
Abstract
Background
Despite international success in reducing ozone-depleting emissions, ultraviolet radiation (UV) is not expected to decrease for several decades. Thus, it is pressing to implement tools that allow investigating the capacity of wildlife to respond to excessive UV, particularly species like cetaceans that lack anatomical or physiological protection. One approach is to examine epidermal expression of key genes involved in genotoxic stress response pathways. However, quantitation of mRNA transcripts requires previous standardization, with accurate selection of control and target genes. The latter is particularly important when working with environmental stressors such as UV that can activate numerous genes.
Results
Using 20 epidermal biopsies from blue, fin and sperm whale, we found that the genes encoding the ribosomal proteins L4 and S18 (RPL4 and RPS18) were the most suitable to use as controls, followed by the genes encoding phosphoglycerate kinase 1 (PGK1) and succinate dehydrogenase complex subunit A (SDHA). A careful analysis of the transcription pathways known to be activated by UV-exposure in humans and mice led us to select as target genes those encoding for i) heat shock protein 70 (HSP70) an indicator of general cell stress, ii) tumour suppressor protein P53 (P53), a transcription factor activated by UV and other cell stressors, and iii) KIN17 (KIN), a cell cycle protein known to be up-regulated following UV exposure. These genes were successfully amplified in the three species and quantitation of their mRNA transcripts was standardised using RPL4 and RPS18. Using a larger sample set of 60 whale skin biopsies, we found that the target gene with highest expression was HSP70 and that its levels of transcription were correlated with those of KIN and P53. Expression of HSP70 and P53 were both related to microscopic sunburn lesions recorded in the whales’ skin.
Conclusion
This article presents groundwork data essential for future qPCR-based studies on the capacity of wildlife to resolve or limit UV-induced damage. The proposed target genes are HSP70, P53 and KIN, known to be involved in genotoxic stress pathways, and whose expression patterns can be accurately assessed by using two stable control genes, RPL4 and RPS18.
Similar content being viewed by others


Background
A current and significant threat to marine ecosystems across the globe is the high level of solar ultraviolet radiation (UV) that continues to reach our biosphere [1], a situation that is not expected to change for several decades [2, 3]. Recently we showed that cetaceans develop macro and microscopic skin lesions as a consequence of solar exposure [4], a study that widened the known range of marine species that can be affected by UV exposure [1, 5]. Taking into consideration that many cetacean species are listed as endangered or threatened by the International Union for the Conservation of Nature, it is pressing to implement tools that allow us to evaluate their capacity to respond to current stressors [6], including excessive UV. One potential avenue for such studies is to examine the expression of key genes involved in genotoxic stress response pathways.
Continuous and unresolved exposure to UV damages DNA, in turn activating a network of interactive pathways that include participation of aberrant DNA sensors, signal transducers and effectors, which interact to execute appropriate responses [7]. Although the identities of the sensors are still unclear, transducers include four sets of conserved proteins, namely phospho-inositide kinases, check point kinases 1 and 2 and the group of BRCT (breast cancer C-terminal repeats) proteins [7]. Effectors involved in DNA repair, transcription regulation and cell cycle control, comprise proteins such as the tumour suppressor protein P53 [7, 8]. This interactive network that involves hundreds of genes is complex [7, 9] and to our knowledge has not been studied in wild mammals.
Real-time or quantitative PCR (hereafter qPCR) is currently the accepted method for quantifying mRNA transcripts [10]. However, despite the technique’s accuracy, sensitivity and speed [11], variations in the amount and integrity of starting material, transcription- and amplification efficiency rates, as well as the occurrence of inhibitors, can lead to quantitation errors. In this context, normalization of data is an essential step that must precede gene expression quantitation [10–12]. Most studies use endogenous reference genes as an internal control to calculate relative expression values of the genes of interest. Such internal controls should not vary in their expression levels amongst individuals, particularly under experimental conditions relevant to the question of interest. Accurate selection of reference genes is therefore central to interpreting quantitative PCR results.
A recent study on free-ranging striped dolphins tested the expression stability of ten commonly used control genes in skin biopsies and found that the genes coding for the tyrosine 3-monoxygenase (YWHAZ) and glyceraldehyde-3P-dehydrogenase (GAPDH) were the most reliable ones followed by those coding for the ribosomal proteins S18 (RSP18) and the ribosomal proteins L4 (RPL4) [13]. However, a second study that evaluated gene expression changes associated with organochlorine exposure in striped dolphin fibroblast cultures showed that the most reliable gene was the one encoding succinate dehydrogenase complex subunit A (SDHA) [14]. These studies illustrate how, depending on the purpose of the study and the target tissue to be analysed, different genes might be better suited as controls. Here, as part of a larger ongoing study on the effects of exposure to UV on whales [4], we examined expression levels and assessed stability of selected reference genes in skin biopsies collected from three species of large whales and used two of the most suitable genes to study expression patterns of key genes involved in genotoxic stress pathways.
Methods
A schematic representation of the general method used in this article is provided in the Additional file 1: Figure S1.
Skin biopsy sampling
Skin biopsies were collected from blue, fin and sperm whales in the Gulf of California, Mexico, between January and June of 2007 to 2009. Samples were collected using a 7 mm stainless-steel dart and a crossbow. An epidermal sub-sample was immediately stabilised in RNA later (Qiagen, UK) and ultrafrozen until processing. This study is registered as project WLE/0405 at the Institute of Zoology and conforms to the regulations on Animal Ethics. Samples were collected under permits SGPA/DGVS/00506/08, SGPA/DGVS/09760/08 and SGPA/DGVS/08021/06 issued by the Mexican Secretaría del Medio Ambiente y Recursos Naturales (SEMARNAT).
