
Abstract
Background
Nematodirus spp. are among the most common nematodes of ruminants worldwide. N. oiratianus and N. spathiger are distributed worldwide as highly prevalent gastrointestinal nematodes, which cause emerging health problems and economic losses. Accurate identification of Nematodirus species is essential to develop effective control strategies for Nematodirus infection in ruminants. Mitochondrial DNA (mtDNA) could provide powerful genetic markers for identifying these closely related species and resolving phylogenetic relationships at different taxonomic levels.
Methods
In the present study, the complete mitochondrial (mt) genomes of N. oiratianus and N. spathiger from small ruminants in China were obtained using Long-range PCR and sequencing.
Results
The complete mt genomes of N. oiratianus and N. spathiger were 13,765 bp and 13,519 bp in length, respectively. Both mt genomes were circular and consisted of 36 genes, including 12 genes encoding proteins, 2 genes encoding rRNA, and 22 genes encoding tRNA. Phylogenetic analyses based on the concatenated amino acid sequence data of all 12 protein-coding genes by Bayesian inference (BI), Maximum likelihood (ML) and Maximum parsimony (MP) showed that the two Nematodirus species (Molineidae) were closely related to Dictyocaulidae.
Conclusions
The availability of the complete mtDNA sequences of N. oiratianus and N. spathiger not only provides new mtDNA sources for a better understanding of nematode mt genomics and phylogeny, but also provides novel and useful genetic markers for studying diagnosis, population genetics and molecular epidemiology of Nematodirus spp. in small ruminants.
Similar content being viewed by others


Background
Nematodirus spp. are among the most common nematodes of ruminants and more than 45 species have been described in the genus Nematodirus Ransom, 1907 [1, 2]. Of these, Nematodirus oiratianus and N. spathiger are widely distributed as highly prevalent gastrointestinal nematodes, mainly inhabiting the small intestines of sheep and goats [3–5]. Although mild, or no symptoms are usually presented during Nematodirus infection in adult ruminants, growth retardation and emaciation of the growing host during infection may result in economic losses. During winter, Nematodirus eggs are able to develop to the gastrula stage within two weeks and then to infective larvae after another 4 weeks, and are strongly resistant to adverse environmental conditions [6]. These larvae therefore represent a potent source of infection during spring leading to high numbers of infected ruminants early in the growing season [6, 7].
Traditionally, Nematodirus species have been identified solely on morphological features of the adults, including characteristics of the spicule tips and copulatory bursae [8]. However, such criteria are often insufficient for specific identification and differentiation of Nematodirus species, especially for eggs, larvae and females [9, 10]. Due to the limitations of morphological approaches, various molecular methods have been used widely for the identification and differentiation of Nematodirus species. The internal transcribed spacer (ITS) of nuclear ribosomal DNA (rDNA) has previously been described as a useful marker for the identification and differentiation of Molineid nematodes [11–13]. At least eight Nematodirus species, including N. battus, N. davtiani alpinus, N. europaeus, N. filicollis, N. helvetianus, N. oiratianus, N. spathiger and N. rupicaprae, could be identified by their ITS rDNA sequences [11–13]. Compared to nuclear rDNA, mitochondrial (mt) DNA (mtDNA) is more reliable for identifying closely related species, particularly cryptic species, since the mtDNA sequences accumulate nucleotide substitutions much more quickly than ITS rDNA [14]. Indeed, various studies have indicated that mt genome sequences provide powerful genetic markers in resolving phylogenetic relationships at different taxonomic levels, particularly when concatenated protein-coding sequences are used for phylogenetic analysis [15–22].
Based on recent progress in Long-range PCR-coupled sequencing and bioinformatic methods [23, 24], the objectives of the present study were to sequence and compare the complete mt genomes of N. oiratianus and N. spathiger. We also assessed phylogenetic relationships of the two Nematodirus species with a range of other Trichostrongyloid nematodes using complete, inferred mt protein sequence data sets.
