
Abstract
Background
Apicomplexan parasites of the genus Babesia, Theileria and Plasmodium are very closely related organisms. Interestingly, their mitochondrial (mt) genomes are highly divergent. Among Babesia, Babesia orientalis is a new species recently identified and specifically epidemic to the southern part of China, causing severe disease to water buffalo. However, no information on the mt genome of B. orientalis was available.
Methods
Four pairs of primers were designed based on the full genome sequence of B. orientalis (unpublished data) and by aligning reported mt genomes of B. bovis, B. bigemina, and T. parva. The entire mt genome was amplified by four sets of PCR. The obtained mt genome was annotated by aligning with published apicomplexan mt genomes and Artemis software v11. Phylogenetic analysis was performed by using cox1 and cob amino acid sequences.
Results
The complete mt genome of B. orientalis (Wuhan strain) was sequenced and characterized. The entire mt genome is 5996 bp in length with a linear form, containing three protein-coding genes including cytochrome c oxidase I (cox1), cytochrome b (cob) and cytochrome c oxidase III (cox3) and six rRNA large subunit gene fragments. The gene arrangement in B. orientalis mt genome is similar to those of B. bovis, B. gibsoni and Theileria parva, but different from those of T. orientalis, T. equi and Plasmodium falciparum. Comparative analysis indicated that cox1 and cob genes were more conserved than cox3. Phylogenetic analysis based on amino acid sequences of cox1, cob and cox1 + cob, respectively, revealed that B. orientalis fell into Babesia clade with the closest relationship to B. bovis.
Conclusions
The availability of the entire mt genome sequences of B. orientalis provides valuable information for future phylogenetic, population genetics and molecular epidemiological studies of apicomplexan parasites.
Similar content being viewed by others


Background
Mitochondria are essential organelles within cells and are responsible for energy transduction, metabolism, cell growth and survival [1–3]. Inside mitochondria, there is a genome called mitochondrial (mt) genome. Mt genomes are present in almost all eukaryotic cells and have remarkable variations in size, structure, and organization [4–6]. The largest mt genome has been found in muskmelons with an estimated size of 2400 kb [7–9]. The smallest mt genome of only 6 kb in length has been reported in an apicomplexan parasite (Plasmodium) [10, 11].
The structure of mt genome contains two major types, the linear form and the circular form. The circular forms are usually present in animal mt genomes with the size ranging from 15 kb to 20 kb, containing 12-13 protein-coding genes, 22 transfer RNA (tRNA) genes and two ribosomal RNA (rRNA) genes, and gene arrangements in the genomes are extremely stable [12]. The linear forms have been documented in many apicomplexan parasites, including Plasmodium, Babesia, Theileria and Eimeria[5, 10, 11, 13]. Compared with animal mt genomes, the mt genomes of apicomplexan parasites encode only three protein-coding genes (cytochrome c oxidase subunits I [cox1] and III [cox3], and cytochrome b [cob]) and six fragments of large subunit rRNA genes [14]. The gene arrangements are also different among animal mt genomes and apicomplexan parasites mt genome.
Meanwhile, in apicomplexan parasites, mitochondrial protein-coding genes have been extensively used as genetic markers for phylogenetic analysis at different taxonomic levels, serving as an ideal model for gene rearrangement, and evolutionary studies [13, 15]. Most of the phylogenies of apicomplexan parasites were constructed using cox1 or cob alone, however, in some cases, a combination of cox1 and cob was employed to evaluate the phylogenetic relationships [13, 16]. In addition, mt genome sequences are also very valuable for population genetic studies [17–19] as reported in Plasmodium vivax, Plasmodium knowlesi, Trypanosoma cruzi[20–22].
