
Abstract
Background
Lophelia pertusa is a keystone cold-water coral species with a widespread distribution. Due to the lack of a mitochondrial marker variable enough for intraspecific analyses, the population structure of this species has only been studied using ITS and microsatellites so far. We therefore decided to sequence and compare complete mitochondrial genomes from two distant L. pertusa populations putatively isolated from each other (in the Barents Sea off Norway and in the Mediterranean Sea off Italy) in the hope of finding regions variable enough for population genetic and phylogeographic studies.
Results
The mitogenomes of two L. pertusa individuals collected in the Mediterranean and Barents seas differed at only one position, which was a non-synonymous substitution, but comparison with another recently published L. pertusa mitochondrial genome sequence from Norway revealed 18 nucleotide differences. These included two synonymous and nine non-synonymous substitutions in protein-coding genes (dN/dS > 1): hence, the mitogenome of L. pertusa may be experiencing positive selection. To test for the presence of cryptic species, the mitochondrial control region and the nuclear ITS2 were sequenced for five individuals from each site: Italian and Norwegian populations turned out to share haplotypes of both markers, indicating that they belonged to the same species.
Conclusions
L. pertusa corals collected 7,500 km apart shared identical nuclear ITS2 and near-identical mitogenomes, supporting the hypothesis of a recent connection between Lophelia reefs in the Mediterranean and in the Northern Atlantic. Multi-locus or population genomic approaches will be required to shed further light on the genetic connectivity between L. pertusa reefs across Europe; nevertheless, ITS2 and the mitochondrial control region may be useful markers for investigating the phylogeography and species boundaries of the keystone genus Lophelia across its worldwide area of distribution.
Similar content being viewed by others


Background
The mitochondrial genomes of cnidarians and sponges are characterized by low rates of evolution that make it often impossible to distinguish species using cox1 sequences [1, 2]. Moreover, the mitochondrial genomes of scleractinian corals exhibit a nearly perfect conservation of a standard gene order [3], in which non-coding regions and additional genes (such as duplicated tRNAs [4–6], putative homing endonucleases [7] and unknown ORFs [8]) are sometimes found inserted. The order of the basic complement of 13 protein-coding genes, 2 rRNAs and 2 tRNAs, all located on the same strand in scleractinian mitochondrial genomes, has been found to be identical in every species investigated so far except in the cold-water coral Lophelia pertusa (Linnaeus, 1758) [9, 10].
L. pertusa, an azooxanthellate scleractinian coral, is an important deep-sea reef builder with an almost cosmopolitan distribution [11]. The reefs it builds host highly diverse and rich faunas [12–14], but these fragile ecosystems are being severely impacted by destructive fishing practice such as bottom trawling [15, 16]; furthermore, CO2-induced ocean acidification may affect their future distribution [17] or even cause their disappearance [18]. Previous analyses of 16S rDNA mitochondrial sequences revealed a very high level of divergence (6.96%) between L. pertusa from the northeast Atlantic and off Brazil, suggesting that these two populations may represent cryptic species [19]. In the northern Atlantic, due to the lack of a mitochondrial marker variable enough for intraspecific studies [20], the population structure of L. pertusa has only been studied using internal transcribed spacer (ITS) sequences [21] and microsatellite markers [21–23]. This is unfortunate as mitochondrial markers present numerous advantages over nuclear ones: haploid markers are cheaper and easier to sequence (no heterozygosity issues), and have a smaller population size resulting in faster coalescence (thereby alleviating the problems posed by shared ancestral polymorphism). Hence we decided to sequence and compare the complete mitogenomes from two individuals originating in locations putatively isolated from each other (in the Barents Sea off Norway and in the Ionian Sea off Italy), in the hope of finding variable mitochondrial regions suitable for population genetic and phylogeographic analyses.
Results and discussion
The mitogenomes of Lophelia pertusa from the Mediterranean and Barents seas are nearly identical and may be experiencing positive Darwinian selection
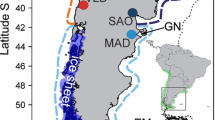
In spite of the geographical distance between them (approximately 7500 km, Figure 1), the two L. pertusa individuals sequenced had nearly identical mitochondrial genomes that differed only by a nucleotide substitution at position 11,876. This single mutation (out of a total of 16,149 bp) is a transition; it is not silent but entails the substitution of a tyrosine (in individual #362 from Italy) with a histidine (in individual #302 from Norway) in the NAD6 protein. As such a mutation may be adaptive, we tested whether it was fixed in the two populations investigated by sequencing the corresponding DNA region in four additional individuals from each location (Table 1): the sequences of all eight individuals were identical to the one of individual #362, whereas the rare sequence variant of individual #302 was confirmed by resequencing.

Topographic map of Europe showing sampling locations. The two populations sampled are Korallen off Norway and Santa Maria di Leuca off Italy. One field picture from each of the latter two sites was included in the figure (above, photograph from Korallen by Pål Buhl-Mortensen, Institute of Marine Research; below, photograph from Santa Maria di Leuca by Marum, University of Bremen). The published L. pertusa mitogenome [10] came from Nord-Leksa. The shaded relief comes from the ETOPO2v2 2-minute grid [24], which north of 64°N is based on the International Bathymetric Chart of the Arctic Ocean (IBCAO) [25].