RNA extraction and cDNA transformation
Total RNA was extracted using the RNeasy® Mini Kit (Qiagen, UK) according to the manufacturer’s instructions. The quantity of RNA obtained was determined for each sample by measuring optical density (OD) with a Nanodrop® ND-1000 UV–vis spectrophotometer (Thermo Scientific, UK). OD 260/280 and 260/230 ratios were used to evaluate RNA purity. Presence of intact RNA subunits 28S and 18S were checked by automated capillary-electrophoresis, using QIAxcel (Qiagen,UK; see Additional file 1: Figure S2). Before performing reverse transcription, all samples were diluted to a final concentration of 50 ng/μl as done previously [15]. Complementary DNA (cDNA) was obtained by reverse transcription using the QuantiTect® Reverse Transcription Kit (Qiagen, UK). This procedure includes a first step of DNA digestion. Only one retro-transcription was run per sample. Prior to use, cDNA was diluted 1:25 with nuclease free water and conserved at −20°C.
Twenty individual RNA samples (belonging to 7 blue whales, 7 fin whales and 6 sperm whales) were selected to measure expression of internal control genes. A total of 60 samples (belonging to 22 blue whales, 22 fin whales and 16 sperm whales) that included the 20 samples selected for the previously described analysis, were used to measure expression of target genes. The maximum and minimum RNA concentrations obtained for the samples selected were 634 ng/μl and 51 ng/μl (mean for all samples: 233 ng/μl ± 42.38 SE). The samples showed an absorbance ratio at 260/280 nm between 2.1 and 1.81, and an absorbance ratio at 260/230 nm greater than 0.95 except for four samples with an absorbance ratio of 0.85, 0.75, 0.74 and 0.59. Criteria for inclusion of the sample in the study include the presence of one or two intact bands during electrophoresis (see Additional file 1: Figure S2).
Internal control gene candidates
We identified potentially-suitable internal control genes as those whose levels of expression are known to not be involved in any process related to UV exposure and that have been shown to be “stable” in other marine mammal species. The primer sets of the four control genes that fitted these criteria were obtained from a previous study conducted on striped dolphins [13], being RSP18, RPL4, SDHA and PGK1 (phosphoglycerate kinase 1). Although GAPDH and YWHAZ have been reported as reliable control genes in dolphins [13] and were used recently in a study that assessed toxicological stress in fin whales [16], these genes were not included in our study because their expression has been shown to be affected directly or indirectly by UV exposure [17–19]. In vitro and in vivo studies that have examined human keratinocyte responses to solar irradiation showed that levels of GAPDH expression shifted significantly 24 h post irradiation [19, 20]. Another study demonstrated low stability of GAPDH in UV irradiated keratinocytes [21], and, perhaps more importantly, the heat shock protein 70, an indicator of cellular stress [22], is known to alter the cellular amount of GAPDH[23]. Finally, YWHAZ not only interacts with the process of apoptosis [24] but has also been proved to be involved with the skin carcinogenesis process [17]. Instead, we selected RSP18 and RPL4, the next most stable genes in dolphin skin [13]. Furthermore, although PGK1 was previously considered less suitable as a control gene in striped dolphin skin [13], we selected it for our study because, together with SDHA, it is considered the most reliable control gene when studying the effects of exposure to UV-B radiation on human keratinocytes [21]. The selected primer pairs were commercially synthesized and tested for specificity in the three whale species as described below.
Target gene candidates
Important genes involved in the complex UV-response pathway are those encoding the heat shock proteins (HSPs), also called stress proteins. HSPs are involved in the recovery of proteins that can unfold under stress [22, 25]. HSPs can either repair the damaged proteins by refolding their structure, or can degrade them if damage is too extensive. The HSPs are also involved in intracellular protein transport between compartments and disposal of old proteins as well as in generating an immune response as they participate in the presentation of abnormal peptides (i.e. antigens) to immune effectors on the surface of abnormal cells [26]. The different families of HSPs, classified according to their structure, function and weight (in kilodaltons), include HSP100, HSP90, HSP70, HSP60, HSP40 and the small heat shock proteins family. One of the most studied HSPs is HSP70, which is the major stress-induced member of the family, specifically involved in protein-folding and protein membrane transport [22]. Humans and laboratory animals studies have showed that under severe UV irradiance, the gene coding for HSP70 is over-expressed and helps to protect against UV-induced epidermal damage, including apoptosis and DNA damage [27].
To minimize the number of heritable mutations transferred from one cell to its daughters, the structure of chromosomes is continuously under surveillance. When damage is detected, repair and cell-cycle progression are coordinated [7]. Protein P53, also termed ‘tumour suppressor protein’ because mutations of this gene can promote cancer [8, 28], is actively involved in different response pathways including cell cycle arrest, DNA repair and, when unrepairable, apoptosis [8]. P53 is a central transcription factor in cellular stress responses and its synthesis is controlled by dozens of other proteins [29]. One of P53’s most important transcriptional targets is the cyclin-dependent kinase inhibitor p21, which can provoke cell-cycle arrest at G1 phase [29]. P53 also participates, via transcriptional regulation and direct interaction, in DNA repair mechanisms such as nucleotide excision repair (NER), although it has been shown that P53 is not always essential to NER [29]. P53 also induces the expression of DDB2 and XPC genes, which encode factors of the global genome repair mechanism (GGR) [9, 29]. While programmed cell death can occur independently of P53, this protein is involved, via various routes, with apoptosis, its most important suppressive function [8, 29]. Finally, P53 is also involved in the tanning response [30].
