
Abstract
Background
One of the concerns of assembling de novo transcriptomes is determining the amount of read sequences required to ensure a comprehensive coverage of genes expressed in a particular sample. In this report, we describe the use of Illumina paired-end RNA-Seq (PE RNA-Seq) reads from Hevea brasiliensis (rubber tree) bark to devise a transcript mapping approach for the estimation of the read amount needed for deep transcriptome coverage.
Findings
We optimized the assembly of a Hevea bark transcriptome based on 16 Gb Illumina PE RNA-Seq reads using the Oases assembler across a range of k-mer sizes. We then assessed assembly quality based on transcript N50 length and transcript mapping statistics in relation to (a) known Hevea cDNAs with complete open reading frames, (b) a set of core eukaryotic genes and (c) Hevea genome scaffolds. This was followed by a systematic transcript mapping process where sub-assemblies from a series of incremental amounts of bark transcripts were aligned to transcripts from the entire bark transcriptome assembly. The exercise served to relate read amounts to the degree of transcript mapping level, the latter being an indicator of the coverage of gene transcripts expressed in the sample. As read amounts or datasize increased toward 16 Gb, the number of transcripts mapped to the entire bark assembly approached saturation. A colour matrix was subsequently generated to illustrate sequencing depth requirement in relation to the degree of coverage of total sample transcripts.
Conclusions
We devised a procedure, the “transcript mapping saturation test”, to estimate the amount of RNA-Seq reads needed for deep coverage of transcriptomes. For Hevea de novo assembly, we propose generating between 5–8 Gb reads, whereby around 90% transcript coverage could be achieved with optimized k-mers and transcript N50 length. The principle behind this methodology may also be applied to other non-model plants, or with reads from other second generation sequencing platforms.
Similar content being viewed by others


Background
Transcriptome analysis has become increasingly powerful through advances in second generation sequencing technologies from companies such as Illumina, Roche and Life Technologies. Improvements in sequencing chemistry and read length have enabled unprecedented depth of sequencing, limited only by cost and availability of biological material. In particular, RNA sequencing (RNA-Seq), also known as whole transcriptome shotgun sequencing, has emerged as a valuable tool for profiling expressed genes in plants and other organisms [1–3]. The depth of transcriptome sequencing provided by RNA-Seq has thus provided a cost-effective means of qualitative and quantitative analyses of gene transcripts in many non-model plant species including the rubber tree, Hevea brasiliensis, the subject of the present study.
Parallel with progress in sequencing technologies, numerous softwares have been developed to assemble de novo transcriptomes. Among the most commonly used sofwares are Velvet [4], Oases [5], SOAPdenovo [6], ABySS [7], Trinity [8], MIRA [9], Newbler (Roche) and CLC (CLC bio). In the absence of a reference genome, the assemble-then-align approach is used in place of the align-then-assemble approach [10–12]. Additional procedures are often integrated in order to improve the quality of the de novo transcriptome assembly. This includes weighing the relative merits of more than one assembler [13–21], optimizing transcript numbers and lengths across different k-mers and other assembly parameters [16, 17, 22–26], hybrid assembly of data from different sequencing platforms [21, 27–30] and alignment of transcripts to sequences from the same or related species [18, 30–32].
Challenges in de novo transcriptome assembly in higher plants lie in the immense number of gene transcripts, large variations in transcript expression levels, presence of alternatively spliced transcript variants and issues in strand directionality [11, 12]. Owing to such problems, de novo assembly requires significantly greater sequencing depth as compared with reference-based assembly. Especially in the case of non-model plants, few guidelines are available for determining the amount of reads to generate to enable deep coverage of transcripts expressed in a particular sample. The general practice commonly adopted, especially for new entrants in second generation sequencing, is to piggy-back on ballpark estimates adopted for the model species. From a survey of recent publications on de novo transcriptome analysis in non-model plants, the read generation per sample could fall below 100 Mb or it could be high as 7 Gb, with 2–5 Gb being the most common sequencing depths [13, 18, 19, 21, 23, 26–49]. Therefore, there is need for a practical procedure to estimate the reads needed for deep coverage of gene transcripts in de novo assembly where such information is unavailable.
Hevea brasiliensis is the commercial source of natural rubber (cis-polyisoprene). The tree, a diploid species (2n = 36) from the Euphorbiaceae family, has a C value of about 2 pg as estimated by flow cytometery [50]. Compared to model plants, transcript resources in this economically important species have only been initiated relatively recently, with the analysis of Expressed Sequence Tags (ESTs) from latex [29, 51–54]. The first crop of publications on the application of second generation sequencing in Hevea rubber genomics emerged in 2011 (Table 1). In these papers, functional profiling of gene transcripts was reported in latex, leaf and bark tissues, focussing on rubber biosynthesis, molecular markers and transcription factors [29, 55–60]. Overall, the depth of sequencing reported in these papers was between 0.2-5 Gb per sample based on Illumina PE-RNA-Seq or 454 pyrosequencing platforms (Table 1). Hevea being a tree, analysis of its gene expression is often in RNAs prepared from distinct cells, tissues or organs, including RNAs from the same sample types but under different physiological conditions [61–64]. Having a means of assessing the degree of coverage of genes that are expressed in various samples would be important to achieve a normalized set of Hevea gene transcripts. In this paper, we describe a method to estimate datasize requirement for high transcriptome coverage based on an analysis of assembly statistics of Illumina PE RNA-Seq reads generated from Hevea bark. We first optimized and validated a 16 Gb bark assembly across a range of datasizes and k-mers. Subsequently, we applied a transcript mappability method to illustrate the trend of gene transcript coverage by different read amounts that had been assembled using a range of k-mers.