B. orientalis is a tick-borne, intra-erythrocytic protozoan parasite causing buffalo babesiosis characterized by fever, anemia, icterus, haemoglobinuria and high mortality [23, 24]. This species was first reported in 1987 and then identified as a new species named Babesia orientalis in 1997 [25]. The new species was discovered initially based on the differences in morphology, transmission, pathogenicity and endemic areas, compared to Babesia bigemina and Babesia bovis[25], and later confirmed by the phylogenetic analysis based on 18S rRNA and heat shock protein 70 (HSP70) genes [26, 27]. The disease caused by B. orientalis is one of the most important parasitic diseases of buffalo in central and south China, resulting in enormous economic losses [27, 28]. In spite of its importance, very limited information was available about this parasite, especially at the molecular level, including mt genome sequences and structures.
In the present study, B. orientalis (Wuhan strain) mt genome was determined and annotated. The structure was characterized and compared with those of related species. In addition, the evolution of structural divergence in the apicomplexan mt genomes was discussed.
Methods
Parasite cultivation
Babesia orientalis (Wuhan strain) was cultivated according to the protocol of He et al. [29]. In brief, two, 1-year-old water buffalo, free of B. orientalis infection as confirmed by microscopy and real-time PCR [29], were splenectomized 14 days prior to B. orientalis infection. Each buffalo was subcutaneously injected with 4 ml of B. orientalis-infected blood (Wuhan strain, percentage parasitized erythrocytes, PPE 1%). Blood samples were collected everyday to monitor the parasitemia until PPE reached 3%.
All the experimental animals were housed, fed and given clean drinking water in accordance with the stipulated rules for the regulation of the administration of affairs concerning experimental animals of P.R. China.
B. orientalis mitochondria DNA sequencing
The blood from experimentally infected buffalo was collected in EDTA. Parasite genomic DNA was extracted from B. orientalis-infected blood using QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The terminal inverted repeat (TIR) regions in the beginning and end of B. orientalis mt genome, 1-181 bp and 5835-5996 bp, were obtained by using B. bovis mt genome sequence to blast the full genome sequence of B. orientalis (unpublished data). Four pairs of primers were designed based on these two mtDNA sequences (1-181 bp and 5835-5996 bp) of B. orientalis and by aligning reported mt genomes of B. bovis (EU075182 and AB499088), B. bigemina (AB499085), and T. parva (Z23263 and AB499089) (Table 1 and Figure 1). The entire mt genome was amplified by P1 and R4. In order to confirm the mt genome, four sets of PCR were processed using primers P1 and R1, P2 and R2, P3 and R3, P4 and R4, respectively (Figure 1). Amplified products were purified and then ligated into the pMD19-T vector (TaKaRa Biotechnology), and the recombinant clones were sequenced using the ABI PRISM 377 DNA sequencer by following the manufacturer’s instructions. The vector primers M13 (-47) and M13 (-48), as well as PCR primers, were used for the sequencing of mt genome.

Schematic representation of the mt genome of B. orientalis. Symbol … and ()in the sequence indicates nucleotide omitted. Primer sequences of P1, P2, P3, P4, R1, R2, R3 and R4 are shown in light grey. Three protein-coding genes cox 1, cox 3 and cob are shown in dark grey. The first and last six codons of each protein-coding gene are shown below the sequence. Six LSU rRNA genes from L1–L6 are showed. Terminal inverted repeats (TIR) are represented by underlining. The symbol I above the genome sequence indicates the beginning and the end of the gene fragment. The nucleotide position of each gene fragment is shown above the sequences.
Gene annotation and sequence analysis
Nucleotide sequences of B. orientalis mt genome (GenBank accession no. KF218819) were aligned with published mt genome sequences of Plasmodium falciparum (M76611), B. bovis (AB499088), T. parva (AB499089), and T. annulata (NT167255) by MAFFT (version 7) [30, 31] with manual corrections. Protein-coding genes were predicted by comparing the previously annotated sequences from these four related species. To identify putative rRNA genes, mt DNA sequences or annotated rRNA gene fragments from the four related species were used as queries by pair-wise comparison (Blastn) in NCBI. The entire B. orientalis mt genome was subjected to tRNAscan SE 1.21 (http://lowelab.ucsc.edu/tRNAscan-SE/) using Mito/Chloroplast model option and Nematode Mito model for analyzing the existence of tRNA gene. Results from both models were compared, final annotation was determined according to B. bovis mt genome annotation. B. orientalis mt genome was also annotated using appropriate mitochondrial codes in Artemis software v11 [32, 33].