A gene recently found to be implicated in cellular responses to UV-induced damage is the gene coding for KIN17 protein (hereafter KIN). The KIN gene is expressed in all tissues and its expression significantly increases after UV exposure [31, 32]. Experimental trials have shown that DNA-bound KIN protein accumulates 24 h after irradiation and that KIN can arrest the cell cycle prior to DNA replication [33, 34]. It has been proposed that the KIN protein helps to overcome the perturbation of DNA replication in unrepaired DNA sites [33].
Target gene primer design
Primers were designed for the three selected target genes. For each gene, cDNA sequences listed for other species were searched for in the NCBI GenBank database (http://www.ncbi.nlm.nih.gov). For KIN, primers were designed by aligning highly conserved exonic regions of this gene in cow, horse, chimpanzee, mouse and human genomes (see Additional file 1). For P53, primers were designed by aligning dolphin, cow, pig and human sequences (see Additional file 1). For HSP70 primers, we used the cDNA sequence reported for a north Atlantic Right whale [GenBank: ES556841.1] [35] (see Additional file 1). Sequences were aligned using the free Multiple Alignment software ClustalW (http://www.ebi.ac.uk/Tools/msa/clustalw2/). Primer pairs were designed within conserved regions (see Additional file 1), ideally spanning two exons to avoid DNA amplification. The primers were targeted to amplify 100–200 nucleotides in order to reduce potential noise caused by eventual RNA degradation and to decrease variation during qPCR. Each primer was 18–24 bp length, had 50–55% GC composition, had a melting temperature of 60°C and ended with a G or C base (3’). Occurrence of hairpins, homodimers and heterodimers were checked in the Integrated DNA technology freeware (IDT, http://eu.idtdna.com/analyzer/Applications/OligoAnalyzer). The primers successfully amplified in the three whale species and generated a single and well-defined band and a unique qPCR dissociation curve. Specificity of the primers was further confirmed by bi-directional sequencing of amplified products.
PCR validation
For each species, each primer pair was tested in two randomly selected samples by independent PCRs. Volume per reaction was 12.5 μl and contained 1× PCR buffer (Tris-Cl, KCl, (NH4)2SO4 and MgCl2; Qiagen, UK), 0.2 mM dNTPs (Bioline, UK), 0.4 μM of each primer, 0.325 U of HotStarTaq®Plus DNA polymerase (Qiagen, UK) and 1 μl of cDNA. The PCR conditions were 95°C for 5 min, 35 cycles of 94°C for 1 min, 60°C for 45 s, 72°C for 30 s, and a final extension at 72°C for 10 min. Amplification products were run on a 2% agarose gel stained with 2.5× SYBR® Safe DNA stain gel (corresponding to 0.1 μl per ml; Invitrogen, USA). Fragments were excised and cleaned using the QIAquick® gel extraction kit (Qiagen, UK) before being sent for bi-directional Sanger sequencing (Cogenics, UK). Each sequence obtained (see Additional file 1) was analysed in the Basic Local Alignment Tool (BLAST, http://www.ncbi.nlm.nih.gov/BLAST) and it was confirmed that the PCR product amplified corresponded to the gene targeted.
Standard curve and amplification efficiency
We used PCR products as a template for the construction of the standard curves for each of the genes tested. For this, three amplified products of each gene were run on a 2% agarose gel and the excised bands cleaned using the QIAquick® gel extraction kit (Qiagen, UK). PCR quantity was measured with the Nanodrop® ND-1000 UV–vis spectrophotometer (Thermo Scientific, UK) and dilutions were made to obtain stocks containing 102 to 108 copies of PCR product per μl. Each dilution was run in triplicate in a 7300 Real-Time PCR System (Applied Biosystems, UK) as described below. The logarithm of the product quantity obtained for each threshold value (Ct) was plotted against the Ct values to obtain the linear correlation coefficient (R2) for each gene. The slope of the curve was used to calculate qPCR amplification efficiencies (E=101/-slope-1) for each set of primers [36].
Real-time quantitative PCR (qPCR) using SYBR green
Real-time quantitative PCR (qPCR) was performed in a 7300 Real-Time PCR System (Applied Biosystems, UK) using Power SYBR Green PCR Master Mix (Applied Biosystems, UK). The total volume of each qPCR reaction was 10 μl, which included forward and reverse primers (500 nM), 1× Power SYBR Green PCR Master Mix (Applied Biosystems, UK) and the cDNA sample (2 μl of a 1:25 cDNA dilution). For each control gene, a 96-well reaction plate was set up to include all 20 samples, seven serial dilution points (10-fold step) of the same gene and three no-template controls (NTC) to ensure detection of inadvertent contamination. For the target gene analyses, each plate contains all the target genes and control genes set up for four samples and three no-template controls (NTC). Three RT-negative controls were run in the first plate to confirm that DNA elimination was successful. All samples were run in triplicate.
Cycling conditions were an initial 2 min at 50°C, followed by 15 min at 95°C, and 40 cycles of 15 s at 95°C, 1 min at 60°C and 1 min at 72°C. A melting curve analysis (95°C/15 sec; 60°C/1 min; 95°C/15 sec; 60°C/15 sec) was added at the end of the final cycle to detect non specific amplifications (see Additional file 1: Figure S3). Threshold values (Ct) and their transformation to quantities (Qt) were automatically determined with the 7300 Real-Time PCR System software (Applied Biosystems, UK). The mean of the triplicate reactions were calculated for each sample (standard deviation = ± 10% of the mean).
Analysis of internal control gene expression stability
Gene expression values were analyzed using the freeware packages BestKeeper (http://gene-quantification.com/bestkeeper.html), geNorm (http://medgen.ugent.be/~jvdesomp/genorm) and NormFinder (http://www.mdl.dk/publicationsnormfinder.htm). The algorithms used by these packages have been developed for a minimum of three genes [12, 37, 38].
