
Abstract
Background
Cutaneous leishmaniasis is an infectious disease caused by flagellate protozoa of the genus Leishmania. In Morocco, anthroponotic cutaneous leishmaniasis due to Leishmania tropica is considered as a public health problem, but its epidemiology has not been fully elucidated. The main objective of this study was to detect Leishmania infection in the vector, Phlebotomus sergenti and in human skin samples, in the El Hanchane locality, an emerging focus of cutaneous leishmaniasis in central Morocco.
Methods
A total of 643 sand flies were collected using CDC miniature light traps and identified morphologically. Leishmania species were characterized by ITS1 PCR-RFLP and ITS1-5.8S rRNA gene nested-PCR of samples from 123 females of Phlebotomus sergenti and 7 cutaneous leishmaniasis patients.
Results
The sand flies collected consisted of 9 species, 7 of which belonged to the genus Phlebotomus and two to the genus Sergentomyia. Phlebotomus sergenti was the most predominant (76.67%).
By ITS1 PCR-RFLP Leishmania tropica was found in three Phlebotomus sergenti females and four patients (4/7). Using nested PCR Leishmania tropica was identified in the same three Phlebotomus sergenti females and all the 7 patients. The sequencing of the nested PCR products recognized 7 haplotypes, of which 6 have never been described.
Conclusions
This is the first molecular detection and identification of Leishmania tropica in human skin samples and Phlebotomus sergenti in support of its vector status in El Hanchane. The finding of seven Leishmania tropica haplotypes underscores heterogeneity of this species at a high level in Morocco.
Similar content being viewed by others


Background
In Morocco, Leishmania major, Leishmania tropica and less frequently Leishmania infantum cause cutaneous leishmaniasis (CL). Zoonotic CL caused by L. major zymodeme MON-25 has been known since 1914 and is largely confined to the arid pre-Saharan regions [1]. In the last decades, outbreaks of CL have been reported repeatedly due to reactivation of old southern foci, amounting to several thousand cases [2]. Amongst the 3 clinically important Leishmania species, L. tropica has the largest geographic distribution in Morocco and is considered as a public health threat by the Ministry of Health. This anthroponotic form of CL was reported for the first time in 1989 [3]. There after hypoendemic rural foci were identified scattered around in the sub-arid area of central Morocco, caused by a great diversity of this species [4, 5]. Recently, CL caused by L. tropica emerged in several new foci in Northern Morocco. Indeed, in 1995, Taza province registered a rapid expansion of CL due to L. tropica MON-102 [6, 7]. In 2001, about 1600 people were also infected with L.tropica in Fes province [8]. Molecular examination of samples from skin lesions collected from different regions has shown that L. tropica is present in a large part of the kingdom, and for the first time even in regions previously known only as L. major CL foci [8].
The epidemiology of leishmaniasis caused by L.tropica in Morocco has not been fully elucidated. The disease is often described as being anthroponotic and most CL cases occur in and around densely populated cities [6, 8]. However, the low frequency of the disease in semi rural locations and the sudden occurrence of small outbreaks suggest that the disease may be zoonotic in some cases [6]. The rapid spread of the parasite, with the increasing number of cases begs epidemiological investigations.
Molecular methods are increasingly employed for diagnostic and epidemiological purposes in order to confirm Leishmania infection and to characterize the parasites at the species or genotype level in hosts and vectors. The detection of Leishmania parasites by PCR methods is highly specific and sensitive, with values reaching up to 100% [9]. Several techniques have been described, such as PCR-restriction fragment length polymorphism (RFLP), sequence analysis of multicopy genes and intergenic spacer regions (ITS), DNA fingerprinting and randomly amplified polymorphic DNA (RAPD) [10] and multilocus sequencing typing (MLST), which is a powerful technique for phylogenetic studies in Leishmania[11] and may substitute isoenzyme analysis. Accurate and sensitive diagnostic and identification procedures are required to distinguish Leishmania species/strains whose geographic distribution can overlap, which is crucial for adequate treatment and appropriate public health control measures. Based on a literature search, we selected two PCR methods shown to have sensitivity and specificity for detecting Leishmania: ITS1 PCR-RFLP and the nested PCR of ITS rDNA genes. Both methods were used to detect and identify the Leishmania parasite responsible for the recent cases of CL and their putative vector species in El Hanchane district, an emerging focus of CL in central Morocco.
Methods
Study area and sample collection
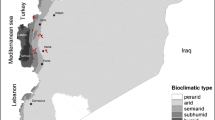
In this study, sand flies were caught in El Hanchane, an emerging focus of CL according to the local health authorities. Since the first case reported in 2006, there has been an average of12 CL cases per year up to 2010. El Hanchane is a semi-rural locality, situated at an altitude of 290–300 m above sea level (31°31’11” N, 9°26’02” W) (Figure 1).