Nucleotide sequence identities were calculated by pairwise comparison between 13 apicomplexan parasites including B. orientalis, another six Babesia species, five Theileria species and P. falciparum for each of the three protein-encoding genes. The data sets of cox1 (472 aa), cox3 (229 aa) or cob (358 aa) were aligned using MAFFT (version 7) employing the FFT-NS-i algorithm [31, 34]. The alignment was manually edited using BioEdit 7.1.11 [35]. The nucleotide identities were determined through BioEdit 7.1.11.
Phylogenetic analysis
The concatenated amino acid sequences of cox1, cox3 and cob from 15 apicomplexan parasites (Table 2) were used for similarity analysis. For phylogenetic analysis, cox3 was not included, because cox3 has been presented in the nuclear genome rather than in mt DNA in some species, such as T. thermophila. A free-living ciliate, Tetrahymena thermophila[36], was used as the outgroup. The best-fit model of cox1, cob, and cox1-cob combined nucleotide substitution was determined by JmodelTest 0.1.1 [37] selected by AIC calculations. A general time reversible model with a proportion of invariable sites and a gamma-shaped distribution of rates across sites (TIM1 + I + G) substitution mode was used in PAUP* v4b10 [38] to explore neighbour-joining, parsimony and maximum likelihood methods. MrBayes v3.1.2 [39, 40] was used to explore Bayesian phylogeny. Consensus trees were edited in MEGA v4.0.2 [41].
All water buffalo studies were carried out in compliance with the regulations (No. 5 proclaim of the Standing Committee of Hubei People's Congress) approved by the Standing Committee of Hubei People's Congress, P. R. China. The animal protocols were approved by Laboratory Animals Research Centre of Hubei province and the ethics committee of Huazhong Agricultural University (Permit number: 4200696657).
Results and discussion
Characterization of B. orientalis mt genome
The full length mt genome of B. orientalis was amplified by P1 and R4, and a 5996 bp fragment was obtained. In order to confirm the mt genome sequence, four overlapping fragments were amplified by using primer sets P1-R1, P2-R2, P3-R3 and P4-R4 with expected sizes of 1749 bp, 1655 bp, 1831 bp and 838 bp, respectively. These four overlapping fragments covered the entire genome of B. orientalis (Figure 1). Each of the PCR products were then cloned into pMD19-T vector, and sequenced. A 5996 bp mt genome sequence of B. orientalis (Wuhan strain) was obtained by assembling all the sequenced fragments.
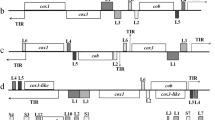
Sequence analysis indicated that B. orientalis mt genome was arranged in a linear form. It containing three protein-coding genes, cox1 (cytochrome c oxidase I), cob (cytochrome b), cox3 (cytochrome c oxidase III) and six large subunit (LSU) rRNA gene fragments, but not any tRNA genes, which is consistent with those of other apicomplexan parasites studied to date (Figures 1 and 2) [11]. The mt genomes of apicomplexan parasites usually contain terminal inverted repeat (TIR) sequences with the size of around 440-450 bp [5]. However, in the mt genome of B. orientalis, 181 bp and 161 bp TIRs were identified from the beginning and the end, respectively (Figure 1). cox1, the first and fourth rRNA large subunit fragments (L1 and L4) are encoded by one strand of the mt genome, whereas cox3, cob, the second, the third, fifth and sixth fragments of large subunit rRNA genes (L2, L3, L5 and L6) are encoded by another strand (Figure 2a). The arrangement and predicted transcriptional direction of three protein-coding genes are the same as that of B. bovis (Figure 2b), B. gibsoni (not shown), and Theileria parva (Figure 2c), however, it greatly differed from that of T. orientalis, T. equi (not shown) and P. falciparum (Figure 2d) [5].