Briefly, BestKeeper ranks the control gene candidates according to the standard deviation of their Ct-value (SDCt value). The correlation (Pearson correlation coefficient and probability) between each gene and index, corresponding to the geometric mean of the Ct-value of all suitable candidate genes, were calculated in order to determine the best suited genes [37]. For this, BestKeeper assumes that the Ct-value for each gene is normally distributed, an assumption that was confirmed by a Shapiro test using the R software [39]. The software geNorm ranks the candidate genes according to their average expression stability M. Briefly, a variation parameter V jk is calculated for every combination of two internal control genes j and k. Vjk is equal to the standard deviation (SD) of the sum of the logarithmic transformed level expression ratio of the two tested genes measured for each sample i (see Equation 1) [12].
M j is determined for each gene j as the arithmetic mean of all V jk [12]. Ideally, the expression ratio of two tested genes is identical in all samples. Increasing variation in ratio corresponds to decreasing expression stability [12]. Finally, normFinder estimates the overall expression variation of the control gene candidates and rank those according to their expression stability [38].
For geNorm and NormFinder, we used transformed Ct values corresponding to the quantities obtained with the standard curve [12, 38], whereas raw Ct values were used for BestKeeper [37].
Analysis of target gene expression
Gene expression levels were analysed using the relative quantification method (level of expression of the target gene relative to internal control genes) that is based on the ΔCt method (Ct target gene- geometric mean Ctcontrol genes) [12, 36]. In order to control for inter-plate qPCR variations, which might reflect unintentional grouping of the data and potentially hide effects on the levels of gene expression, we used linear mixed effect modelling [40] using the lme function in the nlme package [41] and defined ‘plate’ as a random factor. Models were built in R [39] and we used a top-down strategy to determine which variables explained a significant fraction of the data [40]. Violation of normality and homoscedasticity assumptions were corrected by logarithmic transformation of the response variable. As lower ΔCt values represent higher levels of expression, interpretation was easier by using a negative transformation of the response variable (− log (ΔCtgene)).
Results
We successfully amplified the internal control genes RSP18, RPL4, SDHA and PGK1, and the three target genes HSP70, P53 and KIN (see partial gene sequences in Additional file 1). Each primer set (see Table 1) generated a single and well-defined band and had a unique dissociation curve (see Additional file 1: Figure S3). The R2 and amplification efficiency of all genes tested ranged from 0.991 to 1.000 and 0.92 to 101, respectively (see Table 1).
Stability of internal control gene expression
Bestkeeper analysis showed that all the selected genes were stably expressed in the epidermis of all three whale species (SDCt value ≤ 1; Table 2; Additional file 1: Figure S4) and thus can be considered as suitable control genes [37]. When considering all species together, the two most stable genes, according to their SDCt value, were RPL4, and RPS18 followed by SDHA and PGK1 (Table 2). The four candidate genes were used for the calculation of the BestKeeper index. When pooling samples from all three species, the most suitable genes according to their coefficient of correlation were, in order, RPS18, RPL4 or PGK1 and SDHA (Table 2). When looking at each species separately, RPS18 had the highest correlation coefficient in all cases, while the second best candidate gene differed amongst species, being RPL4 for fin and sperm whales and PGK1 for blue whales (Table 3). Sample integrity was of high quality and all intrinsic variances (InVar [±x-fold]) ranged between 0.05 and 0.97. One sample showed a higher InVar value (2.43) but was still within the acceptable range [37].
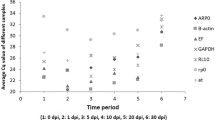
GeNorm analysis showed that the expression of the four selected genes showed strong stability; the highest M value (0.98) detected (SDHA) being lower than the program’s default limit (M=1.5). The two most stable genes for the three species were RPS18 and RPL4 (Figure 1). When looking at each species separately, the best candidate genes for blue and fin whales were RPS18 and RPL4 whereas for sperm whales RPS18 and PGK1 showed higher stability (Table 3). The optimal number of control genes needed for qPCR normalization was more than four genes when pooling the three species (V3/4 = 0.237 > 0.15 default cut-off value), whereas when looking at each species separately, less than three genes were needed.

Gene expression stability of the internal control gene candidates. The average expression stability (M) values of the candidate genes were calculated with geNorm after stepwise exclusion of the least stable gene (left axis; M value from the least stable on the left to the most stable on the right: 0.705, 0.430 and 0.392; dotted line). The right axis corresponds to the stability values calculated with NormFinder (stability value from left, least stable, to right, most stable: 0.501, 0.275, 0.227 and 0.184; plain line).
NormFinder analysis showed that when analysing all three species together, the gene with the lowest (best) stability value was RPL4 (stability value: 0.184; Figure 1) and the most suitable gene combination was RPS18 + RPL4, having a stability value of 0.184. When analysing each species separately, the best gene in all cases was RPS18, concurring with the results generated with the other software (Table 3).
All three software packages used concurred in selecting RPL4 and RPS18 as best intra- and interspecies control genes and these were used for subsequent expression analyses of target genes. The efficiencies of the target and internal control genes were within the range of the accepted 10% of each other (see Table 1), which made it possible to use the Delta Ct method [36].