Map of Morocco showing El Hanchane locality: the study area.
Sandflies were collected using CDC miniature light traps in domestic animal shelters and inside houses during 3 consecutive nights from July to September, 2011. The traps were placed approximately 1.5 meters above ground and set in the late afternoon for collection the following morning. The sand flies were then placed in 1.5 ml Eppendorf tubes and kept frozen at −80°C.
Tissue samples were taken from the edge of the lesions in 7 suspected CL patients at the health center in El Hanchane during 2011. Lesion smears were fixed with absolute methanol, and then stained with Giemsa for CL diagnosis. The whole slides were analyzed twice with a 100X immersion objective.
Microscopically confirmed CL patients received free intra lesional injections of meglumine antimoniate (Glucantime®; Sanofi -Aventis, Bridgewater, NJ, USA) according to the protocol of the Moroccan Ministry of Health [2].
Informed written consent was obtained from patients or from parents if the patient was a minor. Approval for the study was provided by the Ethical Committee of Institut Pasteur of Morocco.
Sand fly identification
Sand fly specimens were washed in sterile distilled water, the head and genitalia of individual sand flies were removed, and the remainder of the body was stored in sterile Eppendorf microtubes for DNA extraction. Specimens were mounted on microscope slides with the solution of Marc-André (30 g of chloral hydrate, 30 mL glacial acetic acid and 30 mL of distilled water) and identified using the taxonomic keys of Moroccan sand flies, particularly those of Boussaa [12].
Extraction of DNA from sand flies and stained smears of human CL lesions
The total DNAs were isolated from the positive stained smears of 7 CL lesions and from 123 unengorged female P. sergenti specimens using the phenol-chloroform method. Then DNA samples were purified using the Qiagen kit (QIAquick PCR Purification Kit, Germany), according to the manufacturer’s instructions and quantitatively determined using NanoDrop (Thermo Scientific) before dilution to a final concentration of 50 ng/μL.
ITS1 PCR-RFLP of Leishmania species
The DNA samples from P. sergenti females and CL patient lesions were examined for the Leishmania-specific ribosomal internal transcribed spacer 1 region (ITS1) by PCR amplification using the primer pair L5.8S and LITSR followed by RFLP (restriction fragment length polymorphism) analysis as described by Schonian et al.[13].
The cycling conditions were 94°C for 2 min followed by 32 amplification cycles, each consisting of three steps: denaturation at 94°C for 20 s, annealing at 53°C for 30 s and extension at 72°C for 1 min, followed by a final extension at 72°C for 6 min in the thermocycler (S1000™ Thermal Cycler, Bio-Rad).
PCR products were digested with the restriction endonuclease Hae III for 2 h at 37°C. Restriction fragments were separated by electrophoresis on a 2% agarose gel and compared with those of WHO reference strains of L. major (MHOM/SU/73/5ASKH), L. tropica (MHOM/SU/74/K27) and L. infantum (MHOM/TN/80/IPT1).
Nested PCR for amplifying ITS1-5.8S rDNA gene of Leishmania species
All female P. sergenti and CL patient lesions were further screened for infection with Leishmania species by nested PCR of the DNA samples for the presence of its ITS-rDNA gene. A two-stage PCR was carried out in two separate tubes [14, 15]. The first-stage PCR used the forward primer IR1 with the reverse primer IR2 [16], and the second-stage PCR used the nested forward primer ITS1F with the nested reverse primer ITS2R4 [15].
The first amplification reaction in a total volume of 20 μL, contained 1X PCR Rxn Buffer (Invitrogen), 1.5 mM MgCl2, 60 μM of each dNTP, 1 μM primer IR1, 1 μM primer IR2, 1 unit Taq DNA polymerase (Invitrogen), and 50 ng template DNA. The mixture was incubated in thermocycler (S1000™ Thermal Cycler, Bio-Rad) under the following conditions: an initial denaturation at 94°C for 3 min, followed by 37 cycles each consisting of three steps: 30 s at 94°C (denaturation), 30 s at 58°C (annealing) and 90 s at 72°C (extension). After the last cycle, the extension step was continued for a further 10 min.
The products obtained were subsequently subjected to nested PCR amplification as follows: the reaction mixtures of 20 μL each contained the same reagents as the first-stage PCR, except that the primers were now primer ITS1F and primer ITS2R4, and the target DNA was 1 μL of the first-stage PCR reaction products. The reaction was programmed, as described for the first-stage PCR. Cross-contamination was monitored by negative controls for sample DNAs and solutions of all PCR reagents used.