Methods
Ethics statement
The performance of this study was strictly according to the recommendations of the Guide for the Care and Use of Laboratory Animals of the Ministry of Health, China, and our protocol was reviewed and approved by the Research Ethics Committee of Northwest A&F University.
Parasites
All the nematode samples were collected from animals with the permission of the Laboratory of Veterinary Parasitology of Northwest A&F University, with no specific permits being required by the authority for the sample collection.
Genomic DNA extraction
Female adults of N. oiratianus (Code: YLF2) and N. spathiger (JYF2) were obtained from the small intestines of naturally-infected goats and sheep, respectively, in Shaanxi Province, China. Adult worms from each host were washed separately in physiological saline, fixed in 70% (v/v) ethanol and stored at -20°C until further study. Nematodirus species were firstly identified morphologically according to characteristics of the spicule tips and copulatory bursae and distribution of bosses on the internal surfaces of the bursae [5, 8]. For each species, individual males were identified on the basis of bursal form, number of cuticular ridges and spicule morphology [25].
The Nematodirus species were further identified using a molecular method based on ITS rDNA. Total genomic DNA was isolated separately from individual worms of each species by proteinase K treatment, column-purification (TIANamp Genomic DNA Purification System, TIANGEN, China) and elution into 40 μl H2O according to the manufacturer’s recommendations. The region spanning ITS-1, 5.8S and ITS-2 rDNA was amplified from each individual using universal primers NC5 and NC2 [26] and sequenced directly. Phylogenetic analyses based on the ITS-2 rDNA sequences, using Maximum parsimony (MP) and Maximum likelihood (ML) methods, were used to further determine the Nematodirus species used in this study.
Long-range PCR-based sequencing of mtDNA
Using primers designed against relatively conserved regions within the cox 1, rrn L and nad 1 regions (Additional file 1) [27], the complete mt genome was amplified from total genomic DNA (from an individual worm) as four overlapping fragments between rrn L and nad 1, nad 1, nad 1 and cox 1, and cox 1 and rrn L (Additional file 1), respectively. Each fragment was amplified by long-range PCR using LA TAQ polymerase (TAKARA, China), following the manufacturer’s recommendations. The cycling conditions used were 92°C for 2 min (initial denaturation), then 92°C for 10 s (denaturation), 45°C for 30 s (annealing), and 60°C for 8 min (extension) for 9 cycles, followed by 92°C for 10 s, 45°C for 30 s, and 60°C for 9 min for 25 cycles, and a final extension at 60°C for 10 min. Each amplicon was represented by a single band in a 1.0% (w/v) agarose gel, following electrophoresis and ethidium-bromide staining. The amplicon was column-purified and then sequenced using a primer walking strategy [23].
Sequence annotation
Sequences were assembled manually and aligned against the complete mt genome sequences of other nematodes (available in GenBank) using the computer program MAFFT 7 [28] to infer gene boundaries. The open-reading frames (ORFs) were predicted by the Open Reading Frame Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) using the invertebrate mitochondrial code and subsequently compared with those of Trichostrongylus axei and Trichostrongylus vitrinus[29]. Each gene was translated into amino acid sequence using the invertebrate mitochondrial genetic code in MEGA 5 [30], and aligned based on its amino acid sequence using default settings. The alignment was back-translated into the corresponding nucleotide sequences. The translation initiation and termination codons were identified to avoid gene overlap and to optimize the similarity between the gene lengths of closely-related nematode mitochondrial genomes. Codon usages were examined based on the relationships between the nucleotide composition of codon families and amino acid occurrence, for which codons are partitioned into AT-rich codons, GC-rich codons and unbiased codons. The secondary structures of 22 tRNA genes were predicted using tRNAscan-SE [31] and/or manual adjustment [32], and the rRNA genes were predicted by comparison with those of closely-related nematodes (T. axei and T. vitrinus[29]) and their secondary structures.