Findings
Generation of Illumina PE RNA- Seq Hevea tissue libraries and de novo assembly
The development of an approach to determine the datasize required for deep coverage of a de novo transcriptome arose from the generation of three Hevea tissue read libraries as part of a programme in developing genomic resources for the rubber tree. Considerably more PE RNA-Seq raw reads were available for bark (16.9 Gb) as compared with latex and leaf tissues (4.9-5 Gb each) (Table 2; see Materials and methods). Hitherto, similar work on the rubber tree involving second generation sequencing (Table 1) has far lower transcriptome coverage per sample compared to what we have generated. Taking cognizance of the immensity of the bark read library, we asked the following questions: (a) What is the trend of assembly characteristics of a significantly larger read library? and (b) Can we devise a method to relate datasize requirement to the degree of coverage of transcripts expressed in the sample?
We first processed the bark, latex and leaf raw reads for quality, and found a very high percentage of clean reads (approximately 96-98%) (Table 2; see Materials and methods). The distribution of read length categories also indicated high PE RNA-Seq quality based on the fact that a vast majority of clean paired reads was in the largest size category (100 nt) (Figure 1). Henceforth in this report, the clean paired reads from bark are referred to as the 16 Gb read set while those from latex or leaf as the 5 Gb read set (Table 2). A number of plant de novo transcriptome projects have reported the use of multiple k-mers in assembly optimization [13, 21, 27, 65]. Therefore, we analyzed the effect of two parameters on transcriptome assembly, namely read amount and k-mer size.

Size distributions of clean paired reads from latex, leaf and bark libraries.
To do this, the 16 Gb bark read set was assembled in incremental quantums of 1, 3, 5, 8, 10, 13 and 16 Gb using the Oases assembler and a k-mer range of 51–77 (see Materials and methods). As shown in Table 3, the higher the k-mer, the lower was the number of transcripts generated by a particular datasize. At the same time, the larger the dataset size, the greater the number of transcripts generated using a particular k-mer. On the other hand, the transcript N50 length (see Materials and methods) showed a bell shape distribution from k-mer 51 to 77 in all datasize sub-assemblies with the exception of the 1 Gb datasize (N50 peak values are highlighted in Table 3). The k-mer at which the N50 value peaked in this bell shape distribution was thus referred to as the “optimized k-mer” of assembly for a particular datasize (and accordingly the “optimized N50”). As seen in Table 3, the optimized N50 became larger with increasing read depth, and this corresponded also with increment in the size of optimized k-mers. Hence, larger data sizes facilitated not only an increase in the number of transcripts assembled, but also an increase in transcript length. We therefore propose that by a judicious combination of datasize and k-mer range, the ensuing N50 trend may serve as a criterion for determining optimal de novo assembly.
Validation of the bark transcriptome
In this study, a k-mer of 73 assembled 87,612 transcripts from the 16 Gb read set with a transcript N50 value of 2,068 bp (Table 3). At this stage, this assembly was referred to as the optimized bark assembly for 16 Gb reads. Due to the importance of quality de novo assembly, we performed three mapping analyses to validate the 16 Gb bark transcriptome (see Materials and methods). First of all, 255 publicly available Hevea cDNAs which had been verified to contain complete open reading frames or ORFs were mapped to 87,612 bark transcripts using the Megablast software. In the absence of more cDNAs containing complete Hevea proteins, rubber-specific ORF quality of bark transcripts could only be evaluated using these sequences which also included isoforms for several gene families (see Additional file 1: Table S1). The results showed that 250 Hevea ORFs (of 255) had hits to bark transcripts with sequence identity match ranging from 86-100% (Table 4 and Additional file 1: Table S1). Also, 80% of the 250 ORFs showed a minimum of 70% utilization of ORF sequence length in their match alignments with bark transcripts (Table 4 and Additional file 1: Table S1). These observations indicated that a high proportion of transcripts within the optimized bark assembly encoded complete or near-complete Hevea proteins.
Secondly, completeness of gene representation in the 16 Gb transcriptome was assessed by mapping 87,612 bark transcripts to a set of 248 core eukaryotic genes (CEGs) that had been shown to be a reliable indicator of completeness of gene space in eukaryotic species [66]. Although initially used to assess gene space in newly sequenced genomes, the approach was recently applied to transcriptomes and it also complements other transcript metrics such as N50 length. Using BlastX, 87,612 bark transcripts detected 247 out of 248 CEG proteins (98%) (e-value ≤ 1e-10). Thus, these results support the completeness or depth of bark gene representation in this transcriptome.
Thirdly, the 87,612 bark transcripts were validated by using the Exonerate software to map them to rubber genome scaffolds (BioProject ID: PRJNA80191). Figure 2 shows the number of bark transcripts which were mapped to categories of transcript-to-scaffold coverage. The higher the percentage of transcript-to-scaffold coverage, the greater the significance of match alignment. A large proportion of bark transcripts (84,471 or 96.41% of total) could be mapped to the scaffolds and of these, almost 70% showed transcript-to-scaffold coverage of 90-100% (Figure 2). Therefore, this indicated the presence of a sizeable proportion of high quality bark transcripts.

Number of transcripts from the optimized 16 Gb bark assembly with hits to rubber genome scaffolds. Hits are classified into transcript-to-scaffold coverage categories describing the extent of alignment from 10-100%. A total of 84,471 bark transcripts (all categories) were mapped to genome scaffolds. The proportion of hits from 84,471 in each category is shown in brackets.
As a whole, results of the three mapping analyses carried out provided sufficient validation for the quality of 87,612 bark transcripts assembled from the 16 Gb read set. Fragmented or erroneous transcripts could still be present to some extent in any assembly but we think that the proportion of bark transcripts which did not show meaningful mapping or alignment with Hevea transcripts or genome scaffolds could also be explained by reasons such as inherent variations between sequences derived from different tree clonal varieties.