Another mitochondrial genome of L. pertusa from Norway was recently published [10]. The individual sequenced in that study was sampled inside Trondheimfjord, a fjord known to harbor subpopulations of L. pertusa genetically differentiated from the subpopulations on the continental margin [21]. Indeed, there were 18 differences between our sequences and the published one, allowing us to assess the variability of the various stretches of the mitochondrial genome of L. pertusa (Table 2). The most variable region was a 730-bp long segment between the nad5 and cob genes that had been previously singled out as the putative control region [10] as it was the longest non-coding stretch and contained three tandem repeats of a 51-bp pattern (consensus: CCTCCATCTATGCATGTGGAACCAGTTCCGGAGCTTTCTCAGGGTTTGATC). Hence, the nad5-cob intergenic region of the L. pertusa mitogenome matches all three criteria used to identify mitochondrial control regions: length, variability, and the presence of tandem repeats [8].
Among the 11 nucleotide substitutions found in coding DNA regions, nine were non-synonymous and only two were synonymous (Table 2). We calculated dN/dS using ten different approaches (seven approximate and three based on maximum likelihood [26]). The values obtained ranged from 1.04 to 5.24 (Table 3), which is much higher than the value of 0.39 previously calculated in the shallow-water scleractinian coral Pocillopora[8]. Hence, the mitochondrial genome of L. pertusa appears to be experiencing positive (diversifying) Darwinian selection, whereas the mitochondrial genome of Pocillopora seems rather under negative (purifying) selection. However, this intriguing result will require further confirmation: the number of differences in coding sequences was so small that the support for positive selection was not statistically significant using Fisher’s exact test (other, more sensitive statistical procedures such as the Z test were not attempted as they require at least 10 synonymous and 10 non-synonymous mutations for their assumptions to be met [27]).
European populations of Lophelia pertusa are comprised of two mitochondrial control region haplogroups but a single ITS2 field for recombination
Mitogenome divergence was higher between two individuals from Norway (our sequence and the published one) rather than between Norway and Italy (the two individuals we sequenced): as this raised the possibility that L. pertusa in Europe comprises two sympatric cryptic lineages, we sequenced the nuclear ITS2 and the mitochondrial control region, two markers of choice for species delimitation in corals [37–41], in a total of 10 individuals: five from Italy and five from Norway (including the two individuals whose complete mitochondrial genomes were sequenced in the present study).
There were three mitochondrial control region haplotypes among our 10 individuals (Figure 2a): the two major haplotypes, found in 5 and 4 individuals respectively, occurred both in Norway and in Italy, whereas a rarer haplotype was only found in one individual in Italy. The published sequence of L. pertusa[10] was one mutation away from one of the two most frequent haplotypes; overall, two haplogroups could be distinguished, comprising respectively one and three control region haplotypes.

Phylogeographic analysis of 10 Lophelia pertusa samples from Italy and Norway. (a) Haplotype network (haplonet) of control region (CR) sequences. Green represents Italy and red Norway, whereas the numbers on the lines between haplotypes indicate the position of the corresponding mutations in the alignment (circle sizes are proportional to the number of individuals harboring each haplotype). Dashed ovals delineate two sympatric CR haplogroups (comprising respectively one and three haplotypes). The two arrows in the upper right corner point at the two individuals whose complete mitochondrial genomes were sequenced in the present study, whereas the bottom left arrow shows the CR haplotype in the published sequence of L. pertusa[10]. (b) Haplotype web (haploweb) of ITS2 sequences. The color code is the same as in (a), and curves connecting haplotypes represent heterozygous individuals harboring two different ITS2 types [39]. Circle sizes are proportional to the number of individuals harboring each type. The arrow points at the common ITS2 type shared by all individuals analyzed in the present study as well as by the one sequenced in [10, 42], whereas the dashed oval delineates the resulting field for recombination [39, 43].
There were five ITS2 sequence types among the 10 individuals analyzed: two of these types were shared by Italian and Norwegian populations, whereas the three others were only found in Norway. Several L. pertusa individuals contained two different ITS2 sequence types and one ITS2 type was shared by all individuals sequenced (Figure 2b), confirming that they belonged to a single field for recombination sensu Doyle [43] and were therefore conspecific following the criterion of mutual exclusivity [39].
Comparison of the ITS2 sequences obtained in the present study with those available online did not reveal any clear geographical pattern. The ITS2 type shared by all our samples (see arrow on Figure 2b) as well as by the coral colony whose mitochondrial genome had previously been sequenced [10, 44] was also the most frequent type in [21] (found in 34 of the 77 samples of this study, from Norway to Spain). Our second most common type (on the right of Figure 2b), found in two heterozygotes from Italy and Norway, had been previously reported from France [21]. A rare ITS2 type that we found only in one heterozygous individual from Norway had been already observed in that country [21]. Only two rare ITS types from the present study, each found in Norway, had never been published until now.
