibits ERK1/2 phosphorylation at subnanomolar concentrations, and exerts antiproliferative effects in multiple tumor cell lines harboring KRAS or BRAF mutations. In vivo pharmacodynamic studies have shown that a single oral dose of GDC-0973 inhibits phospho-ERK1/2 in tumors for up to 48 hours, translating into potent inhibition of tumor growth in human xenograft models. Notably, GDC-0973 appears to have reduced activity in the brain, which may reduce the potential of central nervous system side effects. A phase I dose-escalating study of GDC-0973 was initiated in subjects with solid tumors. Preliminary results from 13 patients indicates that GDC-0973 is well tolerated with no drug-related serious adverse events being reported [96]. One patient with non-small cell lung cancer had stabilization of disease for 7 months and continues on treatment. Another phase I trial of GDC-0973 in combination with the phosphatidylinositol 3-kinase (PI3K) inhibitor GDC-0941 is planned.
RDEA119 (BAY 869766)
RDEA119 (Ardea Biosciences/Bayer) is another orally available, allosteric inhibitor of MEK1/2 [97]. In vitro, RDEA119 selectively inhibits MEK1 (IC50 of 19 nM) and MEK2 (IC50 of 47 nM) in a non-ATP competitive manner. Cellular assays showed that RDEA119 potently inhibits ERK1/2 phosphorylation (IC50 from 2.5 to 16 nM) and cell proliferation in a panel of human cancer cell lines. In vivo, RDEA119 exhibits potent antitumor activity in xenograft models of human melanoma, colon and epidermal carcinoma. Interestingly, pharmacodynamic studies have revealed that the compound has low central nervous system penetration. RDEA119 is currently being evaluated as single agent in a phase I study in advanced cancer patients, and in a phase I/II study in combination with the multikinase and Raf inhibitor sorafenib.
GSK1120212
GSK1120212 (GlaxoSmithKline) is an orally available, selective inhibitor of MEK1/2 with reported antitumor activity in mouse xenograft models [98]. A phase I study of GSK1120212 was undertaken in 2008 in patients with solid tumors and lymphoma. Preliminary evaluation of 6 patients treated at four dose levels indicates that GSK1120212 is well tolerated with no dose-limiting toxicity reported so far [98]. Dose escalation is ongoing. Two other phase I/II studies of GSK1120212 have been recently initiated in subjects with relapsed or refractory leukemias, and in combination with everolimus in patients with solid tumors.
OTHER MEK1/2 INHIBITORS
Five other MEK1/2 inhibitors are currently being evaluated in phase I clinical trials in advanced cancer patients. These are AZD8330 (Array BioPharma/AstraZeneca), RO5126766 and RO4987655 (Hoffmann La Roche), TAK-733 (Millenium Pharmaceuticals) and AS703026 (EMD Serono). Other novel MEK1/2 inhibitors such as RO4927350 and RO5068760 have recently been reported but have not yet passed the pre-clinical stage [99, 100].
Concluding remarks and challenges
Despite strong rationale for the clinical development of drugs targeting the ERK1/2 MAP kinase pathway in cancer, the effectiveness of this approach in cancer therapy remains to be validated. The first and only inhibitor of the ERK1/2 pathway that has received regulatory approval for the treatment of advanced renal cell carcinoma and hepatocellular carcinoma is the Raf inhibitor sorafenib (Nexavar) [101]. However, sorafenib is a multikinase inhibitor that also inhibits the vascular endothelial growth factor and platelet-derived growth factor receptor tyrosine kinases, as well as Flt-3 and c-Kit receptors. To what extent the inhibition of Raf signaling contributes to the clinical activity of the drug is not clear. Future clinical trials of more selective Raf inhibitors will help determine whether blocking the pathway at the level of Raf is a clinically viable approach. Inhibitors of MEK1/2 are highly selective for their targets. However, results from the first clinical trials have been disappointing. New MEK1/2 inhibitors with improved pharmaceutical properties and reduced central nervous system activity are promising and results of ongoing trials are anxiously awaited.