Mapping saturation test for bark transcript accumulation
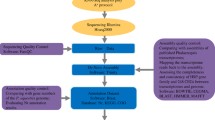
Next, we addressed the question of read depth requirement by mapping the series of incremental bark transcripts to total transcripts from the optimized 16 Gb bark assembly. The principle behind mapping subsets of bark transcripts to total transcripts from the full 16 Gb bark transcriptome is that the number of the former aligning to the latter should follow a saturation pattern. As outlined in Figure 3, transcript sets from 1, 3, 5, 8, 10 and 13 Gb bark reads that had been assembled independently across k-mers 51–77 were mapped to 87,612 bark transcripts. The extent of transcript mapping, expressed as a percentage of total bark transcripts (or the full transcriptome) was taken as a measure of transcript representation by each sub-assembly.

Methodology of the transcript mapping saturation test.
Results of the number of mappings to 87,612 bark transcripts were displayed as a colour matrix as shown in Figure 4 and Additional file 2: Table S2. Overall, transcript mapping by incremental subsets of bark transcripts approached full saturation (i.e. 100%) as datasize increased (for any k-mer) or as k-mer decreased (for any datasize). Transcript representation was high in all cases, being more than 80% transcript mapping level with the exception of three higher k-mer assemblies (73–77) of 3 Gb reads (Figure 4 and Additional file 2: Table S2). At the lower end of the saturation spectrum, 3–8 Gb reads would already be sufficient to obtain even 80-85% mapping level or transcript coverage. This datasize range could also readily generate transcript coverage exceeding 90% if the k-mer range for assembly was decreased. At the upper end of the saturation spectrum, transcripts from nearly all of the 10–13 Gb assemblies across all k-mers showed mapping levels greater than 90%.

Colour matrix representing the transcript mapping saturation test results. An asterisk corresponds to the assembly with the optimized k-mer and transcript N50 length for a particular datasize (1 Gb assembly transcripts were not included since the transcript N50 length was not optimized by any k-mer for this datasize). See Additional file 2: Table S2 for full details of BlastN matches by subsets of bark transcripts to the optimized 16 Gb bark transcriptome.
However, although the colour matrix indicated generally high transcript coverage by assemblies of 3–13 Gb reads, it is important to select a datasize that would also produce the optimal transcript N50 length at the desired transcript coverage level. As determined previously, the optimized N50 increased with datasize (Table 3). In the optimized 3, 5, 8, 10 and 13 Gb assemblies, this corresponded with 87.21, 89.48, 91.46, 92.60 and 92.12% representation of the 16 Gb bark transcripts respectively (Additional file 2: Table S2). Therefore, based on the colour matrix, the optimized 3–5 Gb assemblies would fall within the 85-90% transcript coverage bracket and the optimized 8–13 Gb assemblies within the 90-95% coverage bracket (Figure 4 and Additional file 2: Table S2). This also indicated that based on the mapping saturation test in this study, a shift in bark transcript coverage bracket by the incremental assemblies occurred between 5–8 Gb.
In essence, the amount of reads for optimal coverage of a tissue transcriptome should take into consideration the requirements for transcript coverage level (reflected by percentage of mapping saturation) and for N50 length. In order to attain both high transcript representation and best N50 length, our general recommendation for Hevea is to generate between 5–8 Gb reads for de novo assembly. Firstly, as observed in the shift in transcript coverage bracket, a minimum of nearly 90% (i.e. 89.48%) transcript coverage could already be achieved by an optimized 5 Gb assembly (Figure 4 and Additional file 2: Table S2), a level that is within range of the following coverage bracket (90-95%). Secondly, it is noted from Table 3 that the improvement in optimized N50 length was more rapid from 3–8 Gb than from 8–16 Gb assemblies. Therefore, generating less than 5 Gb reads may lead to reduction in complete transcripts, and sequencing beyond 8 Gb reads may not yield significantly more new or complete transcripts other than the rarely expressed ones.
Analysis of latex and leaf transcriptome assembly
RNAs from different types of tissues and growth conditions are of interest in Hevea transcriptome profiling studies. Application of the recommended 5–8 Gb sequencing depth for Hevea would assume the assembly trends to be the same between reads generated from bark and from other tissues. To validate this, we performed Oases assembly of 5 Gb latex and 5 Gb leaf read sets using the same parameters as those for bark assembly (see Materials and methods). Assemblies were performed in incremental read amounts of 1, 3 and 5 Gb across a k-mer range of 51–77. Table 5 shows the statistics of latex and leaf assemblies which correspond to the same quantums of read assemblies in Table 3. As with bark assembly, the transcript N50 length showed a peaking trend only in the 3 and 5 Gb leaf read assemblies across the k-mer range but not in the 1 Gb assembly. On the other hand, the transcript N50 length showed a peak for all three latex assemblies across the k-mers. For the 5 Gb datasize assemblies, the optimized N50 was highest for bark (1,900 bp) followed by latex (1,281 bp) and leaf (1,086 bp) (Tables 3 and 5). Differences in statistics such as total assembled transcripts and optimized N50 values could be due to the presence of tissue-specific genes and variation in dynamic range of transcript levels in tissues, all of which have effect on the outcome of assembly. However, similar to that for bark, the optimized N50 of latex and leaf assemblies increased with read amounts. In conclusion, assembly of reads generated from different tissues did not display major or unexplainable differences between one another, and therefore, the mapping saturation test should be applicable to other Hevea tissues.