Phylogenetic analyses
Amino acid sequences inferred from the 12 protein-coding genes were concatenated into a single alignment, and then aligned with those of eight other Trichostrongyloid nematodes (Cooperia oncophora, NC_004806 [33]; Haemonchus contortus, NC_010383 [24]; T. axei, NC_013824 [29]; T. vitrinus, NC_013807 [29]; Teladorsagia circumcincta, NC_013827 [29]; Mecistocirrus digitatus, NC_013848 [29]; Dictyocaulus viviparus, NC_019810 [34]; Dictyocaulus eckerti, NC_019809) [34]), using the Strongyloid nematode Oesophagostomum quadrispinulatum (NC_014181) as the outgroup [35]. Any regions of ambiguous alignment were excluded using Gblocks online server [36] (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) using the options for less stringent selection. Phylogenetic analyses were conducted using three methods: Bayesian inference (BI), Maximum parsimony (MP) and Maximum likelihood (ML). The MtArt + G + F model of amino acid evolution was selected as the most suitable model of evolution by ProtTest 2.4 [37] based on the Akaike information criterion (AIC). As the MtArt model is not implemented in the current version of MrBayes, an alternative model, MtREV, was used in BI and four chains (three heated and one cold) were run simultaneously for the Monte Carlo Markov Chain. Two independent runs were performed for 1,000,000 metropolis-coupled MCMC generations, sampling a tree every 100 generation in MrBayes 3.1.1 [38]; the first 2,500 trees represented burn-in and the remaining trees were used to calculate Bayesian posterior probabilities (Bpp). MP analysis was conducted using PAUP 4.0 Beta 10 program [39], with indels treated as missing character states; 1,000 random additional searches were performed using TBR. Bootstrap frequency (Bf) was calculated using 1,000 bootstrap replicates, and 100 random taxon additions in PAUP. ML analysis was performed using PhyML 3.0 [40]. Bf was calculated using 100 bootstrap replicates. Phylograms were drawn using the program Tree View v.1.65 [41].
Results and discussion
Both nematode species have cephalic vesicles, two sets of reproductive organs, a single distribution of large black eggs, and a conical end with a transparent thin spine. Usually, species identification mostly depends on male morphological features, but in this study, identification of the parasites was mainly dependent on ITS rDNA sequences as all worms used herein were female [8, 42]. The region spanning ITS-1, 5.8S and ITS-2 rDNA was amplified from each individual worm using universal primers NC5 and NC2 [26] and sequenced directly. The ITS-1 rDNA sequences (GenBank accession nos. KC580735 and KC580745, respectively) of the N. oiratianus and N. spathiger samples had 96% and 99% identity to those of N. oiratianus and N. spathiger in public databases (HQ389233 and AF194144, respectively). The ITS-2 rDNA sequences (KC580735 and KC580745, respectively) of the N. oiratianus and N. spathiger samples had 94% and 100% identity to those of N. oiratianus and N. spathiger in public databases (HQ389233 and KC998746, respectively) [2]. Phylogenetic analysis based on the ITS-2 rDNA sequences, using Maximum parsimony (MP) and Maximum likelihood (ML), further showed that the Nematodirus isolates represented N. oiratianus and N. spathiger, respectively (Additional file 2).