As for other targeted therapies, several outstanding questions remain to be addressed before MEK1/2 inhibitors join the arsenal of anticancer drugs. Which patients are more likely to benefit from MEK1/2 inhibitors? Pre-clinical studies suggest that patients harboring activating mutations in RAS or BRAF genes are better candidates for treatment with these kinase inhibitors. Thus, selection of appropriate patient populations based on genetic lesions or validated biochemical markers will be critical for future clinical trial evaluation. Is the therapeutic efficacy of MEK1/2 inhibitors hampered by dose-limiting toxicity? The ubiquitous involvement of the ERK1/2 MAP kinase pathway in cellular responses has raised concern about the potential toxicity of drugs blocking this pathway. The ocular toxicity observed with PD0325901 and AZD6244 suggests the existence of mechanism-based adverse effects. Interestingly, new MEK1/2 inhibitors such as GDC-0973 and RDEA119 have reduced activity in the brain, which may increase their therapeutic window. What are the most rationale and best combination therapies with MEK1/2 inhibitors? The multigenetic nature of advanced cancers suggests that MEK1/2 inhibitors will likely find their therapeutic utility in combination with other targeted agents or conventional cytotoxic drugs. Pre-clinical studies have shown that PI3K pathway activation, through PIK3CA activating mutations or PTEN loss of function, significantly decreases the response of KRAS mutant cancer cells to MEK1/2 inhibitors [102]. Importantly, simultaneous inhibition of the ERK1/2 and PI3K pathways was found to exert a marked synergistic effect on tumor regression [102, 103]. These observations have provided a strong rationale for the combination of MEK1/2 and PI3K inhibitors in cancers that harbor concurrent activating mutations in these signaling pathways. In that context, Merck and AstraZeneca have recently announced their plan to collaborate in testing a combination therapy of AZD6244 and the Akt inhibitor MK-2206 in cancer [104]. Finally, is the acquisition of resistance mutations in MEK1/MEK2 going to limit the clinical utility of these small molecule inhibitors? A recent study has reported the identification of a resistant MEK1 mutation in a metastatic tumor that emerged in a melanoma patient treated with AZD6244 [105]. Therefore, it may prove necessary to target other components of the ERK1/2 pathway in patients who develop resistance or, eventually, to combine MEK1/2 inhibitors with Raf inhibitors to slow down the emergence of resistance. A phase I/II study of RDEA119 in combination with the multikinase Raf inhibitor sorafenib is currently ongoing.
References
Hanahan D, Weinberg RA: The hallmarks of cancer. Cell. 2000, 100: 57-70. 10.1016/S0092-8674(00)81683-9.
Ray LB, Sturgill TW: Characterization of insulin-stimulated microtubule-associated protein kinase. Rapid isolation and stabilization of a novel serine/threonine kinase from 3T3-L1 cells. J Biol Chem. 1988, 263: 12721-12727.
Raman M, Chen W, Cobb MH: Differential regulation and properties of MAPKs. Oncogene. 2007, 26: 3100-3112. 10.1038/sj.onc.1210392.
Yoon S, Seger R: The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006, 24: 21-44. 10.1080/02699050500284218.
Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH: Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001, 22: 153-183. 10.1210/er.22.2.153.
Brancho D, Tanaka N, Jaeschke A, Ventura JJ, Kelkar N, Tanaka Y, Kyuuma M, Takeshita T, Flavell RA, Davis RJ: Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003, 17: 1969-1978. 10.1101/gad.1107303.
Ganiatsas S, Kwee L, Fujiwara Y, Perkins A, Ikeda T, Labow MA, Zon LI: SEK1 deficiency reveals mitogen-activated protein kinase cascade crossregulation and leads to abnormal hepatogenesis. Proc Natl Acad Sci USA. 1998, 95: 6881-6886. 10.1073/pnas.95.12.6881.
Fukuda M, Gotoh I, Gotoh Y, Nishida E: Cytoplasmic localization of mitogen-activated protein kinase kinase directed by its NH2-terminal, leucine-rich short amino acid sequence, which acts as a nuclear export signal. J Biol Chem. 1996, 271: 20024-20028. 10.1074/jbc.271.33.20024.