Discussion and conclusions
Knowing whether transcriptome sequencing and assembly have substantially captured all the genes expressed in a sample is an important consideration for plants having limited genomic resources as reference. Generally, the amount of reads for comprehensive coverage of a de novo transcriptome is often determined by a balance of budget, capacity of sequencing platform and guesstimates or “best practices” based on other species. In this work, we report a systematic approach which we name the “transcript mapping saturation test” to assess the amount of reads required for optimal transcriptome coverage in the Hevea rubber tree. This was made possible by the availability of 16 Gb Illumina PE RNA-Seq reads from Hevea bark which enabled us to map transcripts from incremental sub-assemblies to transcripts from the entire assembly (or the full transcriptome) in order to detect the mapping saturation point. The workflow of this methodology is outlined in Figure 3, beginning with assembly optimization and validation of the full transcriptome, followed by the mapping saturation test.
Because sequencing has become increasingly affordable, obtaining as much as 16 Gb reads per sample as a starting point is not insurmountable. Using our approach, sequencing to this extent has to be done only once in the beginning, after which the user is equipped with a guide (the colour matrix) to estimate optimal coverage of expressed genes in the plant species of interest. In developing this approach, we used the Oases assembler because Velvet, for which Oases is an extension, had previously been found to be suitable for producing quality transcripts from Hevea short reads [29]. Thus, we progressed to Oases, which additionally has the ability to resolve alternatively spliced transcripts [5]. We would suggest that a de novo project intending to adopt our approach should first test if the assembler of choice is suited to their transcriptome. Even though our method development is based on Illumina PE RNA-Seq reads, the principle behind this approach should also be applicable to other plant species and to reads from other sequencing platforms.
Although the transcript mapping approach is based on mapping saturation, this does not reduce the need to validate the assembly quality of the full transcriptome. In this work, the N50 trend was used in the initial selection of best k-mer for assembly. Subsequently, the completeness and correctness of assembled transcripts were supported by results of mapping to rubber genome scaffolds [60] whereby a significant proportion showed transcript-to-scaffold coverage of 90% and above. This was also supported by detection of all but one of 248 core genes expressed in eukaryotes [66] and significant alignments with known Hevea protein coding frames. However, we should point out that what this paper proposes is essentially a methodology; we do not specifically assert that a 5–8 Gb read depth would be sufficient for optimal transcriptome coverage universally. The optimal read depth may differ in other species depending on factors such as genome size and transcript complexity.
Materials and methods
Plant material and RNA isolation
Latex and bark shavings were obtained from 15-year old RRIM 928 Hevea brasiliensis trees growing in the Rubber Research Institute of Malaysia Research Station, Sungai Buloh. Equal volumes of latex were tapped from three trees and collected directly into 2× RNA extraction buffer [67]. The bark just below the tapping cut of the trees was scraped to remove surface matter before bark shavings (approximately 1 cm depth) were excised with a tapping knife. Young leaves of RRIM 928 trees were collected from the source bush nursery in the Rubber Research Institute of Malaysia Research Station, Sungai Buloh.
Total RNA was isolated from latex and leaf tissues using the phenol-chloroform method [67]. Bark total RNA was isolated using a modified procedure of the Qiagen RNeasy Plant Mini Kit [68]. RNA samples were assessed for quality and quantity using the Nanodrop spectrophotometer (Thermo Scientific).
Sequence generation and quality assessment
Bark, latex and leaf total RNAs (20 μg each) were sent to the Illumina Fast Track sequencing service in San Diego, USA where 200 bp fragment size libraries were produced for paired-end RNA sequencing (PE RNA-Seq). Each RNA-Seq sample was sequenced 100 nucleotides at each end (2 × 100 nt), resulting in about 50 million raw reads each from latex and leaf and nearly 170 million raw reads from the bark (Table 2). Raw reads from bark, latex and leaf are available from the NCBI Sequence Read Archive (accession nos. SRX278513-5).
Clean reads were obtained by trimming raw reads at a minimum phred score of Q = 20, followed by removal of reads below 30 bp and subsequently reads which contained ‘N’ nucleotides. Clean paired reads from the bark (163,316,702 reads; see Table 2) were referred to as the 16 Gb read set and clean paired read sets from the latex and leaves (48,650,932 and 46,062,766 reads respectively; see Table 2) as the 5 Gb read sets. These read sets were used for subsequent transcriptome assembly. Clean paired reads were classified into arbitrary nucleotide size categories to confirm good PE RNA-Seq data quality (Figure 1).
Transcriptome assembly and transcript mapping
Clean paired reads from the bark, latex and leaf (Table 2) were assembled with the Velvet (Version 1.1.05) [4] and Oases assembler (Version 0.1.22) [5] using default parameters and selection of a minimum transcript length of 100 bp. A range of hash lengths (k-mers 51–77) was used for assembly of a read set to determine the k-mer which produced the highest transcript N50 length. This best N50 value was termed as the “optimized N50” while the hash length which produced it was the “optimized k-mer”. Note: N50 length is the length of the shortest transcript whereby the sum of transcripts of equal length or longer is at least 50% of the total length of all transcripts.
Incremental quantums of bark reads (1, 3, 5, 8, 10, 13 and 16 Gb) were obtained by partitioning the subsets from the 16 Gb read set. Each subset was random as the 16 Gb read set was already fully randomized. (Similarly, 5 Gb read sets from latex and leaf were also fully randomized). Serial mapping of bark transcripts was performed using BlastN [69] and the top hit by any query transcript with e-value ≤ 1.0e-5 was counted as a match. The complete methodology for the transcript mapping saturation test is shown in Figure 3.
