
Abstract
Extensive studies have led to a variety of hypotheses for the molecular basis of depression and related mood disorders, but a definite pathogenic mechanism has yet to be defined. The monoamine hypothesis, in conjunction with the efficacy of antidepressants targeting monoamine systems, has long been the central topic of depression research. While it is widely embraced that the initiation of antidepressant efficacy may involve acute changes in monoamine systems, apparently, the focus of current research is moving toward molecular mechanisms that underlie long-lasting downstream changes in the brain after chronic antidepressant treatment, thereby reaching for a detailed view of the pathophysiology of depression and related mood disorders. In this minireview, we briefly summarize major themes in current approaches to understanding mood disorders focusing on molecular views of depression and antidepressant action.
Similar content being viewed by others

Introduction
Mood disorders such as major depression and bipolar disorders are the most common psychiatric disorders in modern society. About 16% and 1% of the population are estimated to be affected by major depression and bipolar disorder one or more times during their life time, respectively [1]. The presence of the common symptoms of these disorders are collectively called 'depressive syndrome' and includes a long-lasting depressed mood, feelings of guilt, anxiety, and recurrent thoughts of death and suicide [2]. The genetic contribution to the manifestation of depression has been estimated as 40-50% [3]. However, combinations of multiple genetic factors may be involved in the development of depression, because a defect in a single gene usually fails to induce the expression of multifaceted symptoms of depression [4]. Also, various non-genetic factors such as stress, affective trauma, viral infection, and neurodevelopmental abnormalities increase the complexity of the pathogenesis of the disease. Thus, extensive studies have led to a variety of hypotheses for the molecular mechanism of depression, but a definite pathogenic mechanism has yet to be defined.
The 'monoamine hypothesis,' which suggests a deficiency or imbalances in the monoamine neurotransmitters, such as serotonin, dopamine and norepinephrine, as the cause of depression has been the central topic of depression research for approximately the last 50 years. This hypothesis has been initiated and supported by the fact that early versions of antidepressants including tricyclics and monoamine oxidase inhibitors have the common effect of acutely enhancing monoamine function [5–7]. Recent development of the selective serotonin reuptake inhibitors (SSRIs) as effective antidepressants has further strengthened the hypothesis [6, 8]. However, unresolved complexity of the current antidepressants remains. First, antidepressants are effective in less than 50% of patients, and recently discovered drugs have failed to enlarge the extent of applicable patients [2]. Second, chronic treatment with antidepressants is required for clinical effects, and the reason for this is unknown [9]. Third, depression medications as well as mood stabilizers show a wide spectrum of undesired side effects.
In particular, because clinical effects of antidepressants that acutely modify monoamine systems are significantly delayed, it is now believed that an adaptation of downstream events, including lasting changes in gene expression by chronic treatment, underlie the antidepressant efficacy [10]. This phenomenon suggests that there is probably not a simple relationship between biogenic amines and depression postulated by classical monoamine hypothesis. The complexity may be due to multiple factors, which is likely because depression is a group of disorders with several underlying pathologies. Also, expression of depression symptoms may require disturbances in certain neurotransmitter systems that are functionally interconnected to each other at multiple levels. Taken together, while it still has to be emphasized that the initiation of antidepressant efficacy may be mediated by acute changes in monoamine systems, apparently, the focus of current research is moving toward molecular mechanisms that underlie long-lasting downstream changes in the brain after chronic antidepressant treatment, thereby reaching for a detailed view to the pathophysiology of depression and related mood disorders. In this minireview, we summarize major themes in current approaches to understanding depression and related mood disorders.
Gene-environment interactions
As a way to discovering genes predisposing to depression, geneticists have long been searching for gene variants that play a role in the response to life stresses, a critical environmental factor for the onset of depression, which would be an example of 'gene-environment interaction': whereby an environmental factor is filtered through the activity of a gene to confer differential susceptibility to depression among individuals. To this end, polymorphisms in the serotonin transporter (5-hydroxyltryptamine transporter, 5-HTT) gene have been extensively analyzed. It has been reported that the expression level of 5-HTT from the 5-HTT gene is influenced by polymorphisms in the 5'-flanking region (5-HTT gene-linked polymorphic region, 5-HTTLPR) and in the variable number tandem repeat (VNTR) of the second intron [11, 12]. In particular, a short variant of 5-HTTLPR appears to be associated with repressed transcriptional activity of the promoter, decreased 5-HTT expression, and decreased 5-HT uptake when compared with a long variant of 5-HTTLPR [13]. Significantly, genetic studies have shown that these polymorphisms are associated with major depressive disorder in human [14]. Moreover, a longitudinal study with 847 New Zealanders has shown that a short allele of 5-HTTLPR variants is associated with an increase in susceptibility to depression in response to life stresses such as job losses or divorces [15]. Strikingly, in this study, the polymorphism is influential only when the subjects are in significant life stresses, suggesting that 5-HTT may be a connecting point between individual's genetic makeup and environmental triggers of depression. These observations were further strengthened by study showing that increased depression scores in maltreated children without social supports are associated the short allele of 5HTTLPR [16].
However, the insight from these studies does not appear to be fully supported by other studies. The association of allelic variation in VNTR of 5-HTT gene with the susceptibility to depression was not consistently detected in some analyses [17, 18]. A meta-analysis showed that polymorphisms in 5-HTTLPR and the second intron are actually found in depressed patients but the strength of association does not reach a statistical significance [19]. An extensive study using 1206 twins also failed to find a main effect of 5-HTTLPR, or an interaction between the 5-HTTLPR genotype and stressful life events on major depression [20]. Moreover, a recent meta-analysis using 14 comparable studies has yielded no evidence that the serotonin transporter genotype alone or in interaction with stressful life events is associated with an elevated risk of depression [21]. The mixed results from these studies reveal the potential weakness of the 'candidate gene' approach focusing on a specific gene variant to elucidate gene-environment interactions, and thus add importance on unbiased whole-genome scan approach, especially when a disease with polygenic nature, such as depression and related mood disorders, is concerned.
Stress response circuits
Chronic stress is an important component in depression even though it does not seem to function as a necessary or sufficient factor. From this point of view, the hypothalamic-pituitary-adrenal (HPA) axis, a core neuroendocrine circuit for managing stress in the body, has been a topic of interest in depression research [22]. Corticotrophin-releasing factor (CRF) secreted from the paraventricular nucleus of the hypothalamus enhances secretion of adrenocorticotrophin (ACTH) from the pituitary [22, 23], and subsequently, glucocorticoid is secreted from the adrenal cortex, impacting neurobehavioral functions of various brain regions [2]. The HPA axis forms a feedback loop via certain brain regions such as the hippocampus and amygdala [24]. It was reported that hypercortisolemia, a persistent upregulation of blood glucocorticoid levels, increases the excitotoxicity of CA3 pyramidal neurons in the hippocampus, resulting in dendritic atrophy, reduction in spinogenesis, apoptosis of neurons, and possibly inhibition of adult neurogenesis [25]. These functional abnormalities of hippocampal neurons caused by chronic stress can reduce the inhibitory tone on the HPA-axis, which results in hyperactivity of the HPA-axis [23]. Notably, hyperactivity of HPA-axis is evident in approximately half of depressed patients and chronic treatment with antidepressants often reverses this phenomenon [23, 26]. Furthermore, evidence from animal studies suggests that chronic treatment with antidepressants appears to contribute to the recovery of the abnormal function of the hippocampus by increasing neurogenesis [27, 28].