Burgermeister E, Chuderland D, Hanoch T, Meyer M, Liscovitch M, Seger R: Interaction with MEK causes nuclear export and downregulation of peroxisome proliferator-activated receptor gamma. Mol Cell Biol. 2007, 27: 803-817. 10.1128/MCB.00601-06.
Eblen ST, Slack JK, Weber MJ, Catling AD: Rac-PAK signaling stimulates extracellular signal-regulated kinase (ERK) activation by regulating formation of MEK1-ERK complexes. Mol Cell Biol. 2002, 22: 6023-6033. 10.1128/MCB.22.17.6023-6033.2002.
Wu X, Noh SJ, Zhou G, Dixon JE, Guan KL: Selective activation of MEK1 but not MEK2 by A-Raf from epidermal growth factor-stimulated Hela cells. J Biol Chem. 1996, 271: 3265-3271. 10.1074/jbc.271.6.3265.
Xu S, Khoo S, Dang A, Witt S, Do V, Zhen E, Schaefer EM, Cobb MH: Differential regulation of mitogen-activated protein/ERK kinase (MEK)1 and MEK2 and activation by a Ras-independent mechanism. Mol Endocrinol. 1997, 11: 1618-1625. 10.1210/me.11.11.1618.
Catalanotti F, Reyes G, Jesenberger V, Galabova-Kovacs G, de Matos Simoes R, Carugo O, Baccarini M: A Mek1-Mek2 heterodimer determines the strength and duration of the Erk signal. Nat Struct Mol Biol. 2009, 16: 294-303. 10.1038/nsmb.1564.
Scholl FA, Dumesic PA, Khavari PA: Mek1 alters epidermal growth and differentiation. Cancer Res. 2004, 64: 6035-6040. 10.1158/0008-5472.CAN-04-0017.
Voisin L, Julien C, Duhamel S, Gopalbhai K, Claveau I, Saba-El-Leil MK, Rodrigue-Gervais IG, Gaboury L, Lamarre D, Basik M: Activation of MEK1 or MEK2 isoform is sufficient to fully transform intestinal epithelial cells and induce the formation of metastatic tumors. BMC Cancer. 2008, 8: 337-10.1186/1471-2407-8-337.
Scholl FA, Dumesic PA, Barragan DI, Charron J, Khavari PA: Mek1/2 gene dosage determines tissue response to oncogenic Ras signaling in the skin. Oncogene. 2009, 28: 1485-1495. 10.1038/onc.2008.459.
Mody N, Leitch J, Armstrong C, Dixon J, Cohen P: Effects of MAP kinase cascade inhibitors on the MKK5/ERK5 pathway. FEBS Lett. 2001, 502: 21-24. 10.1016/S0014-5793(01)02651-5.
Kamakura S, Moriguchi T, Nishida E: Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signaling pathway to the nucleus. J Biol Chem. 1999, 274: 26563-26571. 10.1074/jbc.274.37.26563.
Ohren JF, Chen H, Pavlovsky A, Whitehead C, Zhang E, Kuffa P, Yan C, McConnell P, Spessard C, Banotai C: Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol. 2004, 11: 1192-1197. 10.1038/nsmb859.
Meloche S, Pouyssegur J: The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007, 26: 3227-3239. 10.1038/sj.onc.1210414.
Meloche S, Seuwen K, Pages G, Pouyssegur J: Biphasic and synergistic activation of p44mapk (ERK1) by growth factors: correlation between late phase activation and mitogenicity. Mol Endocrinol. 1992, 6: 845-854. 10.1210/me.6.5.845.
Jones SM, Kazlauskas A: Growth-factor-dependent mitogenesis requires two distinct phases of signalling. Nat Cell Biol. 2001, 3: 165-172. 10.1038/35055073.
Yamamoto T, Ebisuya M, Ashida F, Okamoto K, Yonehara S, Nishida E: Continuous ERK activation downregulates antiproliferative genes throughout G1 phase to allow cell-cycle progression. Curr Biol. 2006, 16: 1171-1182. 10.1016/j.cub.2006.04.044.
Pages G, Lenormand P, L'Allemain G, Chambard JC, Meloche S, Pouyssegur J: Mitogen-activated protein kinases p42mapk and p44mapk are required for fibroblast proliferation. Proc Natl Acad Sci USA. 1993, 90: 8319-8323. 10.1073/pnas.90.18.8319.