The final PCR products of 400 bp were purified using the Exonuclease I/Shrimp Alkaline Phosphatase (GE Healthcare, US) before sequencing by using BigDye Terminator version 3.1 Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA) and an ABI PRISM 3130 DNA automated sequencer (Applied Biosystems). Sequencing data were analyzed using SeqScape v.2.5 software (Applied Biosystems). Sequences were aligned using the multiple alignment program MEGA5 and GENtle v.1.9.4 software. A phylogenetic tree was constructed by using the Neighbor-Joining method in agreement with Kimura 2-parameter model with uniform rates for transitions and transversions. Bootstrap replicates were performed to estimate the node reliability, and values were obtained from 1,000 randomly selected samples of the aligned sequence data. Sequences were compared with entries retrieved from Genbank.
Results
Morphological identification of sand flies
A total of 643 sand flies were trapped using CDC miniature light traps. All sand flies were morphologically identified using a standard key of Moroccan sand flies modified by Boussaa [12]. Nine species, including seven in the genus Phlebotomus and two in the genus Sergentomyia were identified. P.sergenti was the most prevalent species with 493 specimens (76.67%) (273 females/220 males), followed by P. longicuspis (11.51%) (Table 1).
Leishmania genotyping
The first step of this genotyping was to identify the Leishmania species from sandflies and humans: for this purpose we used the ITS1 PCR-RFLP. The second step by nested PCR will allow enhancing the sensitivity of our diagnosis. Indeed we demonstrated that the nested PCR allowed the detection of 1 pg/μL DNA, whereas the ITS1-PCR detected 10 pg/μL DNA extracted from L.tropica reference strain (MHOM/SU/74/K27) (Figure 2).

Sensitivity analysis of nested PCR and ITS1-PCR. L. tropica DNA was diluted 10-fold starting from 103 pg and tested for amplification. The L. tropica nested PCR amplify the 400 bp product (A), and ITS1-PCR amplify about 320 bp product (B). N: Negative control. 100 bp weight molecular (WM) (Bioline).
Leishmania species identification by ITS1 PCR-RFLP
Three out of 123 female P. sergenti tested for Leishmania infection were positive using the ITS1-PCR, the positive specimens produced a band of 320-bp. The digestion of the PCR product with endonuclease Hae III revealed the profiles of L. tropica (185 and 57/53 bp) for the 3 specimens.
Using ITS1-PCR followed by the digestion of amplicons, Leishmania parasites were successfully identified for 4 of the 7 positive stained slides of human CL lesions, the RFLP profiles are identical for the 4 samples, consisting of two bands (185 and 57/53 bp), identical to that of the L. tropica (MHOM/SU/74/K27).
Leishmania infections of P. sergenti and patients identified by nested PCR of ITS1-5.8S rDNA gene
Only the three female P. sergenti positive for Leishmania by ITS1-PCR were also found positive by the nested PCR of ITS rDNA gene, showing a band of approximately 400 bp; nested PCR of the seven positive CL patients examined for ITS-rDNA gene all produced a single band of 400 bp (Figure 3).

Nested-PCR of ITS1-5.8S rDNA gene of Leishmania DNA from human samples and P. sergenti sand fly. Lines 1–7: human samples; line 8–10: P. sergenti females. Reference strains: L. infantum (MHOM/TN/80/IPT1), L. major (MHOM/SU/73/5ASKH) and L. tropica (MHOM/SU/74/K27); N: Negative control; 1 kb weight molecular (WM) (Invitrogen).
Sequencing and phylogenetic analysis of haplotype sequences of Leishmania ITS-rDNA
Analysis of the ITS1-5.8S rDNA gene sequences obtained for 3 sand flies and 7 patients further confirmed that they all contain L. tropica. The sequences analyzed revealed 217 bp of ITS1 gene and 169 bp of 5.8S gene followed by 15 bp of ITS2. Alignment of these sequences revealed their sequence heterogeneity (Figure 4). The phylogenetic analysis of these sequences segregated seven L. tropica haplotypes: 3 from female P. sergenti and 4 from patients (Figure 5).

ITS1-5.8S rDNA gene sequences alignment from patients and from P. sergenti with Tunisian haplotype. Using GENtle software (v.1.9.4). Identities are denoted by points. Gaps are denoted by dashes.

Neighbor-Joining tree showing the relationships of the haplotypes of the ITS1-5.8S rDNA gene sequences of Leishmania tropica infecting Phlebotomus sergenti and patients, using MEGA 5 software, and it relates ITS1-5.8S rDNA gene haplotypes in GenBank. The tree shown is based on the Kimura 2-parameter model of nucleotide substitution. Bootstrap values are based on 1,000 replicates. L. aethiopica was used as outgroup. Black triangles indicate haplotypes of L. tropica from patients and black disks haplotypes of L. tropica from P. sergenti.
The haplotype KC145159 is identical to the Tunisian strain AJ300485, the haplotype KC145160, predominant in our CL patients (4/7), differs in one character, while KC145158 and KC145161 differ in two or four nucleotide positions, respectively.