Schematic structure of mitochondrial genomes of Babesia orientalis (KF218819) (a) and three apicomplexan parasites including (b) B. bovis (AB499088), (c) Theileriaparva (Z23263) and (d) Plasmodium falciparum (M76611). Gene names above the bold line are transcribed from left to right, and those below are transcribed from right to left. White boxes indicate protein-coding genes, cox 1 (cytochrome c oxidase I), cox 3 (cytochrome c oxidase III), and cob (cytochrome b). Fragments of LSU (L1 – L6, LA – LG, 1, 2, 3, 10 and 13) and SSU (SA, SB, SD – SF, 4, 8 and 9) rRNA genes are shown by dark and light gray boxes. TIR represents terminal inverted repeats.
In this study, BLAST analysis of the entire mt genome sequence of B. orientalis to NCBI databases revealed that the mt genome sequence of B. orientalis was most similar to those of B. bovis (EU075182 and AB499088), with an identity of 87%. The other related species were B. caballi, B. bigemina and B. gibsoni, showing their mt genome sequence identities of 86%, 85% and 84% with that of B. orientalis, respectively. However, nucleotide identities were greatly different for the sequences of three protein-coding genes cox1, cox3 and cob, when compared among different species. Pairwise comparison has been carried out among 13 different apicomplexan species including B. orientialis, another six Babesia species, five Theileria species and P. falciparum (see Table 3). The nucleotide sequence differences among 13 species ranged from 38.7% to 99.8% for cox1 (Table 3a), from 28.8% to 92.5% for cox3 (Table 3b) and 32.7% to 99.9% for cob (Table 3c), respectively. These results indicated that cox1 and cob genes were more conserved than cox3 gene. The different extent of conservation in the protein-coding genes suggested that mt genome sequences could be used as a gene marker for population evolutionary studies.
Phylogenetic analysis
The majority of phylogenetic studies in the phylum apicomplexa have utilized 18S rRNA genes [27, 42], which allow the analysis of the ancient relationships or strains and species differentiation by focusing on the highly conserved regions defining the critical secondary structure, or the more variable internal transcribed spacer regions, respectively. Because of different extent of conservation of three protein-coding genes and important function of mt genome, the extent of sequence difference might reflect the phylogenetic relationship across species of apicomplexan parasites. So the mitochondrion sequences may provide an alternative approach to conduct these studies [5, 10, 13].
To analyze the phylogenetic relationship of B. orientalis with other apicomplexan parasites, phylogenetic trees were constructed with the amino acid sequences of cox1, cob and cox1 + cob using neighbor-joining (NJ), maximum likelihood (ML), maximum parsimony (MP) and Bayesian phylogenetic methods. The trees obtained from three data sets by different methods were consistent with no significant changes in the topology or in the bootstrap values (Figures 3a,b, and c). The NJ trees constructed with cox1, cob and cox1 + cob sequences have been presented as a representative (Figure 3). All three NJ trees displayed the same topology with high bootstrap values. B. orientalis appeared in the Babesia clade, and it’s most close to B. bovis. However, the bootstrap value was higher in the tree constructed by cox1 + cob as compared to those of cox1 and cob. These results suggested that the combined amino acid sequences of cox1 and cob may be more reliable in studying evolutionary relationships than the sequences of single gene. The relationship of B. orientalis with other apicomplexan parasites revealed by all trees from mt genome sequences were consistent with that from the previous phylogenetic trees based on 18S rRNA and heat shock protein 70 (HSP70) gene [26, 27]. These results demonstrated that the mt genome sequences are useful for the phylogenetic studies of apicomplexan parasites.

Phylogenetic analyses of cox 1 (a), cob (b) and cox 1 + cob (c) amino acid sequences of B. orientalis . Each neighbor-joining tree shows the phylogenetic relationship of protein coding genes of Babesiaorientalis mt genome determined in this study with other apicomplexan parasites. Bootstrap values at the nodes are indicating the degree of support for each cluster. Vertical lengths in each tree are not significant and are merely set for clarity. GenBank accession numbers are indicated on the right of each species name. Tetrahymena thermophila was used as an outgroup.