In this regard, one research direction is to evaluate the therapeutic potentials of weakening of the functions of the HPA axis. The obvious targets are CRF receptors expressed in the pituitary and glucocorticoid receptors expressed in the hippocampus and other brain regions, because those receptors are core components in the HPA axis and the associated feedback loop [24, 29–32]. In a similar context, vasopressin receptors have also emerged as alternative targets [33, 34]. Vasopressin is a neuropeptide that enhances CRF function and works through vasopressin receptors expressed in the amygdala and other parts of the limbic system. Also, a single nucleotide polymorphism (SNP) of vasopressin 1b (V1b) receptor has protective effects against major depressive disorder [35]. Intriguingly, antagonism of CRF receptors, glucocorticoid receptors, and vasopressin receptors appear to exhibit antidepressant effects in experimental animals. The applicability to human patients remains to be further refined.
Neurotrophic factors
Long-term stress appears to reduce the expression level of brain derived neurotrophic factor (BDNF) in the hippocampus [36]. Also, in a post-mortem study of depressed patients, a reduction in BDNF expression was reported [37]. In addition, polymorphisms of BDNF gene are associated with neuroticism, a personality trait linked to increased susceptibility to depression [38]. A family-based association study showed that polymorphisms in BDNF genes are related to bipolar disorders [39]. Conversely, a chronic treatment with antidepressants not only enhances the BDNF level but also increases the stress resistance in animals [40, 41]. These observations provided a basis for 'neurotrophism theory' stating that depression is caused by a deficit in neurotrophic factors, and antidepressants neutralize this deficit. This theory may be intimately related to neuronal damages in the hippocampal region caused by hyperactivity of stress response circuits aforementioned. Because BDNF is known to enhance synaptic plasticity in various brain regions [42, 43], it is reasonable to postulate that improving BDNF function may be beneficial to the hippocampal neurons that are susceptible to stress-induced damages. Supporting this idea, direct injection of BDNF into the hippocampus of experimental animals induces behavioral changes similar to antidepressant treatment [41]. Thus, BDNF and its receptor TrkB, have become promising targets of novel-type anti-depression therapies.
Despite these observations, a possible causative relationship between BDNF function and the pathogenesis of depression or antidepressant efficacy requires further clarification. For example, while the antidepressant efficacy is suppressed in experiments using inducible BDNF knock-out mice, depression-related behaviors are only seen in females, showing significant gender differences [36]. Moreover, forebrain-specific conditional TrkB receptor knockout mice do not exhibit depression-related behaviors such as increased behavioral despair in the forced swim test [44], whereas it has been demonstrated that activation of TrkB receptor is required for antidepressant-induced behavioral effects [45]. Thus, the relationship between the loss of BDNF activity and the expression of depressive symptoms is not in a simple correlation. Nevertheless, the potential value of the neurotrophic theory as a basis for the design of new form of anti-depression therapies cannot be excluded by the complexity of the current experimental results.
Histone modifications
One poorly understood characteristic of antidepressants is the long delay before the onset of positive effects in patients [10]. This phenomenon is often attributed to the slow development of adaptation in the relevant neurons that underlies the beneficial effect of the drugs. The identity of the adaptation is not clear yet, but enduring changes in the state of chromatin are thought to be involved. Chronic electro-convulsive shocks that are effective for some depressed patients also induce changes in wide range of the histone modification patterns in experimental animals [46]. One locus with prominent changes is BDNF, and in conjunction with the suggestion of BDNF as a potential target for design of new antidepressants, the epigenetic control of BDNF expression has been extensively analyzed in the context of the expression of depression and chronic antidepressant treatments. In the rat hippocampus, chronic electro-convulsive shocks increase acetylated histone H3 at the BDNF promoters 3 and 4, and these modifications appear to be correlated with increased expression of BDNF and CREB [46]. This upregulation has been linked to the effects of antidepressants in animal studies [28, 47]. Moreover, chronic defeat stress, an experimental model for depression, elicits selective downregulation of some BDNF splice variants, in the hippocampus [28]. This downregulation appears to be due to induction of H3-K27 dimethylation, a histone code for transcriptional repression [28, 48]. Conversely, an antidepressant treatment reverses repression of BDNF expression likely by inducing H3 acetylation and H3-K4 methylation, acting as histone codes for transcriptional activation, at the BDNF promoter region [49]. During this whole process, roles for histone deacetylases (HDACs) seem to be crucial because chronic antidepressant treatment downregulates HDAC5, and overexpression of HDAC5 in the hippocampus prevents its antidepressant effect [28].
HDAC inhibitors have thus received attention for their potentials as promising therapeutics for depression and related mood disorders. HDAC inhibitors are members of four families: the short chain fatty acids (e.g. sodium butyrate (SB), phenylbutyrate, and valproic acid (VPA)), the hyroxamic acids (e.g. TSA and suberoylanilide hydroxamic acid (SAHA)), the epoxyketones (e.g. trapoxin), and the benzamides. One of the most widely used mood stabilizers is VPA. As VPA is known to have an inhibitory activity on HDAC1 and presumably other HDACs [50], it has been proposed that its mood stabilizing efficacy may be mediated at least in part by histone modifications. Another study showed that HDAC inhibitors such as VPA, SB, and TSA increase BDNF expression in the brain [51]. Thus, epigenetic mechanisms, especially histone modification, seem to have the potential to provide new mechanistic insights into the expression of depression and novel treatments for depression and related mood disorders.
Adult hippocampal neurogenesis
Brain imaging studies showing reduced hippocampal volume in depressed patients have provided a platform for investigating adult neurogenesis in the context of the pathogenesis of depression [52]. The hypothesis states that chronic stresses and other depression-inducing stimuli decrease neurogenesis [53–55], whereas antidepressant efficacy may rely on an increase in neurogenesis [54–56]. Adult neurogenesis is restricted to the subventricular zone and subgranular zone of the hippocampus [57], and this emphasizes the potential importance of hippocampal neurogenesis during the onset as well as during the treatment of depression. Supporting this idea, various animal models of depression, such as learned helplessness, chronic mild stress, and psychosocial stress, are associated with reductions in hippocampal neurogenesis [58–60]. Conversely, chronic antidepressant treatment not only increases neurogenesis but also supports survival of newborn neurons [61]. It has also been shown that the antidepressant efficacy of tricyclics, imipramine, and SSRIs requires hippocampal neurogenesis in rodents [58, 62, 63]. Furthermore, chronic fluoxetine treatment appears to increase the number of synapses in the pyramidal cell layers and block the decrease in spine density in the dentate gyrus and other hippocampal cell layers [64]. Notably, enriched environments, which is known to enhance hippocampal neurogenesis [65], decrease depression-related behaviors in rodents [66, 67].
The expression level of BDNF deserves attention when examining the molecular mechanisms underlying the antidepressant-mediated increase in neurogenesis. As described above, in various animal models of depression, the BDNF level is decreased [40], whereas chronic antidepressant medication and electro-convulsive shocks increase the levels in the hippocampus [28, 46]. A recent study showed that CREB, a transcription factor that regulates expression of CRE-containing target genes including BDNF, is also upregulated and activated in hippocampus by chronic antidepressant treatment [2, 53, 68, 69]. However, the cause and effect relationship among the induction of CREB and BDNF, the neurogenesis, and behavioral effects of antidepressants remains to be further investigated.