L. tropica haplotypes KC145155; KC145156 and KC145157 from P. sergenti showed 95 to 98% similarity to the Tunisian haplotype AJ300485 showing 20, 10 and 7 base substitutions, respectively (Figure 4).
Discussion
The main objectives of this study were to identify the sand fly vector and Leishmania species responsible for the recent outbreaks of CL in El Hanchane locality, a semi rural area in central Morocco where the first cases of CL appeared in 2006. Two molecular techniques were used for this purpose, ITS1 PCR-RFLP and nested PCR. Of the 7 samples of CL patients, 4 were successfully amplified and characterized by PCR-RFLP. The three remaining specimens were possibly either inhibited, or failed to amplify because of the low amount of DNA in stained slides. Several studies have exploited the amplification of the ITS-1 region for identifying Leishmania spp. in bone marrow aspirates or stained smears [13, 17–19]. The major advantage of the ITS1-PCR is that species identification can be achieved by simple digestion of the PCR products. Thus, all clinically important species can be distinguished by their RFLP patterns. However, previous reports have shown that ITS1 PCR-RFLP is not sensitive enough for diagnostic purposes [20, 21]. Using nested PCR, the 7 patients were all positive, confirming the better sensitivity of this technique [22].
In this survey, a total of 643 sand flies were collected indoors from July to September 2011 in an emerging focus of CL. The sandfly populations were mainly represented by P. sergenti (76.67%) and P. longicuspis (11.51%). Three unengorged female P. sergenti were infected by Leishmania, similar to L. tropica as demonstrated first by their ITS1PCR-RFLP profiles and then confirmed by the sequence identity of ITS-rDNA gene. In Morocco, L. tropica was first isolated from P. sergenti over three decades ago [4]. Since then and despite the large number of foci of CL due to L. tropica in Morocco, the vectors have never been identified with certainty because most epidemiological studies have focused on the identification of the parasite isolated from humans. P.sergenti is suggested as a vector based only on the circumstantial evidence of its ecology and its high frequency in an endemic area.
Leishmania infection of sand flies has classically been examined by dissecting freshly caught individual sand flies. This method allows the culture of Leishmania strains found in the sand fly gut; but it is time-consuming, needs dissecting expertise and a large number of specimens, since the Leishmania infection rate in sand flies is usually very low even in the highly endemic areas [23]. In recent years, molecular techniques have been used increasingly to identify Leishmania infection both in experimentally infected and field captured phlebotomine sand flies, as a sensitive and effective tool useful in epidemiological studies to facilitate strategic planning for the control of human leishmaniasis [24–26]. The increasing application of molecular techniques in this field has considerably reduced the time needed to obtain results; but such methods are underused in Morocco, despite the existence of CL foci in large areas and the lack of information for its vectors. This is the first report that L. tropica DNA was found in naturally infected P. sergenti from a CL focus of this country. The high density of P.sergenti indoors (i.e., anthropophilic) and their infection with L. tropica among unengorged females, suggest that this species plays a major role as the principal, if not the only vector in this locality.
P. sergenti is the confirmed vector of L. tropica throughout North Africa, Middle East and Central Asia [27–31]. However, P. chabaudi and the closely related species P. riouxi were also suspected as vectors based on their abundance in Tunisian and Algerian foci of CL due to L. tropica[32, 33]. In Israel, in addition to P. sergenti, P. arabicus has been involved in the transmission of L. tropica[34, 35]. None of these sand fly species were found in the present CL focus, where P. sergenti represents 76.67% of the total number of sand flies collected. On the other hand, the screening for Leishmania infection of the other sand fly species, separated into 5 monospecific pools, showed no PCR positives (data no shown), suggesting that P. sergenti is the only vector in this locality.
L. tropica is classically considered to be anthroponotic, however, in the present emerging focus, an anthroponotic transmission is very improbable, since the number of CL cases is too small, and their geographical distribution is too sparse and sporadic to consider humans as an adequate reservoir. Zoonotic outbreaks have been reported in many countries. Rock hyraxes were incriminated as reservoir hosts of L. tropica in Kenya and Namibia [36, 37], in Israel L. tropica was isolated from rock hyrax (Procavia capensis): the strains were identical to those obtained from humans and sand flies in the same focus [38, 39]. In southeast Tunisia gundi (Ctenodactylus gundi) was supported as a reservoir host of L. tropica[40, 41], whereas in Algeria human CL caused by L. killicki is considered as a zoonotic disease with P. sergenti sand flies acting as vectors and gundi rodents (Massoutiera mzabi) as reservoirs [30]. In Morocco L. tropica has also been isolated from dogs in Azilal, a southern CL focus [42], but the reported infections in dogs were strictly cutaneous, the evolution duration of lesions was too short to consider dogs as the reservoir of the parasite.