The respective lengths of the four overlapping fragments amplified by long-PCR covering the entire mt genomes of N. oiratianus and N. spathiger were 4765 bp and 4741 bp for rrn L-nad 1, 511 bp and 510 bp for partial nad 1, 7084 bp and 7060 bp for nad 1-cox 1, and 2330 bp and 2320 bp for cox 1-rrn L, respectively. After sequence splicing, the complete mt genomes of N. oiratianus and N. spathiger (KF573750 and KF573749) were 13,765 bp and 13,519 bp in length, respectively (Figure 1). Both mt genomes contained 12 protein-coding genes (cox 1-3, nad 1-6, nad 4L, atp 6 and cyt b), 22 transfer RNA genes, two ribosomal RNA genes and two non-coding regions, but lacked an atp 8 gene (Table 1). These circular genomes are typical mt genomes of Chromadorea nematodes, such as Ascaris suum[15], Enterobius vermicularis[43], Oesophagostomum spp.[44] and Spirocerca lupi[45]. All mt genes were transcribed in the same direction, with same gene order as Gene arrangement 3 (GA3): nad 6 > nad 4L > tRNA-Trp (W) > tRNA-Glu (E) > rrn S > tRNA-SerUCN (S2) > tRNA-Asn (N) > tRNA-Tyr (Y) > nad 1 > atp 6 > tRNA-Lys (K) > tRNA-LeuUUR (L2) > tRNA-SerAGN (S1) > nad 2 > tRNA-Ile (I) > tRNA-Arg (R) > tRNA-Gln (Q) > tRNA-Phe (F) > cyt b > tRNA-LeuCUN (L1) > cox 3 > tRNA-Thr (T) > nad 4 > Non-coding region (NC1) > cox 1 > tRNA-Cys (C) > tRNA-Met (M) > tRNA-Asp (D) > tRNA-Gly (G) > cox 2 > tRNA-His (H) > rrn L > nad 3 > nad 5 > tRNA-Ala (A) > Non-coding region (NC2) > tRNA-Pro (P) > tRNA-Val (V) (Figure 1, Table 1).The nucleotide compositions of the completemt DNA sequences for N. oiratianus and N. spathiger are biased toward T and A, with T being the most favored nucleotide and C as the least favored. The contents of A + T were 76.14% (T = 47.69%; A = 28.46%; G = 16.8%; C = 7.06%) and 75.04% (T = 48.29%; A = 26.75%; G = 17.69%; C = 7.27%) for N. oiratianus and N. spathiger, respectively. The magnitude of sequence difference across the complete mt genome was 16.29% between N. oiratianus and N. spathiger.

Structure of the mitochondrial genomes of Nematodirus oiratianus and Nematodirus spathiger. Genes are designated according to standard nomenclature, except for the 22 tRNA genes, which are designated using one-letter amino acid codes, with numerals differentiating each of the two leucine- and serine-specifying tRNAs (L1 and L2 for codon families CUN and UUR, respectively; S1 and S2 for codon families AGN and UCN, respectively). “NC1” refers to a small non-coding region; “NC2” refers to a large non-coding region.
All protein-coding genes used ATA, ATT, ATG or TTG as their initiation codons, and TAA or TAG as their termination codons. Incomplete termination codons (TA and T) were also present (Table 1), which was consistent with studies of some other nematodes [15, 16, 44]. Excluding the termination codons, a total of 3419 and 3416 amino acids were encoded by the N. oiratianus and N. spathiger mt genomes, respectively. The codon usages for the two mt genomes are listed in Table 2. Codons composed of A and T were predominantly used, reflecting the high content of A + T in the complete mt genomes of the two Nematodirus species. The most frequent usage of codon was TTT (Phenylalanine) for both two species, with frequencies of 12.61% and 12.88% for N. oiratianus and N. spathiger, respectively, followed by TTA (Leucine) in the two species (10.95% and 9.17%, respectively). Codon CGC (Arginine) was not used in the mt genomes of either species, and codon TGC (Cysteine) was not used in the mt genome of N. spathiger.
Twenty-two tRNA genes were predicted from the mt genomes of N. oiratianus and N. spathiger and varied from 51 to 63 bp in length. The secondary structures predicted for the genes were similar to those of T. axei and T. vitrinus[29]. Twenty tRNA genes (excluding two tRNA-Ser) had a predicted secondary structure with a 3-5 bp DHU arm and a DHU loop of 7-9 bases, in which the variable TψC arm and loop were replaced by a “TV-replacement loop” of 8-10 bases. As in almost all other nematode mtDNA sequences [29], the tRNA-Ser genes of both Nematodirus mt genomes are equipped with a TψC arm and loop but lacked the DHU arm and loop, consisting of a 6-8 bp TψC arm, TψC loop of 4-6 bases and a variable loop of 4 bases.