Recent studies demonstrated that long-term administration of mood stabilizers such as lithium, valproic acid, and carbamazepine also enhances adult hippocampal neurogenesis [70–72]. Lithium directly inhibits glycogen synthase kinase-3 (GSK-3) and inositol signaling [73]. VPA enhances gene expression likely by inhibiting HDACs, indirectly blocks GSK-3 activity, and suppresses inositol signaling [71, 74–76]. Although it remains unclear whether the GSK-3 and inositol signaling are actually linked with clinical effects of mood stabilizers, the data suggest a common molecular pathway constituting the pathophysiology of depression and related mood disorders that converges on adult hippocampal neurogenesis.
Substance withdrawal
Various drugs such as alcohol, psychostimulants, opiates and N-methyl-D-aspartate (NMDA) receptor antagonists generate a physiological response called withdrawal symptoms during abstinence in humans and experimental animals [77–80]. The characteristics of affective symptoms caused by drug withdrawal and major depressive disorder are strikingly similar [80]. Depressed mood and anhedonia are commonly present with both drug abstinence and depressive disorders [81]. Hyperphagia, hypersomnia, feelings of fatigue, and suicidal ideation are also observed in both conditions [82, 83]. Disruptions of the HPA axis are also seen during drug withdrawal, and are accompanied by increased levels of cortisol and elevated cerebrospinal levels of CRF [84]. In addition, elevated levels of cortisol, ACTH and β-endorphin during early cocaine withdrawal resemble those in depressed patients [85]. Brain-imaging studies using positron emission tomography (PET) and functional magnetic resonance imaging (fMRI) have revealed that methamphetamine withdrawal induces decreased glucose metabolism in the anterior cingulate cortex and insula, and increased metabolic activity in the amygdala and orbitofrontal cortex, all of which are frequently observed in clinical depression [86].
Much evidence shows that depression and related mood disorders are accompanied by abnormalities in dopaminergic transmission in the nucleus accumbens (NAc) and ventral tegmental area (VTA), regions that are core parts of the brain reward circuit [87]. It is well established that depressed patients have difficulties in the expression of pleasure and acquisition of motivation, which are mainly governed by a normal NAc-VTA dopamine circuit [88]. Consistently, it has been shown that a deregulation of dopamine D2 receptor signaling results in depression-like behaviors in experimental animals [89], and that neuronal nitric oxide synthase (nNOS) knockout mice with altered dopamine D1 receptor signaling exhibit decreased depression-related behaviors [90]. Because nearly all drugs of abuse directly or indirectly activate monoaminergic neurotransmission in the limbic system, resulting in reward sensations [91, 92], it has been postulated that counter-adaptations may occur in opposition to the reward effects with chronic drug intake, generating cognitive, motivational, and affective impairments, including depression-like symptoms during the drug withdrawal period [93].
As described above, in many ways, depressive mood subsequent to drug withdrawal shares common characteristics, such as neuro-hormonal changes, regional brain activity, and pharmacological responses, with clinical depression. However, it needs to be emphasized that the onset, course, duration, and other factors such as involvement of substances diagnostically distinguishes substance-induced mood disorders from major depressive disorders [94, 95]. Some experimental data also hint at differences between these conditions at the molecular level, demanding cautions when interpreting the related observations. For example, dopamine transporter densities are increased in the striatum in both cases [96], but serotonin transporter densities are elevated in the brainstem during the early stage of cocaine abstinence [97], but not in clinical depression [98]. Also, some abstinent drug addicts have been treated with antidepressant drugs to reduce drug craving, but the positive effect of these drugs needs further validation [99]. Nonetheless, insights from these views not only tell us that brain reward circuits composed of the mesolimbic system are potentially important in understanding depression, but also provide a useful behavioral readout for depressive mood in experimental animals.
Circadian rhythms
Circadian rhythm is a roughly 24-hour cycle of biochemical, physiological, and behavioral processes under control of internal clock [100–102]. From the clinical point of view, a potential link between circadian rhythms and depression or related mood disorders has long been postulated. For example, it is relatively well known that insufficient length of light phase to entrain the circadian rhythm can be causative for the development of seasonal affective disorders [103, 104]. Also, abnormal regulation of sleep/wake cycles, body temperature, blood pressure, and various endocrine functions under the control of circadian clock are prominent symptoms of mood disorders [102, 105–110]. However, molecular mechanisms underlying the link are still largely unknown.
Recently, interesting observations have been made in the mutant mouse that has a deletion of 19th exon of Clock gene, a core component of molecular clock. The mouse exhibits hyperactive VTA dopaminergic neurons and behavioral phenotypes that are reminiscence of mania seen in bipolar disorder patients [111, 112]. Moreover, lithium, a mood stabilizer for bipolar depression patients, effectively inhibits GSK3β, a core regulatory component in the molecular clock. Lithium also has an effect on the nuclear entry of Period-Cryptochrome heterodimers, a key process to form a negative loop in the molecular clock, likely through an inhibition of GSK3β activity. Furthermore, lithium appears to regulate activity of Rev-erb α that links the negative loop to the positive loop in the biological clock [113–116].
Potential links between circadian rhythm and the monoamine system are also reported. The synthesis and/or secretion of monoamine neurotransmitters and the function of their receptors are under influence of circadian rhythms. The circadian rhythmicity of dopamine transporter and tyrosine hydroxylase expression in dopaminergic neurons is also disrupted when the suprachiasmatic nucleus of the hypothalamus, the central part of endogenous clock, is damaged [117]. Moreover, monoamine oxidase-A (MAO-A) expression is regulated by dimer formation of Clock and Bmal1, and MAO-A activity accordingly shows a circadian rhythmicity [118]. Conversely, the expression of circadian genes such as Clock, Per1, and Bmal1 is stimulated when dopamine D1 receptor is activated, and suppressed when dopamine D2 receptor is activated in the limbic area [119]. Collectively, the molecular clock appears to be tightly interconnected with monoamine systems, which might explain symptomatic correlation between circadian rhythm and depression at the molecular level.
Although the relationship among the daily variations of mood, endogenous molecular clock, and the expression of depressive symptoms is complicated, normalization of the biological rhythms of a depressive individual could have a beneficial effect. In this regard, the recent development of agomelatine as an antidepressant is of great interest. Agomelatine is a potent agonist for melatonin receptors and has capacity to reset the internal circadian clock [120, 121]. Intriguingly, it also exhibits antagonistic activity on 5-HT2Creceptor, thereby indirectly enhancing the dopamine and norepinephrine neurotransmission [122–124]. Moreover, agomelatine affects differentially various stages of neurogenesis in the dorsal and ventral hippocampus [125]. Further understanding of the molecular basis of agomelatine action and its efficacy may provide interesting insight into the interface between circadian rhythm and pathophysiology of depression.