Levels of target gene expression
The gene with the highest expression level was the gene coding for the heat shock protein 70 (HSP70; ΔCt mean = 5.22 ± 0.21 SE, n = 60). Expression levels for HSP70 were 1.31 times the levels observed for the tumour protein P53 gene (ΔCt mean = 6.72 ± 0.12 SE, n = 59) and 1.69 times that of the gene coding for the KIN protein (ΔCt mean = 8.85 ± 0.11 SE, n = 60) (Figure 2A). To investigate whether gene expression levels were correlated, we fitted three mixed effects models, one for each of the target genes (Table 4). Direct relationships were observed between the expressions of KIN and HSP70 and the expression of P53 and HSP70 (Table 4; Figure 2B and C, respectively). To investigate whether gene expression levels were related to the presence of UV-induced microscopic damage such as intracellular oedema and cytoplasmic vacuolation previously recorded (see [4] for details), we constructed three mixed effect models, one for each of the target genes (Table 5). Interestingly, the models showed that expression of P53 and HSP70 was lower when oedema was present (Table 5; Figure 3A). Vacuolation did not significantly predict gene expression and thus was not retained in the final models (Table 5). However, when observed graphically, there appears to be a slight trend where higher levels of gene expression tend to be observed when vacuoles are present (Figure 3B). KIN expression was not significantly correlated with any of the epidermal lesions included in the full model (Table 5).

HSP70, P53 and KIN expression levels and correlations. A) Mean levels of expression (in ΔCt, y axis inverted) of HSP70 (n=60), P53 (n=59) and KIN (n=60). Bars = ± 95% CI. B) Correlation between KIN and HSP70 expression levels (in ΔCt). C) Correlation between P53 and HSP70 expression levels (in ΔCt). The lines show regression lines. Lower ΔCt values represent higher levels of expression.

Relation between gene expression levels and the presence of UV-induced microscopic damage. A) Relationship between mean expression levels of P53 and HSP70 genes (in ΔCt, y axis is inverted) and occurrence of intracellular oedema (absence: n = 20; 1; presence: n = 37). B) Variation in HSP70 and P53 expression levels (in ΔCt, y axis inverted) for each level of cytoplasmic vacuolation (level 0 corresponding to absence and level 3 to high abundance; n = 5, 14, 20, 18 for level 0, 1, 2, 3 respectively). Bars ± 95% CI.
Discussion
To study the genotoxic stress pathways used by cetaceans in response to solar UV exposure, we proposed quantifying changes in the expression of key genes, namely those encoding the heat shock protein 70 (HSP70), an indicator of cell stress [22, 25], the tumour protein 53 (P53), involved in most of the UV-induced gene transcription [8, 29], and the KIN17 protein (KIN), a cell cycle control protein up-regulated by UV [32, 34]. We chose qPCR to quantify mRNA transcripts because of the technique’s accuracy, sensitivity, replicability and potential to produce results rapidly [11]. However, despite its advantages, normalization of the technique, including selection of suitable internal control genes, is vital to obtain meaningful results [12]. This is particularly true when working in non-controlled field conditions, as it is extremely difficult to ensure that RNA quality will be equal for all samples.
The genes that encode the ribosomal proteins S18 (RSP18), ribosomal proteins L4 (RPL4), succinate dehydrogenase complex subunit A (SDHA) and phosphoglycerate kinase 1 (PGK1) were stably expressed and thus considered as suitable control genes. The optimal number of control genes needed for qPCR normalization was fewer than three genes when looking at each species separately and more than four genes when pooling the three study species. Although it could well be argued that our selected control gene panel is not optimal in number, due to our limited amount of sample tissue available per whale, we considered it impractical to quantify more than four control genes for studying three target genes. Financial or logistical constraints generally restrict the use of more than two internal control genes, and various studies that examine variation in target gene expression generally use two internal control genes [14, 16]. In this sense, we were similarly constrained by practical issues and aimed to make the best use of our samples by incorporating genes that were relatively stable and that were known to not have a biological association with UV-related pathways as control genes. The number of house-keeping genes we selected for this study was the result of a trade-off between practical considerations and accuracy [12].
The three software packages used concurred in selecting RSP18 and RPL4 as best internal control genes. The combination of these two genes also presented the highest stability value. RSP18 and RPL4 are useful not only for studies that focus on blue, fin or sperm whales independently, but also for studies that aim to compare gene expression between the three species. It is possible that these genes are useful as control genes for other species. For instance, RSP18 and RPL4 showed a similar behaviour in striped dolphin skin samples and were proposed as useful reference genes [13].
In contrast to the previously published study on striped dolphins [13], the gene coding for PGK1 was found to be a better control gene in our study than SDHA, which is probably due to species differences in gene expression. While GAPDH and YWHAZ were the most reliable internal control genes in striped dolphin skin [13], SDHA was the most reliable gene in a study that evaluated gene expression changes associated with organochlorine exposure in fibroblast cultures of striped dolphin [14]. Thus, it is clear that depending on the purpose of the study, species and target tissue to be analyzed, different genes might result more reliable as controls and thus it is essential that each study includes its own internal control gene analysis.
Although it was not possible to ascertain whether gene expression levels were constitutive or induced, HSP70 was found to be the gene with the highest level of expression (1.3 and 1.7 times more than P53 and KIN, respectively). Similar results have been observed in human melanocytes, where HSP70 was expressed at least 2.2 fold higher than the other 11 genes involved in different pathways of DNA repair mechanisms when under UV irradiance [42]. Over-expression of HSP70 might help initially to restore unstable or denatured proteins affected by UV stress [22]. It might also protect the cells against UV-induced damage, including apoptosis and DNA lesions [27].