The rrn L and rrn S genes of N. oiratianus and N. spathiger were identified by comparison with those of T. axei and T. vitrinus[29]. The rrn L gene was located between tRNA-His and nad 3, and rrn S was located between tRNA-Glu and tRNA-SerUCN (Table 1). The length of rrn L was 962 bp for N. oiratianus and 958 bp for N. spathiger, with 5.2% of sequence difference. The length of rrn S was 696 bp for N. oiratianus and 695 bp for N. spathiger, with 9.05% of sequence difference. The A + T contents of rrn L for N. oiratianus and N. spathiger were 80.98% and 79.85%, respectively. The A + T contents of rrn S for N. oiratianus and N. spathiger were 76.29% and 77.70%, respectively.
Two non-coding regions (designated as NC1 and NC2) were inferred in the mt genomes of both N. oiratianus and N. spathiger. For both mt genomes, the AT-rich regions were located between nad 4 and cox 1, tRNA-Ala and tRNA-Pro (Figure 1; Table 1), with A + T contents of 81.25% for N. oiratianus and 81.07% for N. spathiger.
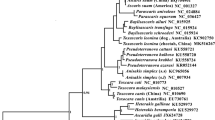
Phylogenetic analysis (Figure 2) based on concatenated amino acid sequence data of all 12 mt proteins showed that nematodes in families Molineidae and Dictyocaulidae grouped together, and parasites in families Trichostrongylidae (T. vitrinus, T. axei and T. circumcincta), Cooperiidae (Cooperia oncophora) and Haemonchidae (M. digitatus and H. contortus) clustered together. N. oiratianus and N. spathiger samples in the present study clustered together and were highly closely related to the family Dictyocaulidae with high clade support (Bpp = 1.00; Bf = 100), confirming the results of previous studies using the morphological features and ITS rDNA as the genetic marker [2, 9, 42]. However, amino acid sequence-based inferences of the relationship between the two Nematodirus species gave slightly different results to those using ITS rDNA data in which they clustered in sister clades with high support, possibly due to the different taxa inclusions and types of analysis performed [2, 11–13, 46].

Genetic relationships of Nematodirus nematodes with other selected Trichostrongyloid nematodes inferred by Bayesian inference (BI) based on mitochondrial sequence data. Phylogenetic analyses based on the concatenated amino acid sequence data representing 12 protein-coding genes were conducted using Bayesian inference (BI), Maximum likelihood (ML) and Maximum parsimony (MP), with Oesophagostomum quadrispinulatum (NC_014181) as the outgroup. The scale bar indicated Posterior Probability.
Molecular analyses have provided new insights into population structure and species identification of parasites [15, 16, 34, 44]. In the present study, we utilized long-range PCR-coupled sequencing and bioinformatic methods [23] to characterize the complete mt genomes of two Nematodirus species from small ruminants and to assess the phylogenetic relationships of these Nematodirus species in relation to representative Trichostrongyloid nematodes. Given that there are no morphological characteristics to allow the explicit specific identification and differentiation of Nematodirus species (Nematoda: Molineidae) at some developmental stages, such as larvae and egg [9, 10], the use of mt DNA markers to investigate genetic composition in these species is a significant advance. To date, more than 45 Nematodirus species have been described based on their morphological features [1, 2], however, no information is yet available about their mt genomes. Although previous studies have indicated that ITS rDNA provides a useful marker for identification and differentiation of Nematodirus species [2, 11, 13], mtDNA in nematodes is usually more variable in sequences within a species than the nuclear ribosomal DNA [14].
The characterization of the mt genomes of N. oiratianus and N. spathiger also allows a reassessment of the systematic relationships of nematodes within the Trichostrongyloidea using mt proteomic datasets. For many years, there has been considerable debate surrounding the systematics of members within the Trichostrongyloidea (including Haemonchidae, Cooperiidae, Trichostrongylidae, Molineidae and Dictyocaulidae) [47]. Given the controversy, concatenated mt proteomic/genomic datasets might be applied effectively to genetically characterize and compare Trichostrongyloid nematodes. The current work has provided insight into the phylogenetic relationships among Trichostrongyloid nematodes, however, many species of Trichostrongyloid nematodes are not represented. Expanding taxon sampling is therefore necessary for future phylogenetic studies of a wide range of Trichostrongyloidea using mt proteomic/genomic datasets.