Conclusion
In this study, we first reported the 5996 bp linear mitochondrion genome of B. orientalis. This mt genome contains 6 LSU rRNA gene fragments and three protein-coding genes, but no tRNA gene. Gene arrangement in the mt genome of B. orientalis is similar to those of B. bovis, B. gibsoni, and T. parva, but different from those of T. orientalis, T. equi and P. falciparum. Phylogenies based on the amino acid sequences of cox1 or cob alone and cox1 + cob combined all indicated that B. orientalis is closest to B. bovis, which is in agreement with the previous phylogenetic studies of B. orientalis. The availability of the entire mt genome sequences of B. orientalis provides valuable information for future phylogenetics, population genetics and molecular epidemiological studies of apicomplexan parasites.
References
Frederick RL, Shaw JM: Moving mitochondria: establishing distribution of an essential organelle. Traffic. 2007, 8 (12): 1668-1675. 10.1111/j.1600-0854.2007.00644.x.
Taylor-Brown E, Hurd H: The first suicides: a legacy inherited by parasitic protozoans from prokaryote ancestors. Parasit Vectors. 2013, 6 (1): 108. 10.1186/1756-3305-6-108.
Kaczanowski S, Sajid M, Reece SE: Evolution of apoptosis-like programmed cell death in unicellular protozoan parasites. Parasit Vectors. 2011, 4: 44. 10.1186/1756-3305-4-44.
Gray G, Maxwell D, Villarimo A, McIntosh L: Mitochondria nuclear signaling of alternative oxidase gene expression occurs through distinct pathways involving organic acids and reactive oxygen species. Plant Cell Rep. 2004, 23 (7): 497-503. 10.1007/s00299-004-0848-1.
Hikosaka K, Watanabe Y-I, Tsuji N, Kita K, Kishine H, Arisue N, Palacpac NMQ, Kawazu S-i, Sawai H, Horii T: Divergence of the mitochondrial genome structure in the apicomplexan parasites, Babesia and Theileria. Mol Biol Evol. 2010, 27 (5): 1107-1116. 10.1093/molbev/msp320.
Feagin JE: Mitochondrial genome diversity in parasites. Int J Parasitol. 2000, 30 (4): 371-390. 10.1016/S0020-7519(99)00190-3.
Lung B, Zemann A, Madej MJ, Schuelke M, Techritz S, Ruf S, Bock R, Hüttenhofer A: Identification of small non-coding RNAs from mitochondria and chloroplasts. Nucleic Acids Res. 2006, 34 (14): 3842-3852. 10.1093/nar/gkl448.
Ward BL, Anderson RS, Bendich AJ: The mitochondrial genome is large and variable in a family of plants (Cucurbitaceae). Cell. 1981, 25 (3): 793-803. 10.1016/0092-8674(81)90187-2.
Palmer DN, Fearnley IM, Walker JE, Hall NA, Lake BD, Wolfe LS, Haltia M, Martinus RD, Jolly RD: Mitochondrial ATP synthase subunit c storage in the ceroid‒lipofuscinoses (Batten disease). American J Medical Genet. 1992, 42 (4): 561-567. 10.1002/ajmg.1320420428.
Hikosaka K, Tsuji N, Watanabe Y-I, Kishine H, Horii T, Igarashi I, Kita K, Tanabe K: Novel type of linear mitochondrial genomes with dual flip-flop inversion system in apicomplexan parasites, Babesia microti and Babesia rodhaini. BMC Genomics. 2012, 13 (1): 1-9. 10.1186/1471-2164-13-1.
Hikosaka K, Watanabe Y-I, Kobayashi F, Waki S, Kita K, Tanabe K: Highly conserved gene arrangement of the mitochondrial genomes of 23 Plasmodium species. Parasitol Int. 2011, 60 (2): 175-180. 10.1016/j.parint.2011.02.001.
Boore JL: Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27 (8): 1767-1780. 10.1093/nar/27.8.1767.