Liu X, Yan S, Zhou T, Terada Y, Erikson RL: The MAP kinase pathway is required for entry into mitosis and cell survival. Oncogene. 2004, 23: 763-776. 10.1038/sj.onc.1207188.
Fremin C, Ezan F, Boisselier P, Bessard A, Pages G, Pouyssegur J, Baffet G: ERK2 but not ERK1 plays a key role in hepatocyte replication: An RNAi-mediated ERK2 knockdown approach in wild-type and ERK1 null hepatocytes. Hepatology. 2007, 45: 1035-1045. 10.1002/hep.21551.
Lefloch R, Pouyssegur J, Lenormand P: Single and combined ERK1/ERK2 silencing reveals their positive contribution to growth signaling depending on their expression levels. Mol Cell Biol. 2007
Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR: A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995, 92: 7686-7689. 10.1073/pnas.92.17.7686.
DeSilva DR, Jones EA, Favata MF, Jaffee BD, Magolda RL, Trzaskos JM, Scherle PA: Inhibition of mitogen-activated protein kinase kinase blocks T cell proliferation but does not induce or prevent anergy. J Immunol. 1998, 160: 4175-4181.
Sebolt-Leopold JS, Dudley DT, Herrera R, Van Becelaere K, Wiland A, Gowan RC, Tecle H, Barrett SD, Bridges A, Przybranowski S: Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med. 1999, 5: 810-816. 10.1038/10533.
Brunet A, Pages G, Pouyssegur J: Constitutively active mutants of MAP kinase kinase (MEK1) induce growth factor-relaxation and oncogenicity when expressed in fibroblasts. Oncogene. 1994, 9: 3379-3387.
Cowley S, Paterson H, Kemp P, Marshall CJ: Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994, 77: 841-852. 10.1016/0092-8674(94)90133-3.
Seger R, Seger D, Reszka AA, Munar ES, Eldar-Finkelman H, Dobrowolska G, Jensen AM, Campbell JS, Fischer EH, Krebs EG: Overexpression of mitogen-activated protein kinase kinase (MAPKK) and its mutants in NIH 3T3 cells. Evidence that MAPKK involvement in cellular proliferation is regulated by phosphorylation of serine residues in its kinase subdomains VII and VIII. J Biol Chem. 1994, 269: 25699-25709.
Pages G, Guerin S, Grall D, Bonino F, Smith A, Anjuere F, Auberger P, Pouyssegur J: Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science. 1999, 286: 1374-1377. 10.1126/science.286.5443.1374.
Saba-El-Leil MK, Vella FD, Vernay B, Voisin L, Chen L, Labrecque N, Ang SL, Meloche S: An essential function of the mitogen-activated protein kinase Erk2 in mouse trophoblast development. EMBO Rep. 2003, 4: 964-968. 10.1038/sj.embor.embor939.
Samuels IS, Karlo JC, Faruzzi AN, Pickering K, Herrup K, Sweatt JD, Saitta SC, Landreth GE: Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J Neurosci. 2008, 28: 6983-6995. 10.1523/JNEUROSCI.0679-08.2008.
D'Souza WN, Chang CF, Fischer AM, Li M, Hedrick SM: The Erk2 MAPK regulates CD8 T cell proliferation and survival. J Immunol. 2008, 181: 7617-7629.
Roovers K, Assoian RK: Integrating the MAP kinase signal into the G1 phase cell cycle machinery. Bioessays. 2000, 22: 818-826. 10.1002/1521-1878(200009)22:9<818::AID-BIES7>3.0.CO;2-6.
Lavoie JN, L'Allemain G, Brunet A, Muller R, Pouyssegur J: Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem. 1996, 271: 20608-20616. 10.1074/jbc.271.34.20608.
Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR: Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 14: 2501-2514. 10.1101/gad.836800.
Yang JY, Zong CS, Xia W, Yamaguchi H, Ding Q, Xie X, Lang JY, Lai CC, Chang CJ, Huang WC: ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008, 10: 138-148. 10.1038/ncb1676.