Conclusions
The present study determined the complete mt genome sequences of N. oiratianus and N. spathiger. The mtDNA data presented here not only provide new mtDNA resources for a better understanding of nematode mt genomics and phylogeny, but also provide novel and useful genetic markers for studying diagnosis, population genetics, and molecular epidemiology of Nematodirus spp. in small ruminants.
References
Kulmamatov A: Ovidovom sostave roda Nematodirus Ransom, 1907. Materialy Nauchnykh Konferentsii Vsesouznogo Obshchestva Gel’mintologev. 1974, 26: 137-140.
Nadler SA, Hoberg EP, Hudspeth DS, Rickard LG: Relationships of Nematodirus species and Nematodirus battus isolates (Nematoda: Trichostrongyloidea) based on nuclear ribosomal DNA sequences. J Parasitol. 2000, 86: 588-601.
Tariq KA, Chishti MZ, Ahmad F, Shawl AS: Epidemiology of gastrointestinal nematodes of sheep managed under traditional husbandry system in Kashmir valley. Vet Parasitol. 2008, 158: 138-143.
Domke AV, Chartier C, Gjerde B, Leine N, Vatn S, Stuen S: Prevalence of gastrointestinal helminths, lungworms and liver fluke in sheep and goats in Norway. Vet Parasitol. 2013, 194: 40-48.
Becklund WW, Walker ML: Nematodirus of domestic sheep, Ovisaries, in the United States with a key to the species. J Parasitol. 1967, 53: 777-781.
Zheng RK, Zhao SH, Gao HY, Geng WH, Hu M: The ecological study of larva of Nematodirus spp. in grazing sheep. Inner Mongolian J Anim Sci Prod. 1997, 1: 9-14. (in Chinese)
Wang JF, Cao RF: The survey of gastrointestinal nematode infection in goats in Laiyang, Shangdong province. Shangdong J Anim Sci Vet Med. 2008, 29: 42-(in Chinese)
Lichtenfels JR, Pilitt PA: Cuticular ridge patterns of Nematodirus (nematoda: trichostrongyloidea) parasitic in domestic ruminants of North America, with a Key to species. J Helminthol. 1983, 50: 261-274.
Newton LA, Chilton NB, Beveridge I, Hoste H, Nansen P, Gasser RB: Genetic markers for strongylid nematodes of livestock defined by PCR-based restriction analysis of spacer rDNA. Acta Trop. 1998, 69: 1-15.
Gasser RB, Bott NJ, Chilton NB, Hunt P, Beveridge I: Toward practical, DNA-based diagnostic methods for parasitic nematodes of livestock — bionomic and biotechnological implications. Biotechnol Adv. 2008, 26: 325-334.
Newton LA, Chilton NB, Beveridge I, Gasser RB: Differences in the second internal transcribed spacer of four species of Nematodirus (Nematoda: Molineidae). Int J Parasitol. 1998, 28: 337-341.
Gasser RB, Rossi L, Zhu X: Identification of Nematodirus species (Nematoda: Molineidae) from wild ruminants in Italy using ribosomal DNA markers. Int J Parasitol. 1999, 29: 1809-1817.
Audebert F, Durette-Desset MC, Chilton NB: Internal transcribed spacer rDNA can be used to infer the phylogenetic relationships of species within the genus Nematodirus (Nematoda: molineoidea). Int J Parasitol. 2000, 30: 187-191.
Blouin MS: Molecular prospecting for cryptic species of nematodes: mitochondrial DNA versus internal transcribed spacer. Int J Parasitol. 2002, 32: 527-531.
Liu GH, Wu CY, Song HQ, Wei SJ, Xu MJ, Lin RQ, Zhao GH, Huang SY, Zhu XQ: Comparative analyses of the complete mitochondrial genomes of Ascaris lumbricoides and Ascaris suum from humans and pigs. Gene. 2012, 492: 110-116.