L. tropica is recognized as a very heterogeneous species of Leishmania and intraspecific heterogeneity has been readily demonstrated by many investigators [43–45]. Microsatellite analysis revealed that two genetically very distinct populations of L. tropica co-exist within the same focus in Morocco: population ‘Morocco A’ is related to population ‘Asia’, whereas population ‘Morocco B’ is genetically closer to the other African populations [46].
In terms of isoenzyme profile variability, Morocco is characterized by the largest number of zymodemes ever described in L. tropica, with 8 zymodemes detected from human CL, dogs and the sandflies, e.g. P. sergenti[4, 5, 47]. In the present study, the DNA sequences of L. tropica isolated from P. sergenti showed a great heterogeneity, that was also observed in a southern Moroccan focus where L.tropica isolates from P. sergenti showed a wider range of zymodemes than isolates collected from the vertebrate hosts (dogs and humans) in the same region [42]: 74 L. tropica strains isolated from P. sergenti females were typed and corresponded to four zymodemes (MON-102: one strain; MON-107: 56 strains, MON-122: two strains; and MON-123: 15 strains). Only the first two zymodemes were also identified in humans and their frequencies were different in humans, dogs and the vector [4]. The zymodeme heterogeneity of strains isolated from vectors was also described with L. infantum isolates from P. perniciosus compared to vertebrate hosts in the same Spanish region [48]. Our results reinforce the observation that L. tropica is polymorphic; especially among those detected in P. sergenti compared to human samples.
Conclusion
In conclusion, the ITS1 PCR-RFLP and the ITS1-5.8S rRNA gene nested PCR enable us to detect and to identify L. tropica in clinical samples and within sand flies. We have demonstrated that L. tropica is the causative agent of CL in this emerging focus of CL and that P. sergenti is the putative, but apparent vector of this species. The sequencing of the ITS1-5.8S rRNA gene amplicons allowed the identification of 6 new previously unknown sequence haplotypes, which under scores specific genetic heterogeneity of this species in Morocco.
References
Rioux JA, Lanotte G, Petter F, Dereure J, Akalay O, Pratlong F, Velez ID, Fikri NB, Maazoun R, Denial M:Les leishmanioses cutanées du bassin méditerranéen occidental: de l'identification enzymatique à l'analyse éco-épidémiologique. L'exemple de trois "foyers", tunisien, marocain et français. Leishmania Taxonomie et phylogenèse Applications éco-épidémiologiques. (CollintCNRS/INSERM) IMEE, Montpellier. 1986, 1986: 365-395.
Moroccan Health Ministry: Etat d'avancement des programmes de lutte contre les maladies parasitaires. 2008, Direction de l'Épidémiologie et de Lutte Contre les Maladies: Rapport annuel d'acitivités
Marty P, Le Fichoux Y, Pratlong F, Rioux JA, Rostain G, Lacour JP:Cutaneous leishmaniasis due to Leishmania tropica in a young Moroccan child observed in Nice, France. Trans R Soc Trop Med Hyg. 1989, 83 (4): 510-10.1016/0035-9203(89)90268-X.
Guilvard E, Rioux JA, Gallego M, Pratlong F, Mahjour J, Martinez-Ortega E, Dereure J, Saddiki A, Martini A: Leishmania tropica in Morocco. III–The vector of Phlebotomus sergenti. Apropos of 89 isolates. Ann Parasitol Hum Comp. 1991, 66 (3): 96-99.
Pratlong F, Rioux JA, Dereure J, Mahjour J, Gallego M, Guilvard E, Lanotte G, Perieres J, Martini A, Saddiki A: Leishmania tropica in Morocco. IV–Intrafocal enzyme diversity. Ann Parasitol Hum Comp. 1991, 66 (3): 100-104.
Guessous-Idrissi N, Chiheb S, Hamdani A, Riyad M, Bichichi M, Hamdani S, Krimech A:Cutaneous leishmaniasis: an emerging epidemic focus of Leishmania tropica in north Morocco. Trans R Soc Trop Med Hyg. 1997, 91 (6): 660-663. 10.1016/S0035-9203(97)90511-3.
Bichichi M, Riyad M, Guessous-Idrissi N:Isoenzyme characterization of Leishmania tropica in the emerging epidemic focus of Taza (north Morocco). Trans R Soc Trop Med Hyg. 1999, 93 (1): 21-22. 10.1016/S0035-9203(99)90164-5.
Rhajaoui M, Nasereddin A, Fellah H, Azmi K, Amarir F, Al-Jawabreh A, Ereqat S, Planer J, Abdeen Z:New clinico-epidemiologic profile of cutaneous leishmaniasis. Morocco. Emerg Infect Dis. 2007, 13 (9): 1358-1360. 10.3201/eid1309.060946.