Hwang CY, Lee C, Kwon KS: Extracellular signal-regulated kinase 2-dependent phosphorylation induces cytoplasmic localization and degradation of p21Cip1. Mol Cell Biol. 2009, 29: 3379-3389. 10.1128/MCB.01758-08.
Chambard JC, Lefloch R, Pouyssegur J, Lenormand P: ERK implication in cell cycle regulation. Biochim Biophys Acta. 2007, 1773: 1299-1310. 10.1016/j.bbamcr.2006.11.010.
Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J: Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci USA. 2004, 101: 13489-13494. 10.1073/pnas.0405659101.
Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP: Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005, 121: 179-193. 10.1016/j.cell.2005.02.031.
Ballif BA, Blenis J: Molecular mechanisms mediating mammalian mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK cell survival signals. Cell Growth Differ. 2001, 12: 397-408.
Balmanno K, Cook SJ: Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 2009, 16: 368-377. 10.1038/cdd.2008.148.
Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995, 270: 1326-1331. 10.1126/science.270.5240.1326.
Parrizas M, Saltiel AR, LeRoith D: Insulin-like growth factor 1 inhibits apoptosis using the phosphatidylinositol 3'-kinase and mitogen-activated protein kinase pathways. J Biol Chem. 1997, 272: 154-161. 10.1074/jbc.272.1.154.
Erhardt P, Schremser EJ, Cooper GM: B-Raf inhibits programmed cell death downstream of cytochrome c release from mitochondria by activating the MEK/Erk pathway. Mol Cell Biol. 1999, 19: 5308-5315.
Le Gall M, Chambard JC, Breittmayer JP, Grall D, Pouyssegur J, Van Obberghen-Schilling E: The p42/p44 MAP kinase pathway prevents apoptosis induced by anchorage and serum removal. Mol Biol Cell. 2000, 11: 1103-1112.
Lips DJ, Bueno OF, Wilkins BJ, Purcell NH, Kaiser RA, Lorenz JN, Voisin L, Saba-El-Leil MK, Meloche S, Pouyssegur J: MEK1-ERK2 signaling pathway protects myocardium from ischemic injury in vivo. Circulation. 2004, 109: 1938-1941. 10.1161/01.CIR.0000127126.73759.23.
Scholl FA, Dumesic PA, Barragan DI, Harada K, Bissonauth V, Charron J, Khavari PA: Mek1/2 MAPK kinases are essential for Mammalian development, homeostasis, and Raf-induced hyperplasia. Dev Cell. 2007, 12: 615-629. 10.1016/j.devcel.2007.03.009.
Grandis JR, Sok JC: Signaling through the epidermal growth factor receptor during the development of malignancy. Pharmacol Ther. 2004, 102: 37-46. 10.1016/j.pharmthera.2004.01.002.
Hynes NE, Lane HA: ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005, 5: 341-354. 10.1038/nrc1609.
Schubbert S, Shannon K, Bollag G: Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007, 7: 295-308. 10.1038/nrc2109.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W: Mutations of the BRAF gene in human cancer. Nature. 2002, 417: 949-954. 10.1038/nature00766.
Garnett MJ, Marais R: Guilty as charged: B-RAF is a human oncogene. Cancer Cell. 2004, 6: 313-319. 10.1016/j.ccr.2004.09.022.
Marks JL, Gong Y, Chitale D, Golas B, McLellan MD, Kasai Y, Ding L, Mardis ER, Wilson RK, Solit D: Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. Cancer Res. 2008, 68: 5524-5528. 10.1158/0008-5472.CAN-08-0099.
Murugan AK, Dong J, Xie J, Xing M: MEK1 mutations, but not ERK2 mutations, occur in melanomas and colon carcinomas, but none in thyroid carcinomas. Cell Cycle. 2009, 8: 2122-2124.
Hoshino R, Chatani Y, Yamori T, Tsuruo T, Oka H, Yoshida O, Shimada Y, Ari-i S, Wada H, Fujimoto J: Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene. 1999, 18: 813-822. 10.1038/sj.onc.1202367.
Sebolt-Leopold JS, Herrera R: Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer. 2004, 4: 937-947. 10.1038/nrc1503.
Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, Woude Vande GF, Ahn NG: Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994, 265: 966-970. 10.1126/science.8052857.
Pinkas J, Leder P: MEK1 signaling mediates transformation and metastasis of EpH4 mammary epithelial cells independent of an epithelial to mesenchymal transition. Cancer Res. 2002, 62: 4781-4790.
Leicht DT, Balan V, Kaplun A, Singh-Gupta V, Kaplun L, Dobson M, Tzivion G: Raf kinases: function, regulation and role in human cancer. Biochim Biophys Acta. 2007, 1773: 1196-1212. 10.1016/j.bbamcr.2007.05.001.
Hobbs RM, Silva-Vargas V, Groves R, Watt FM: Expression of activated MEK1 in differentiating epidermal cells is sufficient to generate hyperproliferative and inflammatory skin lesions. J Invest Dermatol. 2004, 123: 503-515. 10.1111/j.0022-202X.2004.23225.x.
Goel VK, Ibrahim N, Jiang G, Singhal M, Fee S, Flotte T, Westmoreland S, Haluska FS, Hinds PW, Haluska FG: Melanocytic nevus-like hyperplasia and melanoma in transgenic BRAFV600E mice. Oncogene. 2009, 28: 2289-2298. 10.1038/onc.2009.95.
Knauf JA, Ma X, Smith EP, Zhang L, Mitsutake N, Liao XH, Refetoff S, Nikiforov YE, Fagin JA: Targeted expression of BRAFV600E in thyroid cells of transgenic mice results in papillary thyroid cancers that undergo dedifferentiation. Cancer Res. 2005, 65: 4238-4245. 10.1158/0008-5472.CAN-05-0047.
Jeong JH, Wang Z, Guimaraes AS, Ouyang X, Figueiredo JL, Ding Z, Jiang S, Guney I, Kang GH, Shin E: BRAF activation initiates but does not maintain invasive prostate adenocarcinoma. PLoS One. 2008, 3: e3949-10.1371/journal.pone.0003949.
Ji H, Wang Z, Perera SA, Li D, Liang MC, Zaghlul S, McNamara K, Chen L, Albert M, Sun Y: Mutations in BRAF and KRAS converge on activation of the mitogen-activated protein kinase pathway in lung cancer mouse models. Cancer Res. 2007, 67: 4933-4939. 10.1158/0008-5472.CAN-06-4592.
Milella M, Kornblau SM, Estrov Z, Carter BZ, Lapillonne H, Harris D, Konopleva M, Zhao S, Estey E, Andreeff M: Therapeutic targeting of the MEK/MAPK signal transduction module in acute myeloid leukemia. J Clin Invest. 2001, 108: 851-859.
Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, Ye Q, Lobo JM, She Y, Osman I: BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006, 439: 358-362. 10.1038/nature04304.
Collisson EA, De A, Suzuki H, Gambhir SS, Kolodney MS: Treatment of metastatic melanoma with an orally available inhibitor of the Ras-Raf-MAPK cascade. Cancer Res. 2003, 63: 5669-5673.
Kramer BW, Gotz R, Rapp UR: Use of mitogenic cascade blockers for treatment of C-Raf induced lung adenoma in vivo: CI-1040 strongly reduces growth and improves lung structure. BMC Cancer. 2004, 4: 24-10.1186/1471-2407-4-24.
Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR: PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995, 270: 27489-27494. 10.1074/jbc.270.46.27489.
Servant MJ, Giasson E, Meloche S: Inhibition of growth factor-induced protein synthesis by a selective MEK inhibitor in aortic smooth muscle cells. J Biol Chem. 1996, 271: 16047-16052. 10.1074/jbc.271.27.16047.
Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F: Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998, 273: 18623-18632. 10.1074/jbc.273.29.18623.
Williams DH, Wilkinson SE, Purton T, Lamont A, Flotow H, Murray EJ: Ro 09-2210 exhibits potent anti-proliferative effects on activated T cells by selectively blocking MKK activity. Biochemistry. 1998, 37: 9579-9585. 10.1021/bi972914c.
Lorusso PM, Adjei AA, Varterasian M, Gadgeel S, Reid J, Mitchell DY, Hanson L, DeLuca P, Bruzek L, Piens J: Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol. 2005, 23: 5281-5293. 10.1200/JCO.2005.14.415.