Lin R-Q, Qiu L-L, Liu G-H, Wu X-Y, Weng Y-B, Xie W-Q, Hou J, Pan H, Yuan Z-G, Zou F-C: Characterization of the complete mitochondrial genomes of five Eimeria species from domestic chickens. Gene. 2011, 480 (1): 28-33.
Preiser P, Wilson R, Moore P, McCready S, Hajibagheri M, Blight K, Strath M, Williamson D: Recombination associated with replication of malarial mitochondrial DNA. The EMBO J. 1996, 15 (3): 684-
Lei R, Shore GD, Brenneman RA, Engberg SE, Sitzmann BD, Bailey CA, Kimmel LM, Randriamampionona R, Ranaivoarisoa JF, Louis EE: Complete sequence and gene organization of the mitochondrial genome for Hubbard’s sportive lemur (Lepilemur hubbardorum). Gene. 2010, 464 (1): 44-49.
Hikosaka K, Nakai Y, Watanabe Y-I, Tachibana S-I, Arisue N, Palacpac NMQ, Toyama T, Honma H, Horii T, Kita K: Concatenated mitochondrial DNA of the coccidian parasite Eimeria tenella. Mitochondrion. 2011, 11 (2): 273-278. 10.1016/j.mito.2010.10.003.
Hajibabaei M, Singer GA, Hebert PD, Hickey DA: DNA barcoding: how it complements taxonomy, molecular phylogenetics and population genetics. Trends Genet. 2007, 23 (4): 167-172. 10.1016/j.tig.2007.02.001.
Fernando P, Pfrender ME, Encalada SE, Lande R: Mitochondrial DNA variation, phylogeography and population structure of the Asian elephant. Heredity. 2000, 84 (3): 362-372. 10.1046/j.1365-2540.2000.00674.x.
Birungi J, Munstermann LE: Genetic structure of Aedes albopictus (Diptera: Culicidae) populations based on mitochondrial ND5 sequences: evidence for an independent invasion into Brazil and United States. Ann Entomol Soc Am. 2002, 95 (1): 125-132. 10.1603/0013-8746(2002)095[0125:GSOAAD]2.0.CO;2.
Lewis MD, Llewellyn MS, Yeo M, Acosta N, Gaunt MW, Miles MA: Recent, independent and anthropogenic origins of Trypanosoma cruzi hybrids. PLoS Neglect Trop Dis. 2011, 5 (10): e1363. 10.1371/journal.pntd.0001363.
Lee K-S, Divis PC, Zakaria SK, Matusop A, Julin RA, Conway DJ, Cox-Singh J, Singh B: Plasmodium knowlesi: reservoir hosts and tracking the emergence in humans and macaques. PLoS Pathog. 2011, 7 (4): e1002015. 10.1371/journal.ppat.1002015.
Jongwutiwes S, Putaporntip C, Iwasaki T, Ferreira MU, Kanbara H, Hughes AL: Mitochondrial genome sequences support ancient population expansion in Plasmodium vivax. Mol Biol Evol. 2005, 22 (8): 1733-1739. 10.1093/molbev/msi168.
Liu ZL, Ma LH, Zhang GD, Gao XS: An investigation of babesiosis in buffaloes in Hubei province. Acta Vet Zootechnica Sin. 1986, 17 (1): 49-54. (in chinese)
Liu ZL, Ma LH, Gao XS, Cheng XJ: Study on babesiosis of buffaloes in Hubei Province II: experimental infection demonstrated Rhipicephalus haemaphysaloides haemaphysaloides to be the vector of babesiosis in buffaloes. Acta Vet Zootechnica Sin. 1987, 18 (3): 173-178. (in chinese)
Liu Z, Zhao J, Ma L, Yao B: Babesia orientalis sp. nov. parasitized in buffalo bubalus bubalis in China (Piroplasmida: Babesiidae). Acta Vet Zootechnica Sin. 1997, 28 (1): 84-89.