Bark transcript validation
For evaluation of rubber-specific ORF quality of 87,612 bark transcripts from the 16 Gb assembly (k-mer 73), 255 Hevea sequences which were confirmed to encode complete ORFs were selected from the NCBI GenBank non-redundant database (http://www.ncbi.nlm.nih.gov). These Hevea cDNAs, which were isolated by traditional gene cloning approaches such as cDNA library hybridization and PCR, were generally of high quality as they had mainly been obtained by Sanger sequencing (see Additional file 1: Table S1). Megablast [69] was used to map the 255 Hevea ORFs to 87,612 transcripts from the 16 Gb bark assembly. Top hits from this analysis (with 86-100% sequence identity match) were screened for high quality matches based on a minimum of 70% coverage of Hevea ORFs (or query coverage) in their alignments to bark transcripts (the subject) (see Table 4 and Additional file 1: Table S1).
For evaluation of completeness of assembled bark transcripts, 248 core eukaryotic genes (CEGs) of Arabidopsis thaliana were downloaded from the CEGMA resource at http://korflab.ucdavis.edu/Datasets/genome_completeness/. This approach was based on a list of 248 highly conserved but low copy number genes that had been shown to be a reliable indicator of completeness of gene space in eukaryotic species [66]. Using BlastX [69], 87,612 bark transcripts were mapped to the CEGs with any hit of e-value ≤ 1.0e-10 counted as a match.
Rubber genome scaffolds from the BioProject ID: PRJNA80191 (http://www.ncbi.nlm.nih.gov/nuccore/448814761) were used for validating bark transcripts. Bark transcripts were mapped to genome scaffolds by Exonerate (Version 2.2.0) [70] using default settings with the exception of the following parameters: heuristic mode, est2genome model and alignment score of at least 10 percent of the maximal score for each query. The significance of mapped transcripts was evaluated by calculating query coverage which is expressed as percentage of the transcript sequence (query) that overlaps with the scaffold sequence (subject). This percentage reflects the extent of bark transcript coverage in alignments to genome scaffolds. Scaffold hits were classified according to transcript coverage whereby the higher the percentage, the greater the significance of transcript-to-scaffold alignment (see Figure 2).
Availability and requirements
The datasets, SRX278513-5, supporting the results of this article are available in the NCBI Sequence Read Archive:
http://www.ncbi.nlm.nih.gov/sra/?term=SRX278513
References
Wang Z, Gerstein M, Snyder M: RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009, 10: 57-63. 10.1038/nrg2484.
Ozsolak F, Milos PM: RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 2011, 12: 87-98. 10.1038/nrg2934.
Van Verk MC, Hickman R, Pieterse CMJ, Van Wees SCM: RNA-Seq: revelation of the messengers. Trends Plant Sci. 2013, 18: 175-179. 10.1016/j.tplants.2013.02.001.
Zerbino DR, Birney E: Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18: 821-829. 10.1101/gr.074492.107.
Schulz MH, Zerbino DR, Vingron M, Birney E: Oases: robust de novo RNA-Seq assembly across the dynamic range of expression labels. Bioinformatics. 2012, 28: 1086-1092. 10.1093/bioinformatics/bts094.
Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, Li S, Yang H, Wang J, Wang J: De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20: 265-272. 10.1101/gr.097261.109.
Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol İ: ABySS: a parallel assembler for short read sequence data. Genome Res. 2009, 19: 1117-1123. 10.1101/gr.089532.108.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A: Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011, 29: 644-654. 10.1038/nbt.1883.
Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Müller WEG, Wetter T, Suhai S: Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004, 14: 1147-1159. 10.1101/gr.1917404.
Haas BJ, Zody MC: Advancing RNA-Seq analysis. Nat Biotechnol. 2010, 28: 421-423. 10.1038/nbt0510-421.
Martin JA, Wang Z: Next-generation transcriptome assembly. Nat Rev Genet. 2011, 12: 671-682. 10.1038/nrg3068.
Ward JA, Ponnala L, Weber CA: Strategies for transcriptome analysis in non-model plants. Am J Bot. 2012, 99: 267-276. 10.3732/ajb.1100334.
Barrero RA, Chapman B, Yang Y, Moolhuijzen P, Keeble-Gagnère G, Zhang N, Tang Q, Bellgard MI, Qiu D: De novo assembly of Euphorbia fischeriana root transcriptome identifies prostratin pathway related genes. BMC Genomics. 2011, 12: 600-10.1186/1471-2164-12-600.
Bräutigam A, Mullick T, Schliesky S, Weber APM: Critical assessment of assembly strategies for non-model species mRNA-Seq data and application of next-generation sequencing to the comparison of C3 and C4 species. J Exp Bot. 2011, 62: 3093-3102. 10.1093/jxb/err029.
Garg R, Patel RK, Tyagi AK, Jain M: De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 2011, 18: 53-63. 10.1093/dnares/dsq028.
Lin Y, Li J, Shen H, Zhang L, Papasian CJ, Deng HW: Comparative studies of de novo assembly tools for next-generation sequencing technologies. Bioinformatics. 2011, 27: 2031-2037. 10.1093/bioinformatics/btr319.
Zhao QY, Wang Y, Kong YM, Luo D, Li X, Hao P: Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC Bioinformatics. 2011, 12: S2-
Van Bakel H, Stout JM, Cote AG, Tallon CM, Sharpe AG, Hughes TR, Page JE: The draft genome and transcriptome of Cannabis sativa. Genome Biol. 2011, 12: R102-10.1186/gb-2011-12-10-r102.
Wong MML, Cannon CH, Wickneswari R: Identification of lignin genes and regulatory sequences involved in secondary cell wall formation in Acacia auriculiformis and Acacia mangium via de novo transcriptome sequencing. BMC Genomics. 2011, 12: 342-10.1186/1471-2164-12-342.