Functional anatomy
Information on brain regions and neural circuitry responsible for the expression and progression of a disease is an important platform to better diagnose the disease and to properly interpret the observations obtained from molecular, cellular, and tissue experiments in the clinically relevant context. While various brain regions are known to be involved in regulation of mood or emotion, definite information on central neural circuits responsible for mood disorders is still incomplete, mainly because anatomical lesions in patients have been less consistently found relative to other various neurological disorders such as some neurodegenerative diseases. However, there are neuropathological and neuroradiological studies that have established interesting associations between mood disorders and structural abnormalities in the brain. For example, glial reduction was observed in anterior cingulate gyrus and neuronal abnormalities were detected in the dorsolateral prefrontal cortex in post-mortem neuropathological studies of mood disorder patients [126, 127]. Radiological studies using MRI also revealed reduced volumes of orbitofrontal and subgenual anterior cingulate cortex [128–130], electrical stimulation of which correlatively elicits an antidepressant effect [131]. Most notably, reductions in hippocampal volume in depressed elderly patients were reported [132, 133].
Recent brain imaging studies mainly using fMRI are adding information on brain regions that play important roles in depressive symptoms at the functional level [134]. Functional changes in brain regions such as prefrontal/cingulate cortex, hippocampus, striatum, amygdala, and thalamus are correlated with depression [52]. The neocortex and hippocampus also appear to play critical roles in the symptoms related to the cognitive deficits that are prevalent in depressed patients [55], and the nucleus accumbens and amygdala seem to be core regions for anhedonia and emotional memory-related symptoms [135, 136]. The functional changes in the hypothalamus are also linked to sleep- and appetite-associated symptoms [137]. Research on these topics is now being accelerated by fast advances in brain imaging technologies, and the outcome, in combination with the information from the conventional anatomical studies, is driving the generation of a higher-resolution picture of the neural circuitry relevant to depression.
Conclusion
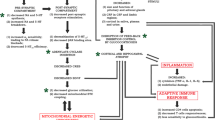
A prerequisite for effective control of depression and related mood disorders is to understand their detailed molecular pathways. Although the classical stress model of depression and current understanding of antidepressant action appears to be partially linked via epigenetic mechanisms and hippocampal neurogenesis (Figure 1), obviously, the current picture of the pathophysiology of depression is largely incomplete, and thus many potential hypotheses are being generated and tested, forming fragmented neurobiological views of depression and related mood disorders. One major task in the field must be to integrate the relevant hypotheses to formulate a bigger picture of the pathophysiology of depression and related disorders. A key step may be to define the high-resolution neural circuitry of depression, which will provide a platform to better interpret the observations obtained from molecular, cellular, and tissue experiments at the organism level. Another critical step will be to identify 'depression genes' that are causative for depression. This will help us generate genetic animal models that may not only be critical for clarifying many issues in depression research using experimental animals, but may also be useful for assessing the potential efficacy of candidate antidepressants. Finally, the most challenging task in the field is to overcome the limitations of current therapies, which are only effective in a fraction of patients. It has long been expected that novel antidepressants targeting non-monoamine systems would enlarge the extent of treatable patients (Figure 1), but the progress still falls short of expectations, thereby leaving it as a pressing task in the field.

Approaches to the development of antidepressants targeting non-monoaminergic components. Chronic stress can cause hypercortisolemia which results in neuronal damages in the hippocampus, thereby weakening the feedback inhibition on HPA axis. Chronic stress also can inhibit the expression of neurotrophic factors through epigenetic mechanisms. On the other hand, chronic treatment of antidepressants and mood stabilizers can establish epigenomic environments that favor the expression of anti-depression genes. The targets may include genes for neurotrophic factors which prevent neuronal damages and enhance hippocampal neurogenesis. Some of approaches to the development of antidepressants targeting non-monoaminergic components are also shown.
References
Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE: Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005, 62: 593-602. 10.1001/archpsyc.62.6.593.
Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM: Neurobiology of depression. Neuron. 2002, 34: 13-25. 10.1016/S0896-6273(02)00653-0.
Fava M, Kendler KS: Major depressive disorder. Neuron. 2000, 28: 335-341. 10.1016/S0896-6273(00)00112-4.
Burmeister M: Basic concepts in the study of diseases with complex genetics. Biol Psychiatry. 1999, 45: 522-532. 10.1016/S0006-3223(98)00316-3.
Manji HK, Drevets WC, Charney DS: The cellular neurobiology of depression. Nat Med. 2001, 7: 541-547. 10.1038/87865.
Ressler KJ, Nemeroff CB: Role of serotonergic and noradrenergic systems in the pathophysiology of depression and anxiety disorders. Depress Anxiety. 2000, 12 (Suppl 1): 2-19. 10.1002/1520-6394(2000)12:1+<2::AID-DA2>3.0.CO;2-4.
Morilak DA, Frazer A: Antidepressants and brain monoaminergic systems: a dimensional approach to understanding their behavioural effects in depression and anxiety disorders. Int J Neuropsychopharmacol. 2004, 7: 193-218. 10.1017/S1461145704004080.
Renard CE, Fiocco AJ, Clenet F, Hascoet M, Bourin M: Is dopamine implicated in the antidepressant-like effects of selective serotonin reuptake inhibitors in the mouse forced swimming test?. Psychopharmacology (Berl). 2001, 159: 42-50. 10.1007/s002130100836.
Hyman SE, Nestler EJ: Initiation and adaptation: a paradigm for understanding psychotropic drug action. Am J Psychiatry. 1996, 153: 151-162.
Mill J, Petronis A: Molecular studies of major depressive disorder: the epigenetic perspective. Mol Psychiatry. 2007, 12: 799-814. 10.1038/sj.mp.4001992.
Lesch KP, Balling U, Gross J, Strauss K, Wolozin BL, Murphy DL, Riederer P: Organization of the human serotonin transporter gene. J Neural Transm Gen Sect. 1994, 95: 157-162. 10.1007/BF01276434.
Heils A, Teufel A, Petri S, Stober G, Riederer P, Bengel D, Lesch KP: Allelic variation of human serotonin transporter gene expression. J Neurochem. 1996, 66: 2621-2624.
Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, Benjamin J, Muller CR, Hamer DH, Murphy DL: Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 1996, 274: 1527-1531. 10.1126/science.274.5292.1527.
Ogilvie AD, Battersby S, Bubb VJ, Fink G, Harmar AJ, Goodwim GM, Smith CA: Polymorphism in serotonin transporter gene associated with susceptibility to major depression. Lancet. 1996, 347: 731-733. 10.1016/S0140-6736(96)90079-3.
Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R: Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003, 301: 386-389. 10.1126/science.1083968.
Kaufman J, Yang BZ, Douglas-Palumberi H, Houshyar S, Lipschitz D, Krystal JH, Gelernter J: Social supports and serotonin transporter gene moderate depression in maltreated children. Proc Natl Acad Sci USA. 2004, 101: 17316-17321. 10.1073/pnas.0404376101.
Kunugi H, Tatsumi M, Sakai T, Hattori M, Nanko S: Serotonin transporter gene polymorphism and affective disorder. Lancet. 1996, 347: 1340-10.1016/S0140-6736(96)90993-9.
Hoehe MR, Wendel B, Grunewald I, Chiaroni P, Levy N, Morris-Rosendahl D, Macher JP, Sander T, Crocq MA: Serotonin transporter (5-HTT) gene polymorphisms are not associated with susceptibility to mood disorders. Am J Med Genet. 1998, 81: 1-3. 10.1002/(SICI)1096-8628(19980207)81:1<1::AID-AJMG1>3.0.CO;2-2.