Expression levels of P53 and of KIN were directly related to expression of HSP70. It is possible that HSP70 induces the expression of P53 and KIN in whales. Alternatively, P53 and/or KIN might induce expression of HSP70. In humans, HSP70 closely interacts with cell-cycle arrest proteins such as P53 protein [43, 44]. Although no studies on the association between HSP70 and KIN have been published, it is possible that, HSP70 may regulate KIN expression as the latter participates in the cell-cycle arrest. Chaperone proteins from the HSP70 family are known to recognize and bind mutant P53 proteins and thus regulate their accumulation and cellular localization [43, 44]. Furthermore, when P53 is mutated, its tertiary structure is modified liberating a binding domain, to which HSP70 can bind [44]. Although HSP70-P53 complexes have been observed in carcinoma cell lines, their biological significance is still unclear [45].
Finally, the observed correlations between transcription levels of genes involved in genotoxic stress response pathways and microscopic lesions associated with excessive acute solar exposure were interesting. For instance, P53 and HSP70 were expressed at lower levels when intracellular oedema, a lesion generally observed in skin inflammation [4, 46]. In humans and laboratory animals, it has been demonstrated that the activation of NF-kappaB, the central director of inflammation, reduces tumor suppressor activity of P53 and, thus, protects cells from P53-mediated death [46]. It is possible that occurrence of intracellular oedema has an inhibitory effect on the expression of P53. Likewise, the anti-inflammatory property of intracellular expression of HSP70[47] might explain the absence of oedema in those cells overexpressing HSP70. Interestingly, the presence of cytoplasmic vacuoles appeared to be directly associated with higher levels of gene expression. Although this pattern was non-significant, it is tempting to say that P53 and HSP70 are molecular tissue guardians [48] as cytoplasmic vacuolation is an alternative to the apoptotic process that aborts precancerous cells [49]. However, the cytoprotective activity of HSP70 overexpression reported in laboratory animals [50] would appear to discard this hypothesis. Exploring these associations in more depth, including protein expression assays, was beyond the scope of our study, but the observed trend suggests exciting possibilities regarding potential pathways used by cetaceans to counteract UV-induced damage.
Conclusion
In this study we successfully standardized the quantitation of mRNA transcripts using qPCR as a proposed method to study cetacean ability to cope with UV-induced damage. Our approach for standardization included selection of the best suitable control genes as the ribosomal proteins S18 (RSP18), ribosomal proteins L4 (RPL4), selection of key genes involved in genotoxic stress pathways, which included genes encoding the heat shock protein 70 (HSP70), an indicator of cell stress, the tumour protein 53 (P53), involved in most of the UV-induced gene transcription, and the KIN17 protein (KIN), a cell cycle control protein up-regulated by UV. Finally, we proposed a robust mixed effect modelling approach, which allowed us to control for interplate experimental variation. In this study we also provided preliminary results that demonstrate an association between the levels of expression of target genes and sunburn microscopic lesions previously recorded in cetacean epidermis [4]. Knowing that the ozone hole over the Arctic has recently recorded its largest size [2], it is pressing to increase the number of studies looking at the effect of UV on vulnerable wildlife.
References
Hader DP, Helbling EW, Williamson CE, Worrest RC: Effects of UV radiation on aquatic ecosystems and interactions with climate change. Photoch Photobio Sci. 2011, 10: 242-260. 10.1039/c0pp90036b.
Manney GL, Santee ML, Rex M, Livesey NJ, Pitts MC, Veefkind P, Nash ER, Wohltmann I, Lehmann R, Froidevaux L, Poole LR, Schoeberl MR, Haffner DP, Davies J, Dorokhov V, Gernandt H, Johnson B, Kivi R, Kyro E, Larsen N, Levelt PF, Makshtas A, McElroy CT, Nakajima H, Parrondo MC, Tarasick DW, von der Gathen P, Walker KA, Zinoviev NS: Unprecedented Arctic ozone loss in 2011. Nature. 2011, 478: 469-475. 10.1038/nature10556.
McKenzie RL, Aucamp PJ, Bais AF, Bjorn LO, Ilyas M, Madronich S: Ozone depletion and climate change: impacts on UV radiation. Photoch Photobio Sci. 2011, 10: 182-198. 10.1039/c0pp90034f.
Martinez-Levasseur LM, Gendron D, Knell RJ, O’Toole EA, Singh M, Acevedo-Whitehouse K: Acute sun damage and photoprotective responses in whales. Proc R Soc B. 2011, 278: 1581-1586. 10.1098/rspb.2010.1903.
Dahms HU, Lee JS: UV radiation in marine ectotherms: molecular effects and responses. Aquat Toxicol. 2010, 97: 3-14. 10.1016/j.aquatox.2009.12.002.
Evans TG, Hofmann GE: Defining the limits of physiological plasticity: how gene expression can assess and predict the consequences of ocean change. Trans R Soc Lond B Biol Sci. 2012, 367 (1596): 1733-45. 10.1098/rstb.2012.0019.
Zhou BB, Elledge SJ: The DNA damage response: putting checkpoints in perspective. Nature. 2000, 408: 433-439. 10.1038/35044005.
Week DW: Tumour suppression by p53: a role for the DNA damage response?. Nat Rev Cancer. 2009, 9: 714-723.
Menendez D, Inga A, Resnick MA: The expanding universe of p53 targets. Nat Rev Cancer. 2009, 9: 724-737. 10.1038/nrc2730.
Derveaux S, Vandesompele J, Hellemans J: How to do successful gene expression analysis using real-time PCR. Methods. 2010, 50: 227-230. 10.1016/j.ymeth.2009.11.001.
Taylor S, Wakem M, Dijkman G, Alsarraj M, Nguyen M: A practical approach to RT-qPCR-Publishing data that conform to the MIQE guidelines. Methods. 2010, 50: S1-5. 10.1016/j.ymeth.2010.01.005.