Mundry M, Bornberg-Bauer E, Sammeth M, Feulner PGD: Evaluating characteristics of de novo assembly software on 454 transcriptome data: a simulation approach. PLoS One. 2012, 7: e31410-10.1371/journal.pone.0031410.
O’Rourke JA, Yang SS, Miller SS, Bucciarelli B, Liu J, Rydeen A, Bozsoki Z, Uhde-Stone C, Tu ZJ, Allan D, Gronwald JW, Vance CP: An RNA-Seq transcriptome analysis of orthophosphate-deficient white lupin reveals novel insights into phosphorus acclimation in plants. Plant Physiol. 2013, 161: 705-724. 10.1104/pp.112.209254.
Collins LJ, Biggs PJ, Voelckel C, Joly S: An approach to transcriptome analysis of non-model organisms using short-read sequences. Genome Inform. 2008, 21: 3-14.
Parchman TL, Geist KS, Grahnen JA, Benkman CW, Buerkle CA: Transcriptome sequencing in an ecologically important tree species: assembly, annotation and marker discovery. BMC Genomics. 2010, 11: 180-10.1186/1471-2164-11-180.
Surget-Groba Y, Montoya-Burgos JI: Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 2010, 20: 1432-1440. 10.1101/gr.103846.109.
Gruenheit N, Deusch O, Esser C, Becker M, Voelckel C, Lockhart P: Cutoffs and k-mers: implications from a transcriptome study in allopolyploid plants. BMC Genomics. 2012, 13: 92-10.1186/1471-2164-13-92.
Gordo SMC, Pinheiro DG, Moreira ECO, Rodrigues SM, Poltronieri MC, de Lemos OF, Da Silva IT, Ramos RTJ, Silva A, Schneider H, Silva WA, Sampaio I, Darnet S: High-throughput sequencing of black pepper root transcriptome. BMC Plant Biol. 2012, 12: 168-10.1186/1471-2229-12-168.
Iorizzo M, Senalik DA, Grzebelus D, Bowman M, Cavagnaro PF, Matvienko M, Ashrafi H, Van Deynze A, Simon PW: De novo assembly and characterization of the carrot transcriptome reveals novel genes, new markers, and genetic diversity. BMC Genomics. 2011, 12: 389-10.1186/1471-2164-12-389.
Su CI, Chao YT, Chang YCA, Chen WC, Chen CY, Lee AY, Hwa KT, Shih MC: De novo assembly of expressed transcripts and global analysis of the Phalaenopsis aphrodite transcriptome. Plant Cell Physiol. 2011, 52: 1501-1514. 10.1093/pcp/pcr097.
Chow K-S, Mat-Isa MN, Bahari A, Ghazali A-K, Alias H, Mohd-Zainuddin Z, Hoh C-C, Wan K-L: Metabolic routes affecting rubber biosynthesis in Hevea brasiliensis latex. J Exp Bot. 2012, 63: 1863-1871. 10.1093/jxb/err363.
Kudapa H, Bharti AK, Cannon SB, Farmer AD, Mulaosmanovic B, Kramer R, Bohra A, Weeks NT, Crow JA, Tuteja R, Shah T, Dutta S, Gupta DK, Singh A, Gaikwad K, Sharma TK, May GD, Singh NK, Varshney RK: A comprehensive transcriptome assembly of pigeonpea (Cajanus cajan L.) using Sanger and second-generation sequencing platforms. Mol Plant. 2012, 5: 1020-1028. 10.1093/mp/ssr111.
Hao DC, Ge GB, Xiao PG, Zhang YY, Yang L: The first insight into the tissue specific Taxus transcriptome via Illumina second generation sequencing. PLoS One. 2011, 6: e21220-10.1371/journal.pone.0021220.
Logacheva MD, Kasianov AS, Vinogradov DV, Samigullin TH, Gelfand MS, Makeev VJ, Penin AA: De novo sequencing and characterization of floral transcriptome in two species of buckwheat (Fagopyrum). BMC Genomics. 2011, 12: 30-10.1186/1471-2164-12-30.
Mizrachi E, Hefer CA, Ranik M, Jourbert F, Myburg AA: De novo assembled expressed gene catalog of a fast-growing Eucalyptus tree produced by Illumina mRNA-Seq. BMC Genomics. 2010, 11: 681-10.1186/1471-2164-11-681.
Franssen SU, Shrestha RP, Bräutigam A, Bornberg-Bauer E, Weber APM: Comprehensive transcriptome analysis of the highly complex Pisum sativum genome using next generation sequencing. BMC Genomics. 2011, 12: 227-10.1186/1471-2164-12-227.
Krishnan NM, Pattnaik S, Deepak SA, Hariharan AK, Gaur P, Chaudhary R, Jain P, Vaidyanathan S, Krishna PGB, Panda B: De novo sequencing and assembly of Azadirachta indica fruit transcriptome. Curr Sci. 2011, 101: 1553-1561.
Natarajan P, Parani M: De novo assembly and transcriptome analysis of five major tissues of Jatropha curcas L. using GS FLX titanium platform of 454 pyrosequencing. BMC Genomics. 2011, 12: 191-10.1186/1471-2164-12-191.
Sui C, Zhang J, Wei J, Chen S, Li Y, Xu J, Jin Y, Xie C, Gao Z, Chen H, Yang C, Zhang Z, Xu Y: Transcriptome analysis of Bupleurum chinense focusing on genes involved in the biosynthesis of saikosaponins. BMC Genomics. 2011, 12: 539-10.1186/1471-2164-12-539.
Duan J, Xia C, Zhao G, Jia J, Kong X: Optimizing de novo common wheat transcriptome assembly using short-read RNA-Seq data. BMC Genomics. 2012, 13: 392-10.1186/1471-2164-13-392.