Garcia AL, Kindt A, Quispe-Tintaya KW, Bermudez H, Llanos A, Arevalo J, Banuls AL, De Doncker S, Le Ray D, Dujardin JC:American tegumentary leishmaniasis: antigen-gene polymorphism, taxonomy and clinical pleomorphism. Infect Genet Evol. 2005, 5 (2): 109-116. 10.1016/j.meegid.2004.07.003.
Talmi-Frank D, Nasereddin A, Schnur LF, Schonian G, Toz SO, Jaffe CL, Baneth G:Detection and identification of old world Leishmania by high resolution melt analysis. PLoS Negl Trop Dis. 2010, 4 (1): e581-10.1371/journal.pntd.0000581.
Schonian G, Mauricio I, Gramiccia M, Canavate C, Boelaert M, Dujardin JC:Leishmaniases in the Mediterranean in the era of molecular epidemiology. Trends Parasitol. 2008, 24 (3): 135-142. 10.1016/j.pt.2007.12.006.
Boussaa S: Epidémiologie des leishmanioses dans la région de Marrakech, Maroc : effet de l’urbanisation sur la répartition spatio-temporelle des Phlébotomes et caractérisation moléculaire de leurs populations. 2008, Strasbourg: Université Louis Pasteur
Schonian G, Nasereddin A, Dinse N, Schweynoch C, Schallig HD, Presber W, Jaffe CL:PCR diagnosis and characterization of Leishmania in local and imported clinical samples. Diagn Microbiol Infect Dis. 2003, 47 (1): 349-358. 10.1016/S0732-8893(03)00093-2.
Parvizi P, Mauricio I, Aransay AM, Miles MA, Ready PD:First detection of Leishmania major in peridomestic Phlebotomus papatasi from Isfahan province, Iran: comparison of nested PCR of nuclear ITS ribosomal DNA and semi-nested PCR of minicircle kinetoplast DNA. Acta Trop. 2005, 93 (1): 75-83. 10.1016/j.actatropica.2004.09.007.
Parvizi P, Ready PD:Nested PCRs and sequencing of nuclear ITS-rDNA fragments detect three Leishmania species of gerbils in sandflies from Iranian foci of zoonotic cutaneous leishmaniasis. Trop Med Int Health. 2008, 13 (9): 1159-1171. 10.1111/j.1365-3156.2008.02121.x.
Cupolillo E, Grimaldi Junior G, Momen H, Beverley SM:Intergenic region typing (IRT): a rapid molecular approach to the characterization and evolution of Leishmania. Mol Biochem Parasitol. 1995, 73 (1–2): 145-155.
Roelfsema JH, Nozari N, Herremans T, Kortbeek LM, Pinelli E:Evaluation and improvement of two PCR targets in molecular typing of clinical samples of Leishmania patients. Exp Parasitol. 2011, 127 (1): 36-41. 10.1016/j.exppara.2010.06.024.
Amro A, Azmi K, Schonian G, Nasereddin A, Alsharabati MB, Sawalha S, Hamarsheh O, Ereqat S, Abdeen Z:Epidemiology of paediatric visceral leishmaniasis in Hebron district, Palestine. Trans R Soc Trop Med Hyg. 2009, 103 (7): 731-736. 10.1016/j.trstmh.2008.10.008.
Hamarsheh O, Nasereddin A, Damaj S, Sawalha S, Al-Jawabreh H, Azmi K, Amro A, Ereqat S, Abdeen Z, Al-Jawabreh A:Serological and molecular survey of Leishmania parasites in apparently healthy dogs in the West Bank, Palestine. Parasit Vectors. 2012, 5: 183-10.1186/1756-3305-5-183.
Bensoussan E, Nasereddin A, Jonas F, Schnur LF, Jaffe CL:Comparison of PCR assays for diagnosis of cutaneous leishmaniasis. J Clin Microbiol. 2006, 44 (4): 1435-1439. 10.1128/JCM.44.4.1435-1439.2006.
Azmi K, Nasereddin A, Ereqat S, Schnur L, Schonian G, Abdeen Z:Methods incorporating a polymerase chain reaction and restriction fragment length polymorphism and their use as a 'gold standard' in diagnosing Old World cutaneous leishmaniasis. Diagn Microbiol Infect Dis. 2011, 71 (2): 151-155. 10.1016/j.diagmicrobio.2011.06.004.
da Silva MA, Pedrosa Soares CR, Medeiros RA, Medeiros Z, de Melo FL:Optimization of single-tube nested PCR for the diagnosis of visceral leishmaniasis. Exp Parasitol. 2013, 134 (2): 206-210. 10.1016/j.exppara.2013.03.003.
Sharma U, Singh S:Insect vectors of Leishmania: distribution, physiology and their control. J Vector Borne Dis. 2008, 45 (4): 255-272.