Lotrich FE, Pollock BG: Meta-analysis of serotonin transporter polymorphisms and affective disorders. Psychiatr Genet. 2004, 14: 121-129. 10.1097/00041444-200409000-00001.
Gillespie NA, Whitfield JB, Williams B, Heath AC, Martin NG: The relationship between stressful life events, the serotonin transporter (5-HTTLPR) genotype and major depression. Psychol Med. 2005, 35: 101-111. 10.1017/S0033291704002727.
Risch N, Herrell R, Lehner T, Liang KY, Eaves L, Hoh J, Griem A, Kovacs M, Ott J, Merikangas KR: Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis. JAMA. 2009, 301: 2462-2471. 10.1001/jama.2009.878.
de Kloet ER, Joels M, Holsboer F: Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005, 6: 463-475. 10.1038/nrn1683.
Parker KJ, Schatzberg AF, Lyons DM: Neuroendocrine aspects of hypercortisolism in major depression. Horm Behav. 2003, 43: 60-66. 10.1016/S0018-506X(02)00016-8.
Brown ES, Varghese FP, McEwen BS: Association of depression with medical illness: does cortisol play a role?. Biol Psychiatry. 2004, 55: 1-9. 10.1016/S0006-3223(03)00473-6.
McEwen BS: Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007, 87: 873-904. 10.1152/physrev.00041.2006.
Raison CL, Miller AH: When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry. 2003, 160: 1554-1565. 10.1176/appi.ajp.160.9.1554.
Scaccianoce S, Del Bianco P, Paolone G, Caprioli D, Modafferi AM, Nencini P, Badiani A: Social isolation selectively reduces hippocampal brain-derived neurotrophic factor without altering plasma corticosterone. Behav Brain Res. 2006, 168: 323-325. 10.1016/j.bbr.2005.04.024.
Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ: Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006, 9: 519-525. 10.1038/nn1659.
Holsboer F: The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000, 23: 477-501. 10.1016/S0893-133X(00)00159-7.
Valdez GR: CRF receptors as a potential target in the development of novel pharmacotherapies for depression. Curr Pharm Des. 2009, 15: 1587-1594. 10.2174/138161209788168083.
Todorovic C, Sherrin T, Pitts M, Hippel C, Rayner M, Spiess J: Suppression of the MEK/ERK signaling pathway reverses depression-like behaviors of CRF2-deficient mice. Neuropsychopharmacology. 2009, 34: 1416-1426. 10.1038/npp.2008.178.
Skelton KH, Oren D, Gutman DA, Easterling K, Holtzman SG, Nemeroff CB, Owens MJ: The CRF1 receptor antagonist, R12 attenuates the severity of precipitated morphine withdrawal. Eur J Pharmacol. 1919, 571: 17-24. 10.1016/j.ejphar.2007.05.041.
Simon NG, Guillon C, Fabio K, Heindel ND, Lu SF, Miller M, Ferris CF, Brownstein MJ, Garripa C, Koppel GA: Vasopressin antagonists as anxiolytics and antidepressants: recent developments. Recent Pat CNS Drug Discov. 2008, 3: 77-93. 10.2174/157488908784534586.
Surget A, Belzung C: Involvement of vasopressin in affective disorders. Eur J Pharmacol. 2008, 583: 340-349. 10.1016/j.ejphar.2007.11.065.
van West D, Del-Favero J, Aulchenko Y, Oswald P, Souery D, Forsgren T, Sluijs S, Bel-Kacem S, Adolfsson R, Mendlewicz J, et al: A major SNP haplotype of the arginine vasopressin 1B receptor protects against recurrent major depression. Mol Psychiatry. 2004, 9: 287-292. 10.1038/sj.mp.4001420.
Monteggia LM, Luikart B, Barrot M, Theobold D, Malkovska I, Nef S, Parada LF, Nestler EJ: Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol Psychiatry. 2007, 61: 187-197. 10.1016/j.biopsych.2006.03.021.
Karege F, Vaudan G, Schwald M, Perroud N, La Harpe R: Neurotrophin levels in postmortem brains of suicide victims and the effects of antemortem diagnosis and psychotropic drugs. Brain Res Mol Brain Res. 2005, 136: 29-37. 10.1016/j.molbrainres.2004.12.020.
Sen S, Nesse RM, Stoltenberg SF, Li S, Gleiberman L, Chakravarti A, Weder AB, Burmeister M: A BDNF coding variant is associated with the NEO personality inventory domain neuroticism, a risk factor for depression. Neuropsychopharmacology. 2003, 28: 397-401. 10.1038/sj.npp.1300053.
Neves-Pereira M, Mundo E, Muglia P, King N, Macciardi F, Kennedy JL: The brain-derived neurotrophic factor gene confers susceptibility to bipolar disorder: evidence from a family-based association study. Am J Hum Genet. 2002, 71: 651-655. 10.1086/342288.
Duman RS, Monteggia LM: A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006, 59: 1116-1127. 10.1016/j.biopsych.2006.02.013.
Shirayama Y, Chen AC, Nakagawa S, Russell DS, Duman RS: Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci. 2002, 22: 3251-3261.
Sairanen M, Lucas G, Ernfors P, Castren M, Castren E: Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. J Neurosci. 2005, 25: 1089-1094. 10.1523/JNEUROSCI.3741-04.2005.
Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M, et al: Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006, 311: 864-868. 10.1126/science.1120972.
Zorner B, Wolfer DP, Brandis D, Kretz O, Zacher C, Madani R, Grunwald I, Lipp HP, Klein R, Henn FA, Gass P: Forebrain-specific trkB-receptor knockout mice: behaviorally more hyperactive than "depressive". Biol Psychiatry. 2003, 54: 972-982. 10.1016/S0006-3223(03)00418-9.
Saarelainen T, Hendolin P, Lucas G, Koponen E, Sairanen M, MacDonald E, Agerman K, Haapasalo A, Nawa H, Aloyz R, et al: Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J Neurosci. 2003, 23: 349-357.
Tsankova NM, Kumar A, Nestler EJ: Histone modifications at gene promoter regions in rat hippocampus after acute and chronic electroconvulsive seizures. J Neurosci. 2004, 24: 5603-5610. 10.1523/JNEUROSCI.0589-04.2004.
Wilkinson MB, Xiao G, Kumar A, LaPlant Q, Renthal W, Sikder D, Kodadek TJ, Nestler EJ: Imipramine treatment and resiliency exhibit similar chromatin regulation in the mouse nucleus accumbens in depression models. J Neurosci. 2009, 29: 7820-7832. 10.1523/JNEUROSCI.0932-09.2009.
Jenuwein T, Allis CD: Translating the histone code. Science. 2001, 293: 1074-1080. 10.1126/science.1063127.
Tsankova N, Renthal W, Kumar A, Nestler EJ: Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007, 8: 355-367. 10.1038/nrn2132.
Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang DM: The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry. 2009, 14: 51-59. 10.1038/sj.mp.4002099.
Schroeder FA, Lin CL, Crusio WE, Akbarian S: Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 2007, 62: 55-64. 10.1016/j.biopsych.2006.06.036.
Drevets WC: Neuroimaging and neuropathological studies of depression: implications for the cognitive-emotional features of mood disorders. Curr Opin Neurobiol. 2001, 11: 240-249. 10.1016/S0959-4388(00)00203-8.