Hyun TK, Rim Y, Jang HJ, Kim CH, Park J, Kumar R, Lee S, Kim BC, Bhak J, Nguyen-Quoc B, Kim SW, Lee SY, Kim JY: De novo transcriptome sequencing of Momordica cochinchinensis to identify genes involved in the carotenoid biosynthesis. Plant Mol Biol. 2012, 79: 413-427. 10.1007/s11103-012-9919-9.
Lulin H, Xiao Y, Pei S, Wen T, Shangqin H: The first Illumina-based de novo transcriptome sequencing and analysis of safflower flowers. PLoS One. 2012, 7: e38653-10.1371/journal.pone.0038653.
Mutasa-Göttgens ES, Joshi A, Holmes HF, Hedden P, Göttgens B: A new RNA Seq-based reference transcriptome for sugar beet and its application in transcriptome-scale analysis of vernalization and gibberellin responses. BMC Genomics. 2012, 13: 99-10.1186/1471-2164-13-99.
Sloan DB, Keller SR, Berardi AE, Sanderson BJ, Karpovich JF, Taylor DR: De novo transcriptome assembly and polymorphism detection in the flowering plant Silene vulgaris (Caryophyllaceae). Mol Ecol Resour. 2012, 12: 333-343. 10.1111/j.1755-0998.2011.03079.x.
Zhang XM, Zhao L, Larson-Rabin Z, Li DZ, Guo ZH: De novo sequencing and characterization of the floral transcriptome of Dendrocalamus latiflorus (Poaceae: Bambusoideae). PLoS One. 2012, 7: e42082-10.1371/journal.pone.0042082.
Zhao Z, Tan L, Dang C, Zhang H, Wu Q, An L: Deep-sequencing transcriptome analysis of chilling tolerance mechanisms of a subnival alpine plant, Chorispora bungeana. BMC Plant Biol. 2012, 12: 222-10.1186/1471-2229-12-222.
Bai S, Saito T, Sakamoto D, Ito A, Fujii H, Moriguchi T: Transcriptome analysis of Japanese pear (Pyrus pyrifolia Nakai.) flower buds transitioning through endodormancy. Plant Cell Physiol. 2013, 54: 1132-1151. 10.1093/pcp/pct067.
Gil-Amado JA, Gomez-Jimenez MC: Transcriptome analysis of mature fruit abscission control in olive. Plant Cell Physiol. 2013, 54: 244-269. 10.1093/pcp/pcs179.
Ramilowski JA, Sawai S, Seki H, Mochida K, Yoshida T, Sakurai T, Muranaka T, Saito K, Daub CO: Glycyrrhiza uralensis transcriptome landscape and study of phytochemicals. Plant Cell Physiol. 2013, 54: 697-710. 10.1093/pcp/pct057.
Van Moerkercke A, Fabris M, Pollier J, Baart GJE, Rombauts S, Hasnain G, Rischer H, Memelink J, Oksman-Caldentey KM, Goossens A: CathaCyc, a metabolic pathway database built from Catharanthus roseus RNA-Seq data. Plant Cell Physiol. 2013, 54: 673-685. 10.1093/pcp/pct039.
Zhang J, Wu K, Zeng S, da Silva JAT, Zhao X, Tian CE, Xia H, Duan J: Transcriptome analysis of Cymbidium sinense and its application to the identification of genes associated with floral development. BMC Genomics. 2013, 14: 279-10.1186/1471-2164-14-279.
Bennett MD, Leitch I: Nuclear DNA amounts in angiosperms-583 new estimates. Ann Bot. 1997, 80: 169-196. 10.1006/anbo.1997.0415.
Han KH, Shin DH, Yang J, Kim IJ, Oh SK, Chow KS: Genes expressed in the latex of Hevea brasiliensis. Tree Physiol. 2000, 20: 503-510. 10.1093/treephys/20.8.503.
Ko J-H, Chow K-S, Han K-H: Transcriptome analysis reveals novel features of the molecular events occurring in the laticifers of Hevea brasiliensis (para rubber tree). Plant Mol Biol. 2003, 53: 479-492.
Chow K-S, Wan K-L, Mat-Isa M-N, Bahari A, Tan S-H, Harikrishna K, Yeang H-Y: Insights into rubber biosynthesis from transcriptome analysis of Hevea brasiliensis latex. J Exp Bot. 2007, 58: 2429-2440. 10.1093/jxb/erm093.
Mat-Isa M-N, Chow K-S, Mohamad A-F-H, Shahrum M-Y, Hoh C-C, Mohd-Amin M-R, Zainal K-A, Yeang H-Y, Wan K-L: NRESTdb: access to the transcriptome of natural rubber latex. J Rubber Res. 2009, 12: 229-238.
Xia Z, Xu H, Zhai J, Li D, Luo H, He C, Huang X: RNA-Seq analysis and de novo transcriptome assembly of Hevea brasiliensis. Plant Mol Biol. 2011, 77: 299-308. 10.1007/s11103-011-9811-z.
Pootakham W, Chanprasert J, Jomchai N, Sangsrakru D, Yoocha T, Therawattanasuk K, Tangphatsornruang S: Single nucleotide polymorphism marker development in the rubber tree, Hevea brasiliensis (Euphorbiaceae). Am J Bot. 2011, 98: e337-e338. 10.3732/ajb.1100228.
Triwitayakorn K, Chatkulkawin P, Kanjanawattanawong S, Sraphet S, Yoocha T, Sangsrakru D, Chanprasert J, Ngamphiw C, Jomchai N, Therawattanasuk K, Tangphatsornruang S: Transcriptome sequencing of Hevea brasiliensis for development of microsatellite markers and construction of a genetic linkage map. DNA Res. 2011, 18: 471-482. 10.1093/dnares/dsr034.