Liu GH, Gasser RB, Su A, Nejsum P, Peng L, Lin RQ, Li MW, Xu MJ, Zhu XQ: Clear genetic distinctiveness between human- and pig-derived Trichuris based on analyses of mitochondrial datasets. PLoS Negl Trop Dis. 2012, 6: e1539-
Liu GH, Shao R, Li JY, Zhou DH, Li H, Zhu XQ: The complete mitochondrial genomes of three parasitic nematodes of birds: a unique gene order and insights into nematode phylogeny. BMC Genomics. 2012, 14: 414-
Liu GH, Chen F, Chen YZ, Song HQ, Lin RQ, Zhou DH, Zhu XQ: Complete mitochondrial genome sequence data provides genetic evidence that the brown dog tick Rhipicephalus sanguineus (Acari: Ixodidae) represents a species complex. Int J Biol Sci. 2013, 9: 361-369.
Plazzi F, Ribani A, Passamonti M: The complete mitochondrial genome of Solemya velum (Mollusca: Bivalvia) and its relationships with Conchifera. BMC Genomics. 2013, 14: 409-
Li H, Liu H, Song F, Shi A, Zhou X, Cai W: Comparative mitogenomic analysis of damsel bugs representing three tribes in the family Nabidae (Insecta: Hemiptera). PLoS One. 2012, 7: e45925-
Wan X, Kim MI, Kim MJ, Kim I: Complete mitochondrial genome of the free-living earwig, Challia fletcheri (Dermaptera: Pygidicranidae) and phylogeny of Polyneoptera. PLoS One. 2012, 7: e42056-
Chai HN, Du YZ, Zhai BP: Characterization of the complete mitochondrial genomes of Cnaphalocrocis medinalis and Chilo suppressalis (Lepidoptera: Pyralidae). Int J Biol Sci. 2012, 8: 561-579.
Hu M, Jex AR, Campbell BE, Gasser RB: Long PCR amplification of the entire mitochondrial genome from individual helminths for direct sequencing. Nat Protoc. 2007, 2: 2339-2344.
Jex AR, Hu M, Littlewood DT, Waeschenbach A, Gasser RB: Using 454 technology for long-PCR based sequencing of the complete mitochondrial genome from single Haemonchus contortus (Nematoda). BMC Genomics. 2008, 9: 11-
Stringfellow F: Bursal bosses as a diagnostic character in nematodirus of domestic sheep, Ovisaries, in the United States. J Parasitol. 1968, 54: 891-895.
Zhu X, Gasser RB, Podolska M, Chilton NB: Characterization of anisakid nematodes with zoonotic potential by nuclear ribosomal DNA sequences. Int J Parasitol. 1998, 28: 1911-1921.
Hu M, Chilton NB, Gasser RB: Long PCR-based amplification of the entire mitochondrial genome from single parasitic nematodes. Mol Cell Probes. 2002, 16: 261-267.
Katoh K, Standley DM: MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013, 30: 772-780.
Jex AR, Hall RS, Littlewood DT, Gasser RB: An integrated pipeline for next-generation sequencing and annotation of mitochondrial genomes. Nucleic Acids Res. 2010, 38: 522-533.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 28: 2731-2739.
Lowe TM, Eddy SR: tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25: 955-964.
Hu M, Chilton NB, Gasser RB: The mitochondrial genomes of the human hookworms, Ancylostoma duodenale and Necator americanus (Nematoda: Secernentea). Int J Parasitol. 2002, 32: 145-158.
Van der Veer M, de Vries E: A single nucleotide polymorphism map of the mitochondrial genome of the parasitic nematode Cooperia oncophora. Parasitology. 2004, 128: 421-431.
Gasser RB, Jabbar A, Mohandas N, Höglund J, Hall RS, Littlewood DT, Jex AR: Assessment of the genetic relationship between Dictyocaulus species from Bos taurus and Cervus elaphus using complete mitochondrial genomic datasets. Parasit Vectors. 2012, 5: 241-
Lin RQ, Liu GH, Hu M, Song HQ, Wu XY, Li MW, Zhang Y, Zou FC, Zhu XQ: Oesophagostomum dentatum and Oesophagostomum quadrispinulatum: characterization of the complete mitochondrial genome sequences of the two pig nodule worms. Exp Parasitol. 2012, 131: 1-7.