Pittenger C, Duman RS: Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology. 2008, 33: 88-109. 10.1038/sj.npp.1301574.
Sahay A, Hen R: Adult hippocampal neurogenesis in depression. Nat Neurosci. 2007, 10: 1110-1115. 10.1038/nn1969.
Dranovsky A, Hen R: Hippocampal neurogenesis: regulation by stress and antidepressants. Biol Psychiatry. 2006, 59: 1136-1143. 10.1016/j.biopsych.2006.03.082.
Hunsberger JG, Newton SS, Bennett AH, Duman CH, Russell DS, Salton SR, Duman RS: Antidepressant actions of the exercise-regulated gene VGF. Nat Med. 2007, 13: 1476-1482. 10.1038/nm1669.
Gage FH: Neurogenesis in the adult brain. J Neurosci. 2002, 22: 612-613.
Surget A, Saxe M, Leman S, Ibarguen-Vargas Y, Chalon S, Griebel G, Hen R, Belzung C: Drug-dependent requirement of hippocampal neurogenesis in a model of depression and of antidepressant reversal. Biol Psychiatry. 2008, 64: 293-301. 10.1016/j.biopsych.2008.02.022.
Lee KJ, Kim SJ, Kim SW, Choi SH, Shin YC, Park SH, Moon BH, Cho E, Lee MS, Chun BG, Shin KH: Chronic mild stress decreases survival, but not proliferation, of new-born cells in adult rat hippocampus. Exp Mol Med. 2006, 38: 44-54.
Vollmayr B, Simonis C, Weber S, Gass P, Henn F: Reduced cell proliferation in the dentate gyrus is not correlated with the development of learned helplessness. Biol Psychiatry. 2003, 54: 1035-1040. 10.1016/S0006-3223(03)00527-4.
Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, Weisstaub N, Lee J, Duman R, Arancio O, et al: Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003, 301: 805-809. 10.1126/science.1083328.
Chen F, Madsen TM, Wegener G, Nyengaard JR: Imipramine treatment increases the number of hippocampal synapses and neurons in a genetic animal model of depression. Hippocampus. 2009
Boldrini M, Underwood MD, Hen R, Rosoklija GB, Dwork AJ, John Mann J, Arango V: Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology. 2009, 34: 2376-2389. 10.1038/npp.2009.75.
Wu X, Castren E: Co-treatment with diazepam prevents the effects of fluoxetine on the proliferation and survival of hippocampal dentate granule cells. Biol Psychiatry. 2009, 66: 5-8. 10.1016/j.biopsych.2009.01.023.
Kempermann G, Kuhn HG, Gage FH: More hippocampal neurons in adult mice living in an enriched environment. Nature. 1997, 386: 493-495. 10.1038/386493a0.
Brenes Saenz JC, Villagra OR, Fornaguera Trias J: Factor analysis of Forced Swimming test, Sucrose Preference test and Open Field test on enriched, social and isolated reared rats. Behav Brain Res. 2006, 169: 57-65. 10.1016/j.bbr.2005.12.001.
Hattori S, Hashimoto R, Miyakawa T, Yamanaka H, Maeno H, Wada K, Kunugi H: Enriched environments influence depression-related behavior in adult mice and the survival of newborn cells in their hippocampi. Behav Brain Res. 2007, 180: 69-76. 10.1016/j.bbr.2007.02.036.
Boer U, Alejel T, Beimesche S, Cierny I, Krause D, Knepel W, Flugge G: CRE/CREB-driven up-regulation of gene expression by chronic social stress in CRE-luciferase transgenic mice: reversal by antidepressant treatment. PLoS One. 2007, 2: e431-10.1371/journal.pone.0000431.
Blendy JA: The role of CREB in depression and antidepressant treatment. Biol Psychiatry. 2006, 59: 1144-1150. 10.1016/j.biopsych.2005.11.003.
Chen G, Zeng WZ, Yuan PX, Huang LD, Jiang YM, Zhao ZH, Manji HK: The mood-stabilizing agents lithium and valproate robustly increase the levels of the neuroprotective protein bcl-2 in the CNS. J Neurochem. 1999, 72: 879-882. 10.1046/j.1471-4159.1999.720879.x.
Leng Y, Liang MH, Ren M, Marinova Z, Leeds P, Chuang DM: Synergistic neuroprotective effects of lithium and valproic acid or other histone deacetylase inhibitors in neurons: roles of glycogen synthase kinase-3 inhibition. J Neurosci. 2008, 28: 2576-2588. 10.1523/JNEUROSCI.5467-07.2008.
Kalemenev SV, Kosacheva ES, Sem'ianov AV, Godukhin OV, Raevskii KS: Effect of anticonvulsants lamotrigine and carbamazepine on the synaptic transmission in CA1 field of the rat hippocampal slices. Biull Eksp Biol Med. 1998, 126: 307-310.
Jope RS: Lithium and GSK-3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol Sci. 2003, 24: 441-443. 10.1016/S0165-6147(03)00206-2.
Kim AJ, Shi Y, Austin RC, Werstuck GH: Valproate protects cells from ER stress-induced lipid accumulation and apoptosis by inhibiting glycogen synthase kinase-3. J Cell Sci. 2005, 118: 89-99. 10.1242/jcs.01562.
Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM: Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther. 2007, 321: 892-901. 10.1124/jpet.107.120188.
Chen G, Huang LD, Jiang YM, Manji HK: The mood-stabilizing agent valproate inhibits the activity of glycogen synthase kinase-3. J Neurochem. 1999, 72: 1327-1330. 10.1046/j.1471-4159.2000.0721327.x.
Koob GF: Animal models of craving for ethanol. Addiction. 2000, 95 (Suppl 2): S73-81.
Barr AM, Markou A, Phillips AG: A 'crash' course on psychostimulant withdrawal as a model of depression. Trends Pharmacol Sci. 2002, 23: 475-482. 10.1016/S0165-6147(02)02086-2.
Taylor JR, Punch LJ, Elsworth JD: A comparison of the effects of clonidine and CNQX infusion into the locus coeruleus and the amygdala on naloxone-precipitated opiate withdrawal in the rat. Psychopharmacology (Berl). 1998, 138: 133-142. 10.1007/s002130050655.
Spielewoy C, Markou A: Withdrawal from chronic phencyclidine treatment induces long-lasting depression in brain reward function. Neuropsychopharmacology. 2003, 28: 1106-1116.
Kampman KM, Volpicelli JR, McGinnis DE, Alterman AI, Weinrieb RM, D'Angelo L, Epperson LE: Reliability and validity of the Cocaine Selective Severity Assessment. Addict Behav. 1998, 23: 449-461. 10.1016/S0306-4603(98)00011-2.
Gawin FH, Kleber HD: Abstinence symptomatology and psychiatric diagnosis in cocaine abusers. Clinical observations. Arch Gen Psychiatry. 1986, 43: 107-113.
Uslaner J, Kalechstein A, Richter T, Ling W, Newton T: Association of depressive symptoms during abstinence with the subjective high produced by cocaine. Am J Psychiatry. 1999, 156: 1444-1446.