Li D, Deng Z, Qin B, Liu X, Men Z: De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST-SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.). BMC Genomics. 2012, 13: 192-10.1186/1471-2164-13-192.
Duan C, Argout X, Gébelin V, Summo M, Dufayard JF, Leclercq J, Hadi K, Piyatrakul P, Pirrello J, Rio M, Champion A, Montoro P: Identification of the Hevea brasiliensis AP2/ERF superfamily by RNA sequencing. BMC Genomics. 2013, 14: 30-10.1186/1471-2164-14-30.
Rahman AY, Usharraj AO, Misra BB, Thottathil GP, Jayasekaran K, Feng Y, Hou S, Ong SY, Ng FL, Lee LS, Tan HS, Muhd Sakaff MKL, Teh BS, Khoo BF, Badai SS, Ab Aziz N, Yuryev A, Knudsen B, Dionne-Laporte A, Mchunu NP, Yu Q, Langston BJ, Freitas TAK, Young AG, Chen R, Wang L, Najimudin N, Saito JA, Alam M: Draft genome sequence of the rubber tree Hevea brasiliensis. BMC Genomics. 2013, 14: 75-10.1186/1471-2164-14-75.
Qin Y, Shi F, Tang C: Molecular characterization and expression analysis of cDNAs encoding four Rab and two Arf GTPases in the latex of Hevea brasiliensis. Plant Physiol Biochem. 2011, 49: 729-737. 10.1016/j.plaphy.2011.03.006.
Ruderman S, Kongsawadworakul P, Viboonjun U, Mongkolporn O, Chrestin H: Mitochondrial/Cytosolic Acetyl CoA and rubber biosynthesis genes expression in Hevea brasiliensis latex and rubber yield. Kasetsart J (Nat Sci). 2012, 46: 346-362.
Piyatrakul P, Putranto RA, Martin F, Rio M, Dessailly F, Leclercq J, Dufayard JF, Lardet L, Montoro P: Some ethylene biosynthesis and AP2/ERF genes reveal a specific pattern of expression during somatic embryogenesis in Hevea brasiliensis. BMC Plant Biol. 2012, 12: 244-10.1186/1471-2229-12-244.
Putranto RA, Sanier C, Leclercq J, Duan C, Rio M, Jourdan C, Thaler P, Sabau X, Argout X, Montoro P: Differential gene expression in different types of Hevea brasiliensis roots. Plant Sci. 2012, 183: 149-158.
Wang Z, Fang B, Chen J, Zhang X, Luo Z, Huang L, Chen X, Li Y: De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweetpotato (Ipomoea batatas). BMC Genomics. 2010, 11: 726-10.1186/1471-2164-11-726.
Parra G, Bradnam K, Ning Z, Keane T, Korf I: Assessing the gene space in draft genomes. Nucleic Acids Res. 2009, 37: 289-297. 10.1093/nar/gkn916.
Kush A, Goyvaerts E, Chye ML, Chua NH: Laticifer-specific gene expression in Hevea brasiliensis (rubber tree). P Natl Acad Sci USA. 1990, 87: 1787-1790. 10.1073/pnas.87.5.1787.
MacKenzie DJ, McLean MA, Mukerji S, Green M: Improved RNA extraction from woody plants for the detection of viral pathogens by reverse transcription-polymerase chain reaction. Plant Dis. 1997, 81: 222-226. 10.1094/PDIS.1997.81.2.222.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215: 403-410.
Slater GS, Birney E: Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005, 6: 31-10.1186/1471-2105-6-31.
Acknowledgements
This work was supported by the Malaysian Rubber Board. The authors thank Siti Zakiah Zailani and V. Mony Rajan for field and laboratory assistance, and Haizarudin Amin Nordin for graphic design.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
A-KG, C-CH and K-SC conceptualized the transcript saturation mapping methodology. A-KG and C-CH performed the transcriptome assembly, transcript mapping and other bioinformatics analysis. K-SC wrote the manuscript with contributions from A-KG and C-CH during final editing. K-SC and ZMZ initiated the tissue transcriptome sequencing project including the selection of tree material, RNA preparations and other supporting analyses. All authors approved the final manuscript.
Electronic supplementary material
13104_2013_2649_MOESM1_ESM.xlsx
Additional file 1: Table S1: Megablast analysis of 255 Hevea complete ORFs against 87,612 bark transcripts from the optimized 16 Gb bark assembly. The last 5 rows show Hevea ORFs which did not have any Megablast hit. Percentage identity of alignments in hits by 250 Hevea ORFs ranged from 86-100% (see column J). Highlighted rows (total of 50) indicate hits which showed less than 70% utilization of ORF sequence length in their alignments (see column K). (XLSX 44 KB)
13104_2013_2649_MOESM2_ESM.docx
Additional file 2: Table S2: Colour matrix (full version) representing the transcript mapping saturation test results. BlastN matches (e-value ≤ 1.0e-5) by subsets of bark transcripts to 87,612 transcripts from the optimized 16 Gb bark assembly are shown as numbers and percentages (in brackets) of the total. Numbers in bold correspond to the assembly with the optimized k-mer and transcript N50 length for a particular datasize. The 1 Gb assembly transcripts are not included since transcript N50 length was not optimized by any k-mer for this datasize. (DOCX 20 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Chow, KS., Ghazali, AK., Hoh, CC. et al. RNA sequencing read depth requirement for optimal transcriptome coverage in Hevea brasiliensis. BMC Res Notes 7, 69 (2014). https://doi.org/10.1186/1756-0500-7-69
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-0500-7-69