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F: Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3 (7): research0034.1–0034.11. http://genomebiology.com/content/3/7/RESEARCH0034
Spinsanti G, Panti C, Lazzeri E, Marsili L, Casini S, Frati F, Maria Fossi CM: Selection of reference genes for quantitative RT-PCR studies in striped dolphin (Stenella coeruleoalba) skin biopsies. BMC Mol Biol. 2006, 7: 32-10.1186/1471-2199-7-32.
Spinsanti G, Panti P, Bucalossi D, Marsili L, Casini S, Frati F, Fossi MC: Selection of reliable reference genes for qRT-PCR studies on cetacean fibroblast cultures exposed to OCs, PBDEs, and 17[beta]-estradiol. Aquat Toxicol. 2008, 87: 178-186. 10.1016/j.aquatox.2008.01.018.
Noutsias M, Rohde M, Block A, Klippert K, Lettau O, Blunert K, Hummel M, Kühl U, Lehmkuhl H, Hetzer R, Rauch U, Poller W, Pauschinger M, Schultheiss HP, Volk HD, Kotsch K: Preamplification techniques for real-time RT-PCR analyses of endomyocardial biopsies. BMC Mol Biol. 2008, 9: 3-10.1186/1471-2199-9-3.
Fossi MC, Urban J, Casini S, Maltese S, Spinsanti G, Panti C, Porcelloni S, Panigada S, Lauriano G, Niño-Torres C, Rojas-Bracho L, Jimenez B, Muñoz-Arnanz J, Marsili L: A multi-trial diagnostic tool in fin whale (Balaenoptera physalus) skin biopsies of the Pelagos Sanctuary (Mediterranean Sea) and the Gulf of California (Mexico). Mar Environ Res. 2010, 69: S17-S20.
Lin M, Morrison CD, Jones S, Mohamed N, Bacher J, Plass C: Copy number gain and oncogenic activity of YWHAZ/14-3-3ζ in head and neck squamous cell carcinoma. Int J Cancer. 2009, 125 (3): 603-611. 10.1002/ijc.24346.
Sirover MA: On the functional diversity of glyceraldehyde-3-phosphate dehydrogenase: Biochemical mechanisms and regulatory control. Biochim Biophys Acta. 2011, 1810: 741-751. 10.1016/j.bbagen.2011.05.010.
Wu YY, Rees JL: Variation in epidermal housekeeping gene expression in different. Acta Derm Venereol. 2000, 80: 2-3. 10.1080/000155500750012397.
Garmyn M, Yaar M, Holbrook N, Gilchrest BA: Immediate and delayed molecular response of human keratinocytes to solar-simulated irradiation. Lab Invest. 1991, 65 (4): 471-478.
Balogh A, Paragh G, Juhász A, Köbling T, Törocsik D, Mikó E, Varga V, Emri G, Horkay I, Scholtz B, Remenyik E: Reference genes for quantitative real time PCR in UVB irradiated keratinocytes. J Photochem Photobiol B. 2008, 93: 133-139. 10.1016/j.jphotobiol.2008.07.010.
Mayer MP, Bukau B: Hsp70 chaperones: Cellular functions and molecular mechanism. CMLS Cell Mol Life Sci. 2005, 62: 670-684. 10.1007/s00018-004-4464-6.
Guzhova IV, Lazarev VF, Kaznacheeva AV, Ippolitova MV, Muronetz VI, Kinev AV, Margulis BA: Novel mechanism of Hsp70 chaperone-mediated prevention of polyglutamine aggregates in a cellular model of huntington disease. Hum Mol Genet. 2011, 20: 3953-3963. 10.1093/hmg/ddr314.
Li Y, Zou L, Li Q, Haibe-Kains B, Tian R, Li Y, Desmedt C, Sotiriou C, Szallasi Z, Iglehart JD, Richardson AL, Wang ZC: Amplification of LAPTM4B and YWHAZ contributes to chemotherapy resistance and recurrence of breast cancer. Nat Med. 2010, 16: 214-218. 10.1038/nm.2090.
Hightower LE: Heat shock, stress proteins, chaperones, and proteotoxicity. Cell. 1991, 66: 191-197. 10.1016/0092-8674(91)90611-2.
Tsang MF, Gao B: Heat shock proteins and immune system. J Leukocyte Biol. 2009, 85: 905-910. 10.1189/jlb.0109005.
Matsuda M, Hoshino T, Yamashita Y, Tanaka K, Maji D, Sato K, Adachi H, Sobue G, Ihn H, Funasaka Y, Mizushima T: Prevention of UVB radiation-induced epidermal damage by expression of heat shock protein 70. J Biol Chem. 2010, 285: 5848-5858. 10.1074/jbc.M109.063453.
Kucab JE, Phillips DH, Arlt VM: Linking environmental carcinogen exposure to TP53 mutations in human tumours using the human TP53 knock-in (Hupki) mouse model. FEBS J. 2010, 277: 2567-2583. 10.1111/j.1742-4658.2010.07676.x.
Latonen L, Laiho M: Cellular UV damage responses–functions of tumor suppressor p53. Biochim Biophys Acta. 2005, 1755: 71-89.
Oren M, Bartek J: The sunny side of p53. Cell. 2007, 128: 826-828. 10.1016/j.cell.2007.02.027.
Biard DS, Saintigny Y, Maratrat M, Paris F, Martin M, Angulo JF: Enhanced expression of the Kin17 protein immediately after low doses of ionizing radiation. Radiat Res. 1997, 147: 442-450. 10.2307/3579501.
Kannouche P, Mauffrey P, Pinon-Lataillade G, Mattei MG, Sarasin A, Daya-Grosjean L, Angulo JF: Molecular cloning and characterization of the human KIN17 cDNA encoding a component of the UVC response that is conserved among metazoans. Carcinogenesis. 2000, 21: 1701-1710. 10.1093/carcin/21.9.1701.