Plotsky PM, Owens MJ, Nemeroff CB: Psychoneuroendocrinology of depression. Hypothalamic-pituitary-adrenal axis. Psychiatr Clin North Am. 1998, 21: 293-307. 10.1016/S0193-953X(05)70006-X.
Deuschle M, Schweiger U, Weber B, Gotthardt U, Korner A, Schmider J, Standhardt H, Lammers CH, Heuser I: Diurnal activity and pulsatility of the hypothalamus-pituitary-adrenal system in male depressed patients and healthy controls. J Clin Endocrinol Metab. 1997, 82: 234-238. 10.1210/jc.82.1.234.
London ED, Simon SL, Berman SM, Mandelkern MA, Lichtman AM, Bramen J, Shinn AK, Miotto K, Learn J, Dong Y, et al: Mood disturbances and regional cerebral metabolic abnormalities in recently abstinent methamphetamine abusers. Arch Gen Psychiatry. 2004, 61: 73-84. 10.1001/archpsyc.61.1.73.
Nestler EJ, Carlezon WA: The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006, 59: 1151-1159. 10.1016/j.biopsych.2005.09.018.
Scully JH: The American Psychiatric Association textbook of psychiatry, 3rd edition. Journal of Clinical Psychiatry. 2000, 61: 306-306.
Park SK, Nguyen MD, Fischer A, Luke MP, Affar el B, Dieffenbach PB, Tseng HC, Shi Y, Tsai LH: Par-4 links dopamine signaling and depression. Cell. 2005, 122: 275-287. 10.1016/j.cell.2005.05.031.
Tanda K, Nishi A, Matsuo N, Nakanishi K, Yamasaki N, Sugimoto T, Toyama K, Takao K, Miyakawa T: Abnormal social behavior, hyperactivity, impaired remote spatial memory, and increased D1-mediated dopaminergic signaling in neuronal nitric oxide synthase knockout mice. Mol Brain. 2009, 2: 19-10.1186/1756-6606-2-19.
Mark GP, Kinney AE, Grubb MC, Keys AS: Involvement of acetylcholine in the nucleus accumbens in cocaine reinforcement. Ann N Y Acad Sci. 1999, 877: 792-795. 10.1111/j.1749-6632.1999.tb09324.x.
Verhoeff NP, Christensen BK, Hussey D, Lee M, Papatheodorou G, Kopala L, Rui Q, Zipursky RB, Kapur S: Effects of catecholamine depletion on D2 receptor binding, mood, and attentiveness in humans: a replication study. Pharmacol Biochem Behav. 2003, 74: 425-432. 10.1016/S0091-3057(02)01028-6.
Solomon RL, Corbit JD: An opponent-process theory of motivation. I. Temporal dynamics of affect. Psychol Rev. 1974, 81: 119-145. 10.1037/h0036128.
DSM-IV. APA-TFo: Diagnostic and statistical manual of mental disorders: DSM-IV-TR. 2000, Washington, DC: American Psychiatric Association, 4 edn
Quello SB, Brady KT, Sonne SC: Mood disorders and substance use disorder: a complex comorbidity. Sci Pract Perspect. 2005, 3: 13-21. 10.1151/spp053113.
Laasonen-Balk T, Kuikka J, Viinamaki H, Husso-Saastamoinen M, Lehtonen J, Tiihonen J: Striatal dopamine transporter density in major depression. Psychopharmacology (Berl). 1999, 144: 282-285. 10.1007/s002130051005.
Jacobsen LK, Staley JK, Malison RT, Zoghbi SS, Seibyl JP, Kosten TR, Innis RB: Elevated central serotonin transporter binding availability in acutely abstinent cocaine-dependent patients. Am J Psychiatry. 2000, 157: 1134-1140. 10.1176/appi.ajp.157.7.1134.
Malison RT, Price LH, Berman R, van Dyck CH, Pelton GH, Carpenter L, Sanacora G, Owens MJ, Nemeroff CB, Rajeevan N, et al: Reduced brain serotonin transporter availability in major depression as measured by [123I]-2 beta-carbomethoxy-3 beta-(4-iodophenyl)tropane and single photon emission computed tomography. Biol Psychiatry. 1998, 44: 1090-1098. 10.1016/S0006-3223(98)00272-8.
Nunes EV, Levin FR: Treatment of depression in patients with alcohol or other drug dependence: a meta-analysis. JAMA. 2004, 291: 1887-1896. 10.1001/jama.291.15.1887.
Reppert SM, Weaver DR: Molecular analysis of mammalian circadian rhythms. Annu Rev Physiol. 2001, 63: 647-676. 10.1146/annurev.physiol.63.1.647.
Lowrey PL, Takahashi JS: Mammalian circadian biology: elucidating genome-wide levels of temporal organization. Annu Rev Genomics Hum Genet. 2004, 5: 407-441. 10.1146/annurev.genom.5.061903.175925.
Bell-Pedersen D, Cassone VM, Earnest DJ, Golden SS, Hardin PE, Thomas TL, Zoran MJ: Circadian rhythms from multiple oscillators: lessons from diverse organisms. Nat Rev Genet. 2005, 6: 544-556. 10.1038/nrg1633.
Dalgleish T, Rosen K, Marks M: Rhythm and blues: the theory and treatment of seasonal affective disorder. Br J Clin Psychol. 1996, 35 (Pt 2): 163-182.
Rosenthal NE, Sack DA, Jacobsen FM, Skwerer RG, Wehr TA: Seasonal affective disorder & light: past, present and future. Clin Neuropharmacol. 1986, 9 (Suppl 4): 193-195.
Atkinson M, Kripke DF, Wolf SR: Autorhythmometry in manic-depressives. Chronobiologia. 1975, 2: 325-335.
Souetre E, Salvati E, Belugou JL, Pringuey D, Candito M, Krebs B, Ardisson JL, Darcourt G: Circadian rhythms in depression and recovery: evidence for blunted amplitude as the main chronobiological abnormality. Psychiatry Res. 1989, 28: 263-278. 10.1016/0165-1781(89)90207-2.
Boivin DB: Influence of sleep-wake and circadian rhythm disturbances in psychiatric disorders. J Psychiatry Neurosci. 2000, 25: 446-458.
Jones EM, Knutson D, Haines D: Common problems in patients recovering from chemical dependency. Am Fam Physician. 2003, 68: 1971-1978.
Lenox RH, Gould TD, Manji HK: Endophenotypes in bipolar disorder. Am J Med Genet. 2002, 114: 391-406. 10.1002/ajmg.10360.
Wasielewski JA, Holloway FA: Alcohol's interactions with circadian rhythms. A focus on body temperature. Alcohol Res Health. 2001, 25: 94-100.
McClung CA, Sidiropoulou K, Vitaterna M, Takahashi JS, White FJ, Cooper DC, Nestler EJ: Regulation of dopaminergic transmission and cocaine reward by the Clock gene. Proc Natl Acad Sci USA. 2005, 102: 9377-9381. 10.1073/pnas.0503584102.
Roybal K, Theobold D, Graham A, DiNieri JA, Russo SJ, Krishnan V, Chakravarty S, Peevey J, Oehrlein N, Birnbaum S, et al: Mania-like behavior induced by disruption of CLOCK. Proc Natl Acad Sci USA. 2007, 104: 6406-6411. 10.1073/pnas.0609625104.