Rinehart J, Adjei AA, Lorusso PM, Waterhouse D, Hecht JR, Natale RB, Hamid O, Varterasian M, Asbury P, Kaldjian EP: Multicenter phase II study of the oral MEK inhibitor, CI- in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 1040, 22: 4456-4462. 10.1200/JCO.2004.01.185.
Barrett SD, Bridges AJ, Dudley DT, Saltiel AR, Fergus JH, Flamme CM, Delaney AM, Kaufman M, LePage S, Leopold WR: The discovery of the benzhydroxamate MEK inhibitors CI-1040 and PD 0325901. Bioorg Med Chem Lett. 2008, 18: 6501-6504. 10.1016/j.bmcl.2008.10.054.
Lorusso P, Krishnamurthi S, Rinehart JR, Nabell L, Croghan G, Varterasian M, Sadis SS, Menon SS, Leopold J, Meyer MB: A phase 1-2 clinical study of a second generation oral MEK inhibitor, PD 0325901 in patients with advanced cancer. J Clin Oncol (abstract). 2005, 23: 3011-
Menon SS, Whitfield LR, Sadis S, Meyer MB, Leopold J, Lorusso PM, Krishnamurthi S, Rinehart JR, Nabell L, Croghan G: Pharmacokinetics (PK) and pharmacodynamics (PD) of PD0325901, a second generation MEK inhibitor after multiple oral doses of PD0325901 to advanced cancer patients. J Clin Oncol (abstract). 2005, 23: 3066-
Wang D, Boerner SA, Winkler JD, LoRusso PM: Clinical experience of MEK inhibitors in cancer therapy. Biochim Biophys Acta. 2007, 1773: 1248-1255. 10.1016/j.bbamcr.2006.11.009.
Lorusso P, Krishnamurthi S, Rinehart J, Nabell L, Croghan G, Chapman P, Selaru P, Kim S, Ricart A, Wliner K: Clinical aspects of a phase I study of PD032 a selective oral MEK inhibitor, in patients with advanced cancer. Mol Cancer Ther (abstract B113). 5901, 6: 3646s-
Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, Parry J, Smith D, Brandhuber BJ, Gross S: Biological Characterization of ARRY-142886 (AZD6244), a Potent, Highly Selective Mitogen-Activated Protein Kinase Kinase 1/2 Inhibitor. Clin Cancer Res. 2007, 13: 1576-1583. 10.1158/1078-0432.CCR-06-1150.
Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, Cockerill M, Cartlidge S, Smith PD: AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther. 2007, 6: 2209-2219. 10.1158/1535-7163.MCT-07-0231.
Huynh H, Soo KC, Chow PK, Tran E: Targeted inhibition of the extracellular signal-regulated kinase kinase pathway with AZD6244 (ARRY-142886) in the treatment of hepatocellular carcinoma. Mol Cancer Ther. 2007, 6: 138-146. 10.1158/1535-7163.MCT-06-0436.
Haass NK, Sproesser K, Nguyen TK, Contractor R, Medina CA, Nathanson KL, Herlyn M, Smalley KS: The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin Cancer Res. 2008, 14: 230-239. 10.1158/1078-0432.CCR-07-1440.
Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, Hanson LJ, Gore L, Chow L, Leong S: Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008, 26: 2139-2146. 10.1200/JCO.2007.14.4956.