Biard DS, Miccoli L, Despras E, Frobert Y, Creminon C, Angulo JF: Ionizing radiation triggers chromatin-bound kin17 complex formation in human cells. J Biol Chem. 2002, 277: 19156-19165. 10.1074/jbc.M200321200.
Masson C, Menaa F, Pinon-Lataillade G, Frobert Y, Chevillard S, Radicella JP, Sarasin A, Angulo JF: Global genome repair is required to activate KIN17, a UVC-responsive gene involved in DNA replication. PNAS. 2003, 100: 616-621. 10.1073/pnas.0236176100.
Ierardi JL, Mancia A, McMillan J, Lundqvist ML, Romano TA, Wise JP, Warr GW, Chapman RW: Sampling the skin transcriptome of the North Atlantic right whale. Comp Biochem Phy D. 2009, 4: 154-158.
Schmittgen TD, Livak KJ: Analyzing real-time PCR data by the comparative CT method. Nat Protoc. 2008, 3: 1101-1108. 10.1038/nprot.2008.73.
Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP: Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper – Excel-based tool using pair-wise correlations. Biotechnol Lett. 2004, 26: 509-515.
Andersen CL, Jensen JL, Orntoft TF: Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64: 5245-5250. 10.1158/0008-5472.CAN-04-0496.
R Development Core Team: R: A language and environment for statistical computing. 2008, Vienna, Austria: R Foundation for Statistical Computing, URL http://www.R-project.org, 3-900051-07-0
Zuur A, Ieno E, Walker N, Saveliev A, Smith G: Mixed effects models and extensions in ecology with R. 2009, New York: Springer
Pinheiro J, Bates D, DebRoy S, Sarkar D, Coreteam R: nlme: Linear and Nonlinear Mixed Effects Models. 2008, R package version 3.1-89
Jean S, Bideau C, Bellon L, Halimi G, De Meo M, Orsiere T, Dumenil G, Berge-Lefranc JL, Botta A: The expression of genes induced in melanocytes by exposure to 365-nm UVA: study by cDNA arrays and real-time quantitative RT-PCR. Biochim Biophys Acta. 2001, 1522: 89-96. 10.1016/S0167-4781(01)00326-8.
Helmbrecht K, Zeise E, Rensing L: Chaperones in cell cycle regulation and mitogenic signal transduction: a review. Cell Prolif. 2000, 33: 341-365. 10.1046/j.1365-2184.2000.00189.x.
Zylicz M, King FW, Wawrzynow A: Hsp70 interactions with the p53 tumour suppressor protein. EMBO J. 2001, 20: 4634-463. 10.1093/emboj/20.17.4634.
Fourie AM, Hupp TR, Lane DP, Sang BC, Barbosa MS, Sambrook JF, Gething MJH: HSP70 Binding sites in the tumor suppressor protein p53. J Biol Chem. 1997, 272: 19471-19479. 10.1074/jbc.272.31.19471.
Gudkov AV, Gurova KV, Komarova EA: Inflammation and p53: A Tale of Two Stresses. Genes Cancer. 2011, 2 (4): 503-16. 10.1177/1947601911409747.
De Jong PR, Schadenberg AW, Jansen NJ, Prakken BJ: Hsp70 and cardiac surgery: molecular chaperone and inflammatory regulator with compartmentalized effects. Cell Stress Chaperon. 2009, 14 (2): 117-31. 10.1007/s12192-008-0066-9.
Ziegler A, Jonason AS, Leffell DJ, Simon JA, Sharma HW, Kimmelman J, Remington L, Jacks T, Brash DE: Sunburn and p53 in the onset of skin cancer. Nature. 1994, 372 (6508): 773-6. 10.1038/372773a0.
Sperandio S, De Belle I, Bredesen DE: An alternative, nonapoptotic form of programmed cell death. P Natl Acad Sci USA. 2000, 97 (26): 14376-14381. 10.1073/pnas.97.26.14376.
Garrido C, Schmitt E, Candé C, Vahsen N, Parcellier A, Kroemer G: HSP27 and HSP70: potentially oncogenic apoptosis inhibitors. Cell Cycle. 2003, 2 (6): 579-84.
Acknowledgements
We thank Kate Ciborowski and Solenn Patalano for their valuables advice on technical aspects of qPCR. L.M. was funded by a NERC PhD Studentship (NE/F00818X/1) awarded at Queen Mary University of London. This work was partially funded by the IPN (Instituto Politecnico National de Mexico) and CONACYT (CB-2006-61982).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare they do not have any competing interests.
Authors’ contributions
LMML and DG collected the samples and conducted fieldwork. LMML performed all laboratory work and analysed the data. LMML and KAW designed the experiments and wrote the manuscript. KAW conceived the study. All authors discussed the study’s results, read and approved the final manuscript.
Electronic supplementary material
13104_2013_2289_MOESM1_ESM.pdf
Additional file 1:The supplementary material is presented together as a pdf file. The content is divided into subheadings: 1. Multiple sequence alignment (CLUSTALW) used to design the pair of primer for the three selected target genes; 2. Gene sequences included in the analyses; 3. Supplementary Figures, which include four figures: Figure S1. Schematic representation of the gene expression protocol; Figure S2. RNA integrity using the QIAxcel system; Figure S3. qPCR dissociation curves of the genes used in the analyses and Figure S4. Expression levels of the internal control gene candidates. (PDF 377 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Martinez-Levasseur, L.M., Gendron, D., Knell, R.J. et al. Control and target gene selection for studies on UV-induced genotoxicity in whales. BMC Res Notes 6, 264 (2013). https://doi.org/10.1186/1756-0500-6-264
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-0500-6-264