He L, Liu Q, Quan M, Zhou D-N, Zhou Y-Q, Zhao J-L: Molecular cloning and phylogenetic analysis of Babesia orientalis heat shock protein 70. Vet Parasitol. 2009, 162 (3): 183-191.
Liu Q, Zhao J, Zhou Y, Liu E, Yao B, Fu Y: Study on some molecular characterization of Babesia orientalis. Vet Parasitol. 2005, 130 (3): 191-198.
He L, Zhou Y-Q, Oosthuizen MC, Zhao J-L: Loop-mediated isothermal amplification (LAMP) detection of Babesia orientalis in water buffalo (Bubalus babalis, Linnaeus, 1758) in China. Vet Parasitol. 2009, 165 (1): 36-40.
He L, Feng H-H, Zhang Q-L, Zhang W-J, Khan MK, Hu M, Zhou Y-Q, Zhao J-L: Development and evaluation of real-time PCR assay for the detection of Babesia orientalis in water buffalo (Bubalus bubalis, Linnaeus, 1758). J Parasitol. 2011, 97 (6): 1166-1169. 10.1645/GE-2819.1.
Katoh K, Frith MC: Adding unaligned sequences into an existing alignment using MAFFT and LAST. Bioinformatics. 2012, 28 (23): 3144-3146. 10.1093/bioinformatics/bts578.
Katoh K, Standley DM: MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013, 30 (4): 772-780. 10.1093/molbev/mst010.
Carver T, Harris SR, Berriman M, Parkhill J, McQuillan JA: Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics. 2012, 28 (4): 464-469. 10.1093/bioinformatics/btr703.
Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream M-A, Barrell B: Artemis: sequence visualization and annotation. Bioinformatics. 2000, 16 (10): 944-945. 10.1093/bioinformatics/16.10.944.
Katoh K, Misawa K, Kuma KI, Miyata T: MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30 (14): 3059-3066. 10.1093/nar/gkf436.
Hall TA: BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Oxford University Press. 1999, 41: 95-98.
Brunk CF, Lee LC, Tran AB, Li J: Complete sequence of the mitochondrial genome of Tetrahymena thermophila and comparative methods for identifying highly divergent genes. Nucleic Acids Res. 2003, 31 (6): 1673-1682. 10.1093/nar/gkg270.
Posada D: jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008, 25 (7): 1253-1256. 10.1093/molbev/msn083.
Swofford D: PAUP 4.0 b10: phylogenetic analysis using parsimony. 2002, Sunderland, MA, USA: Sinauer Associates
Huelsenbeck JP, Ronquist F: MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001, 17 (8): 754-755. 10.1093/bioinformatics/17.8.754.
Ronquist F, Huelsenbeck JP: MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003, 19 (12): 1572-1574. 10.1093/bioinformatics/btg180.
Tamura K, Dudley J, Nei M, Kumar S: MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007, 24 (8): 1596-1599. 10.1093/molbev/msm092.
Allsopp M, Cavalier-Smith T, DeWaal D, Allsopp B: Phylogeny and evolution of the piroplasms. Parasitology. 1994, 108: 147-147. 10.1017/S0031182000068232.
Acknowledgements
The study was partially supported by Special Fund for Agro-scientific Research in the Public Interest (201003060-01-06), China Postdoctoral Science Foundation Grant (2012 M511629) and the National Natural Science Foundation of China (30671575).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All the authors contributed significantly to this study. LH and JLZ designed the experiments, carried out field and laboratory experiments, interpreted the results, and wrote the manuscript. YZ, QLZ, WJZ and HHF participated in cloning experiments and sequence analysis. MKK, MH and YQZ carried out editing and also helped in writing the manuscript. All the authors of the manuscript read and approved the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
He, L., Zhang, Y., Zhang, QL. et al. Mitochondrial genome of Babesia orientalis, apicomplexan parasite of water buffalo (Bubalus babalis, Linnaeus, 1758) endemic in China. Parasites Vectors 7, 82 (2014). https://doi.org/10.1186/1756-3305-7-82
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-3305-7-82