Aransay AM, Scoulica E, Tselentis Y:Detection and identification of Leishmania DNA within naturally infected sand flies by seminested PCR on minicircle kinetoplastic DNA. Appl Environ Microbiol. 2000, 66 (5): 1933-1938. 10.1128/AEM.66.5.1933-1938.2000.
Kato H, Uezato H, Katakura K, Calvopina M, Marco JD, Barroso PA, Gomez EA, Mimori T, Korenaga M, Iwata H:Detection and identification of Leishmania species within naturally infected sand flies in the andean areas of ecuador by a polymerase chain reaction. Am J Trop Med Hyg. 2005, 72 (1): 87-93.
Rossi E, Bongiorno G, Ciolli E, Di Muccio T, Scalone A, Gramiccia M, Gradoni L, Maroli M:Seasonal phenology, host-blood feeding preferences and natural Leishmania infection of Phlebotomus perniciosus (Diptera, Psychodidae) in a high-endemic focus of canine leishmaniasis in Rome province, Italy.Acta Trop. 2008, 105 (2): 158-165. 10.1016/j.actatropica.2007.10.005.
Al-Zahrani MA, Peters W, Evans DA, Chin C, Smith V, Lane RP:Phlebotomus sergenti, a vector of Leishmania tropica in Saudi Arabia. Trans R Soc Trop Med Hyg. 1988, 82 (3): 416-10.1016/0035-9203(88)90142-3.
Guilvard E, Rioux JA, Gallego M, Pratlong F, Mahjour J, Martinez-Ortega E, Dereure J, Saddiki A, Martini A:Leishmania tropica au Maroc. III. Rôle de Phlebotomus sergenti. A propros de 89 isolats. Ann Parasitol Hum Comp. 1991, 66: 96-99.
Killick-Kendrick R:Phlebotomine vectors of the leishmaniases: a review. Med Vet Entomol. 1990, 4 (1): 1-24. 10.1111/j.1365-2915.1990.tb00255.x.
Boubidi SC, Benallal K, Boudrissa A, Bouiba L, Bouchareb B, Garni R, Bouratbine A, Ravel C, Dvorak V, Votypka J:Phlebotomus sergenti (Parrot, 1917) identified as Leishmania killicki host in Ghardaia, south Algeria. Microbes Infect. 2011, 13 (7): 691-696. 10.1016/j.micinf.2011.02.008.
Tabbabi A, Bousslimi N, Rhim A, Aoun K, Bouratbine A:First report on natural infection of Phlebotomus sergenti with Leishmania promastigotes in the cutaneous leishmaniasis focus in southeastern Tunisia. Am J Trop Med Hyg. 2011, 85 (4): 646-647. 10.4269/ajtmh.2011.10-0681.
Bounamous A, Boudabous R, Jouet D, Augot D, Ferte H, Babba H, Berchi S, Depaquit J: [Molecular and morphological characterisation of two closely related species belonging to the subgenus Phlebotomus chabaudi Croset, Abonnenc &Rioux, 1970 et P. riouxi Depaquit, Killick-Kendrick &Leger, 1998 (Diptera: Psychodidae)]. Parasite. 2008, 15 (4): 565-571. 10.1051/parasite/2008154565.
Tabbabi A, Ghrab J, Aoun K, Ready PD, Bouratbine A: Habitats of the sandfly vectors of Leishmania tropica and L. major in a mixed focus of cutaneous leishmaniasis in southeast Tunisia. Acta Trop. 2011, 119 (2–3): 131-137.
Jacobson RL, Eisenberger CL, Svobodova M, Baneth G, Sztern J, Carvalho J, Nasereddin A, El Fari M, Shalom U, Volf P:Outbreak of cutaneous leishmaniasis in northern Israel. J Infect Dis. 2003, 188 (7): 1065-1073. 10.1086/378204.
Schnur LF, Nasereddin A, Eisenberger CL, Jaffe CL, El Fari M, Azmi K, Anders G, Killick-Kendrick M, Killick-Kendrick R, Dedet JP:Multifarious characterization of leishmania tropica from a Judean desert focus, exposing intraspecific diversity and incriminating phlebotomus sergenti as its vector. Am J Trop Med Hyg. 2004, 70 (4): 364-372.
Sang DK, Njeru WK, Ashford RW:A zoonotic focus of cutaneous leishmaniasis due to Leishmania tropica at Utut, Rift Valley Province, Kenya. Trans R Soc Trop Med Hyg. 1994, 88 (1): 35-37. 10.1016/0035-9203(94)90486-3.
Grove SS:Leishmaniasis in South West Africa/Namibia to date. S Afr Med J. 1989, 75 (6): 290-292.
Talmi-Frank D, Jaffe CL, Nasereddin A, Warburg A, King R, Svobodova M, Peleg O, Baneth G:Leishmania tropica in rock hyraxes (Procavia capensis) in a focus of human cutaneous leishmaniasis. Am J Trop Med Hyg. 2010, 82 (5): 814-818. 10.4269/ajtmh.2010.09-0513.