Drummer R, Robert C, Chapman P, Sosman J, Middleton M, Bastholt L, Kemsley K, Cantarini M, Morris C, Kirkwood J: AZD6244 (ARRY-142886) vs Temozolomide in Patients With Advanced Melanoma: an Open-Label, Randomized, Multicenter, Phase II Study. J Clin Oncol (abstract). 2008, 26: 9033-
Tzekova V, Cebotaru C, Ciuleanu TE, Damjanov D, Ganchev V, Kanarev V, Stella PJ, Sanders N, Pover G, Hainsworth JD: Efficacy and safety of AZD6244 (ARRY-142886) as second/third-line treatment of patients (pts) with advanced non-small cell lung cancer (NSCLC). J Clin Oncol (abstract). 2008, 26: 8029-
Lang I, Adenis A, Boer K, Escudero P, Kim T, Valladares M, Sanders N, Pover G, Douillard JY: AZD6244 (ARRY-142886) Versus Capecitabine in Patients With Metastatic Colorectal Cancer Who Have Failed Prior Chemotherapy. J Clin Oncol (abstract). 2008, 26: 4114-
O'Neil BH, Williams-Goff LW, Kauh J, Bekaii-Saab T, Strosberg JR, Lee R, Deal AM, Sullivan D, Sebti SM: A phase II study of AZD6244 in advanced or metastatic hepatocellular carcinoma. J Clin Oncol (abstract). 2009, 27: 15574-
Johnston S: XL518, a potent, selective orally bioavailable MEK1 inhibitor, down-regulates the Ras/Raf/MEK/ERK pathway in vivo, resulting in tumor growth inhibition and regression in preclinical models. 19th AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics. 2007, Abstract C209
Rosen LS, Galatin P, Fehling JM, Laux I, Dinolfo M, Frye J, Laird D, Sikic BI: A phase 1 dose-escalation study of XL518, a potent MEK inhibitor administered orally daily to subjects with solid tumors. J Clin Oncol (abstract). 2008, 26: 14585-
Iverson C, Larson G, Lai C, Yeh LT, Dadson C, Weingarten P, Appleby T, Vo T, Maderna A, Vernier JM: RDEA119/BAY 869766: a potent, selective, allosteric inhibitor of MEK1/2 for the treatment of cancer. Cancer Res. 2009, 69: 6839-6847. 10.1158/0008-5472.CAN-09-0679.
Thompson D, Flaherty K, Messersmith W, Harlacker K, Nallapareddy S, Vincent C, DeMarini D, Cox D, O'Neill V, Burris H: A three-part, phase I, dose-escalation study of GSK1120212, a potent MEK inhibitor, administred orally with solid tumors or lymphoma. J Clin Oncol (abstract). 2009, 27: e14584-
Daouti S, Higgins B, Kolinsky K, Packman K, Wang H, Rizzo C, Moliterni J, Huby N, Fotouhi N, Liu M: Preclinical in vivo evaluation of efficacy, pharmacokinetics, and pharmacodynamics of a novel MEK1/2 kinase inhibitor RO5068760 in multiple tumor models. Mol Cancer Ther. 2010, 9: 134-144. 10.1158/1535-7163.MCT-09-0601.
Daouti S, Wang H, Li WH, Higgins B, Kolinsky K, Packman K, Specian A, Kong N, Huby N, Wen Y: Characterization of a novel mitogen-activated protein kinase kinase 1/2 inhibitor with a unique mechanism of action for cancer therapy. Cancer Res. 2009, 69: 1924-1932. 10.1158/0008-5472.CAN-08-2627.
Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, Schwartz B, Simantov R, Kelley S: Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006, 5: 835-844. 10.1038/nrd2130.
Wee S, Jagani Z, Xiang KX, Loo A, Dorsch M, Yao YM, Sellers WR, Lengauer C, Stegmeier F: PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009, 69: 4286-4293. 10.1158/0008-5472.CAN-08-4765.
Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y: Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008, 14: 1351-1356. 10.1038/nm.1890.
Nature Journal. [http://www.nature.com/news/2009/090602/full/news.2009.536.html]
Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, Hatton C, Chopra R, Oberholzer PA, Karpova MB: MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci USA. 2009, 106: 20411-20416. 10.1073/pnas.0905833106.
Acknowledgements
C. Frémin is recipient of a fellowship from the Cole Foundation. S. Meloche holds the Canada Research Chair in Cellular Signaling. Work in the author's laboratory was supported by grants from the National Cancer Institute of Canada, the Cancer Research Society and the Canadian Institutes for Health Research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
Both authors participated in drafting and editing the manuscript. Both authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Frémin, C., Meloche, S. From basic research to clinical development of MEK1/2 inhibitors for cancer therapy. J Hematol Oncol 3, 8 (2010). https://doi.org/10.1186/1756-8722-3-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-8722-3-8