Abe M, Herzog ED, Block GD: Lithium lengthens the circadian period of individual suprachiasmatic nucleus neurons. Neuroreport. 2000, 11: 3261-3264. 10.1097/00001756-200009280-00042.
Harms E, Kivimae S, Young MW, Saez L: Posttranscriptional and posttranslational regulation of clock genes. J Biol Rhythms. 2004, 19: 361-373. 10.1177/0748730404268111.
Iitaka C, Miyazaki K, Akaike T, Ishida N: A role for glycogen synthase kinase-3beta in the mammalian circadian clock. J Biol Chem. 2005, 280: 29397-29402. 10.1074/jbc.M503526200.
Yin L, Wang J, Klein PS, Lazar MA: Nuclear receptor Rev-erbalpha is a critical lithium-sensitive component of the circadian clock. Science. 2006, 311: 1002-1005. 10.1126/science.1121613.
Sleipness EP, Sorg BA, Jansen HT: Diurnal differences in dopamine transporter and tyrosine hydroxylase levels in rat brain: dependence on the suprachiasmatic nucleus. Brain Res. 2007, 1129: 34-42. 10.1016/j.brainres.2006.10.063.
Hampp G, Ripperger JA, Houben T, Schmutz I, Blex C, Perreau-Lenz S, Brunk I, Spanagel R, Ahnert-Hilger G, Meijer JH, Albrecht U: Regulation of monoamine oxidase A by circadian-clock components implies clock influence on mood. Curr Biol. 2008, 18: 678-683. 10.1016/j.cub.2008.04.012.
Imbesi M, Yildiz S, Dirim Arslan A, Sharma R, Manev H, Uz T: Dopamine receptor-mediated regulation of neuronal "clock" gene expression. Neuroscience. 2009, 158: 537-544. 10.1016/j.neuroscience.2008.10.044.
Martinet L, Guardiola-Lemaitre B, Mocaer E: Entrainment of circadian rhythms by S-2 a melatonin agonist, is dose and plasma concentration dependent. Pharmacol Biochem Behav. 0098, 54: 713-718. 10.1016/0091-3057(95)02221-X.
Redman JR, Guardiola-Lemaitre B, Brown M, Delagrange P, Armstrong SM: Dose dependent effects of S-2 a melatonin agonist, on direction of re-entrainment of rat circadian activity rhythms. Psychopharmacology (Berl). 0098, 118: 385-390. 10.1007/BF02245938.
Chagraoui A, Protais P, Filloux T, Mocaer E: Agomelatine(S 20098) antagonizes the penile erections induced by the stimulation of 5-HT2C receptors in Wistar rats. Psychopharmacology (Berl). 2003, 170: 17-22. 10.1007/s00213-003-1465-3.
Millan MJ, Gobert A, Lejeune F, Dekeyne A, Newman-Tancredi A, Pasteau V, Rivet JM, Cussac D: The novel melatonin agonist agomelatine (S20098) is an antagonist at 5-hydroxytryptamine2C receptors, blockade of which enhances the activity of frontocortical dopaminergic and adrenergic pathways. J Pharmacol Exp Ther. 2003, 306: 954-964. 10.1124/jpet.103.051797.
Millan MJ, Brocco M, Gobert A, Dekeyne A: Anxiolytic properties of agomelatine, an antidepressant with melatoninergic and serotonergic properties: role of 5-HT2C receptor blockade. Psychopharmacology (Berl). 2005, 177: 448-458. 10.1007/s00213-004-1962-z.
Banasr M, Soumier A, Hery M, Mocaer E, Daszuta A: Agomelatine, a new antidepressant, induces regional changes in hippocampal neurogenesis. Biol Psychiatry. 2006, 59: 1087-1096. 10.1016/j.biopsych.2005.11.025.
Ongur D, An X, Price JL: Prefrontal cortical projections to the hypothalamus in macaque monkeys. J Comp Neurol. 1998, 401: 480-505. 10.1002/(SICI)1096-9861(19981130)401:4<480::AID-CNE4>3.0.CO;2-F.
Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY, Overholser JC, Roth BL, Stockmeier CA: Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. 1999, 45: 1085-1098. 10.1016/S0006-3223(99)00041-4.
Ballmaier M, Toga AW, Blanton RE, Sowell ER, Lavretsky H, Peterson J, Pham D, Kumar A: Anterior cingulate, gyrus rectus, and orbitofrontal abnormalities in elderly depressed patients: an MRI-based parcellation of the prefrontal cortex. Am J Psychiatry. 2004, 161: 99-108. 10.1176/appi.ajp.161.1.99.
Coffey CE, Wilkinson WE, Weiner RD, Parashos IA, Djang WT, Webb MC, Figiel GS, Spritzer CE: Quantitative cerebral anatomy in depression. A controlled magnetic resonance imaging study. Arch Gen Psychiatry. 1993, 50: 7-16.
Drevets WC, Price JL, Simpson JR, Todd RD, Reich T, Vannier M, Raichle ME: Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 1997, 386: 824-827. 10.1038/386824a0.
Mayberg HS, Lozano AM, Voon V, McNeely HE, Seminowicz D, Hamani C, Schwalb JM, Kennedy SH: Deep brain stimulation for treatment-resistant depression. Neuron. 2005, 45: 651-660. 10.1016/j.neuron.2005.02.014.
Sheline YI: 3D MRI studies of neuroanatomic changes in unipolar major depression: the role of stress and medical comorbidity. Biol Psychiatry. 2000, 48: 791-800. 10.1016/S0006-3223(00)00994-X.
MacQueen GM, Campbell S, McEwen BS, Macdonald K, Amano S, Joffe RT, Nahmias C, Young LT: Course of illness, hippocampal function, and hippocampal volume in major depression. Proc Natl Acad Sci USA. 2003, 100: 1387-1392. 10.1073/pnas.0337481100.
Sheline YI: Neuroimaging studies of mood disorder effects on the brain. Biol Psychiatry. 2003, 54: 338-352. 10.1016/S0006-3223(03)00347-0.
Schlaepfer TE, Cohen MX, Frick C, Kosel M, Brodesser D, Axmacher N, Joe AY, Kreft M, Lenartz D, Sturm V: Deep brain stimulation to reward circuitry alleviates anhedonia in refractory major depression. Neuropsychopharmacology. 2008, 33: 368-377. 10.1038/sj.npp.1301408.
Phelps EA, LeDoux JE: Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron. 2005, 48: 175-187. 10.1016/j.neuron.2005.09.025.
Willie JT, Chemelli RM, Sinton CM, Yanagisawa M: To eat or to sleep? Orexin in the regulation of feeding and wakefulness. Annu Rev Neurosci. 2001, 24: 429-458. 10.1146/annurev.neuro.24.1.429.
Acknowledgements
This work was supported by a grant (2009K001271) from Brain Research Center of the 21st Century Frontier Research Program and by grants, 331-2007-1-C00213, 3-200900000001605, and 20090076351, funded by the Ministry of Education, Science, and Technology, the Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SL, JJ, and YK collected information and participated in drafting the manuscript. SKP wrote the manuscript and coordinated the drafting process. All authors read and approved the final form of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lee, S., Jeong, J., Kwak, Y. et al. Depression research: where are we now?. Mol Brain 3, 8 (2010). https://doi.org/10.1186/1756-6606-3-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-6606-3-8