Svobodova M, Votypka J, Peckova J, Dvorak V, Nasereddin A, Baneth G, Sztern J, Kravchenko V, Orr A, Meir D:Distinct transmission cycles of Leishmania tropica in 2 adjacent foci, Northern Israel. Emerg Infect Dis. 2006, 12 (12): 1860-1868. 10.3201/eid1212.060497.
Bousslimi N, Ben-Ayed S, Ben-Abda I, Aoun K, Bouratbine A:Natural infection of North African gundi (Ctenodactylus gundi) by Leishmania tropica in the focus of cutaneous leishmaniasis, Southeast Tunisia. Am J Trop Med Hyg. 2012, 86 (6): 962-965. 10.4269/ajtmh.2012.11-0572.
Jaouadi K, Haouas N, Chaara D, Gorcii M, Chargui N, Augot D, Pratlong F, Dedet JP, Ettlijani S, Mezhoud H:First detection of Leishmania killicki (Kinetoplastida, Trypanosomatidae) in Ctenodactylus gundi (Rodentia, Ctenodactylidae), a possible reservoir of human cutaneous leishmaniasis in Tunisia. Parasit Vectors. 2011, 4: 159-10.1186/1756-3305-4-159.
Dereure J, Rioux JA, Gallego M, Perieres J, Pratlong F, Mahjour J, Saddiki H:Leishmania tropica in Morocco: infection in dogs. Trans R Soc Trop Med Hyg. 1991, 85 (5): 595-
Azmi K, Schnur L, Schonian G, Nasereddin A, Pratlong F, El Baidouri F, Ravel C, Dedet JP, Ereqat S, Abdeen Z:Genetic, serological and biochemical characterization of Leishmania tropica from foci in northern Palestine and discovery of zymodeme MON-307. Parasit Vectors. 2012, 5: 121-10.1186/1756-3305-5-121.
Schonian G, Schnur L, el Fari M, Oskam L, Kolesnikov AA, Sokolowska-Kohler W, Presber W:Genetic heterogeneity in the species Leishmania tropica revealed by different PCR-based methods. Trans R Soc Trop Med Hyg. 2001, 95 (2): 217-224. 10.1016/S0035-9203(01)90173-7.
Le Blancq SM, Peters W: Leishmania in the Old World: 2 Heterogeneity among L. tropica zymodemes. Trans R Soc Trop Med Hyg. 1986, 80 (1): 113-119. 10.1016/0035-9203(86)90208-7.
Schwenkenbecher JM, Wirth T, Schnur LF, Jaffe CL, Schallig H, Al-Jawabreh A, Hamarsheh O, Azmi K, Pratlong F, Schonian G:Microsatellite analysis reveals genetic structure of Leishmania tropica. Int J Parasitol. 2006, 36 (2): 237-246. 10.1016/j.ijpara.2005.09.010.
Lemrani M, Nejjar R, Pratlong F:A new Leishmania tropica zymodeme–causative agent of canine visceral leishmaniasis in northern Morocco. Ann Trop Med Parasitol. 2002, 96 (6): 637-638. 10.1179/000349802125001645.
Martin-Sanchez J, Guilvard E, Acedo-Sanchez C, Wolf-Echeverri M, Sanchiz-Marin MC, Morillas-Marquez F:Phlebotomus perniciosus Newstead, 1911, infection by various zymodemes of the Leishmania infantum complex in the Granada province (southern Spain). Int J Parasitol. 1994, 24 (3): 405-408. 10.1016/0020-7519(94)90089-2.
Acknowledgements
This work was supported by EMRO-COMSTECH grant; RAB&GH 10-11/09.
We would like to thank the team of the Department of Parasitology, Direction d’Epidémiologie et de Lutte Contre les Maladies, Moroccan Ministry of Health. The authors are also very grateful to Doctors and nurses of the El Hanchane Health center and the Health representation of the MOH in Essaouira.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
Authors declare that they have no competing interests.
Authors’ contributions
MA: collected sandflies carried out the laboratory work of entomological and molecular studies, analyzed data, and prepared the manuscript. NE: contributed to the entomological studies and data analysis. SH: was involved in the collection of samples. AAE: is involved in the study design. MR: was involved in the review of the manuscript. ML: designed the study, contributed to interpretation, analyzed and finalized the manuscript. All the authors read and approved the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ajaoud, M., Es-sette, N., Hamdi, S. et al. Detection and molecular typing of Leishmania tropica from Phlebotomus sergenti and lesions of cutaneous leishmaniasis in an emerging focus of Morocco. Parasites Vectors 6, 217 (2013). https://doi.org/10.1186/1756-3305-6-217
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-3305-6-217