Talavera G, Castresana J: Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007, 56: 564-577.
Abascal F, Zardoya R, Posada D: ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005, 21: 2104-2105.
Ronquist F, Huelsenbeck JP: MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003, 19: 1572-1574.
Swofford DL: PAUP*: Phylogenetic analysis using parsimony (and other methods). 2002, Sunderland, MA: Sinauer Associates
Guindon S, Gascuel O: A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003, 52: 696-704.
Page RD: TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci. 1996, 12: 357-358.
Liang JS, Ma ZQ, Lin XM: Nematodirus hsui, a new nematode from the sheep of Kansu province. Acta Vet Et Zootechnica Sinica. 1958, 3: 143-147. (in Chinese)
Kang S, Sultana T, Eom KS, Park YC, Soonthornpong N, Nadler SA, Park JK: The mitochondrial genome sequence of Enterobius vermicularis (Nematoda: Oxyurida)--an idiosyncratic gene order and phylogenetic information for chromadorean nematodes. Gene. 2009, 429: 87-97.
Zhao GH, Hu B, Cheng WY, Jia YQ, Li HM, Yu SK, Liu GH: The complete mitochondrial genomes of Oesophagostomum asperum and Oesophagostomum columbianum in small ruminants. Infect Genet Evol. 2013, 19: 205-211.
Liu GH, Wang Y, Song HQ, Li MW, Ai L, Yu XL, Zhu XQ: Characterization of the complete mitochondrial genome of Spirocerca lupi: sequence, gene organization and phylogenetic implications. Parasit Vectors. 2013, 6: 45-
Zhao GH, Jia YQ, Bian QQ, Nisbet AJ, Cheng WY, Liu Y, Fang YQ, Ma XT, Yu SK: Molecular approaches to differentiate three species of Nematodirus in sheep and goats from China based on internal transcribed spacer rDNA sequences. J Helminthol. 2013, 16: 1-8.
Nadler SA, Carreno RA, Mejía-Madrid H, Ullberg J, Pagan C, Houston R, Hugot JP: Molecular phylogeny of clade III nematodes reveals multiple origins of tissue parasitism. Parasitology. 2007, 134: 1421-1442.
Acknowledgements
This work was supported by grant from Fund of the “Special Fund for Agro-scientific Research in the Public Interest” (Grant No. 201303037). The authors thank Dr. Alasdair Nisbet of the Moredun Research Institute, UK, for help in editing the English in this manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
GHZ and GHL conceived and designed the study, and critically revised the manuscript. GHZ, YQJ, WYC, WZ and QQB performed the experiments, analyzed the data and drafted the manuscript. All authors read and approved the final manuscript.
Guang-Hui Zhao, Yan-Qing Jia contributed equally to this work.
Electronic supplementary material
13071_2014_1509_MOESM1_ESM.docx
Additional file 1: Sequences of primers used to amplify PCR fragments from Nematodirus oiratianus and N. spathiger .(DOCX 14 KB)
13071_2014_1509_MOESM2_ESM.tiff
Additional file 2: Phylogenetic relationships of Nematodirus spp. inferred by maximum parsimony (MP) and maximum likelihood (ML) analyses based on ITS-2 rDNA sequences. Ostertagia sp. is used as the outgroup. Posterior probabilities/bootstrap values (in percentage) above 50% from 1,000 pseudo-replicates are shown for the MP (the first value), and ML analyses (the second value). MP analysis was performed using PAUP* 4.0 Beta10 program with default parameters. ML analyses were performed using PhyML 3.0 with the GTR model. (TIFF 1 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Zhao, GH., Jia, YQ., Cheng, WY. et al. Characterization of the complete mitochondrial genomes of Nematodirus oiratianus and Nematodirus spathiger of small ruminants. Parasites Vectors 7, 319 (2014). https://doi.org/10.1186/1756-3305-7-319
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-3305-7-319