Shared mitochondrial and nuclear haplotypes support the hypothesis of a recent connection between Atlantic and Mediterranean Lophelia pertusa populations
The mitochondrial genomes of corals are known to be very stable compared with those of most other metazoans [1, 2]. Identical sequences of a 630-bp fragment of the cox1 gene was reported for populations of another azooxanthellate coral species, Balanophyllia elegans, sampled 3000 km apart [45], but it is the first time that such an extremely low level of variation across several thousand kilometers is reported using complete mitochondrial genome sequences. In contrast, the only other study that compared complete mitogenomes from conspecific corals reported between 3 and 18 intraspecific nucleotide differences in three Montastraea species analyzed at a single location [46].
The fact that L. pertusa populations located 7,500 km apart in different ocean basins share nuclear ITS2 and mitochondrial control region sequences contrasts with the results of previous population genetic studies using ITS and microsatellites that reported differentiation and even genetic discontinuities among northern Atlantic populations of this species [21, 22]. A possible explanation could be a difference in time scale: at ecological time scale, differences in gene frequencies would indicate a low connectivity and/or predominantly asexual mode of reproduction of L. pertusa in the northern Atlantic, whereas at evolutionary time scale, the identical sequences found in individuals collected along the European shelf and in the Mediterranean Sea would point at a recent connection between these population. Indeed, the hypothesis of a Mediterranean origin for East Atlantic L. pertusa reefs was proposed by De Mol et al. [47, 48] based on the correspondence between the depth distribution of coral mounds in the Porcupine Seabight and the depth of the Mediterranean outflow water: deep-sea reefs proliferated in the Mediterranean during the Younger Dryas (12,900 to 11,700 years BP) [49], a period during which the Mediterranean outflow was at its maximum [50] and could have carried coral larvae into the Atlantic. Our molecular results bring support to this hypothesis that will have to be further tested using multi-locus or population genomic approaches.
Conclusions
L. pertusa individuals collected 7,500 km apart in the Barents and Mediterranean seas shared identical nuclear ITS2 and mitochondrial control region sequences, suggesting recent gene flow between Lophelia reefs in the Mediterranean and in the North Atlantic Ocean. Multi-locus or population genomic approaches will be required to shed further light on the genetic connectivity between L. pertusa reefs across Europe; nevertheless, ITS2 and the mitochondrial control region may be useful markers for investigating the phylogeography and species boundaries of the keystone genus Lophelia across its worldwide area of distribution.
Methods
Sample collection
Lophelia corals were sampled during research cruises at two distant localities (Table 1): Korallen (70°55′N & 22°12′E) is located northwest of the Norwegian island Sørøya in the Barents Sea, at the northern biogeographic limit for distribution of cold-water coral ecosystems [51], whereas the Santa Maria di Leuca coral province (39°33′N & 18°26′E) is found in the northern part of the Ionian Sea in the Mediterranean [52–54] (Figure 1). Both cruises (R/V Meteor Cruise 70/1 off Italy and R/V G.O. Sars Cruise No. 2006112 off Norway) were conducted in compliance with local legislations and with the Convention on Biological Diversity (CBD); samples were transported to Sweden with appropriate export and import permits following the Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES).
Mitogenome sequencing and annotation
Frozen coral tissues were preserved in buffered guanidium thiocyanate solution [55, 56] and their DNA purified on an ABI Prism 6100 Nucleic Acid PrepStation. PCR primers covering the entire genome of L. pertusa were defined using Primer3 [57] (Table 4). To prevent cross-contamination, the two mitochondrial genomes were not sequenced simultaneously but one after the other; moreover, we used filter tips and included negative controls in all our PCRs. PCR amplifications were performed in 25 μl reaction mixes containing 1x Red Taq buffer, 264 μM dNTP, 5% DMSO, 0.3 μM PCR primers, 0.3 units Red Taq (Sigma), and 10–50 ng DNA. PCR conditions comprised an initial denaturation step of 60 s at 94°C, followed by 45–55 cycles (30 s denaturation at 94°C, 30 s annealing, 1–3 min elongation at 72°C) and a final 5-min elongation step at 72°C. All PCR products were Sanger-sequenced using the same primers as for amplification (for long PCR products, internal sequencing primers were also defined using Primer3).
As there was no published mitogenome of L. pertusa available at that time, the genes present in the two L. pertusa mitogenomes sequenced were identified using BLAST [59] and ORFfinder (available online at http://www.ncbi.nlm.nih.gov/gorf/orfig.cgi). One exception was the atp8 gene that often cannot be annotated using BLAST since its aminoacid sequence is extremely variable; however, this gene always starts with the characteristic aminoacid sequence (M)PQ [60] and was therefore easily detected among translated ORF sequences. tRNA genes were localized using tRNA-scan [61], and tandem repeats were detected using the online program Tandem Repeats Finder [62]. Except for some details of gene boundaries, our annotation was consistent with the L. pertusa mitogenome that was published in the meantime [10].
Haplonet and haploweb analyses
We sequenced the mitochondrial control region of four additional individuals from each site (Korallen and Santa Maria di Leuca), as well as the nuclear ITS2 of all ten individuals studied (see Table 5 for a list of the primers used). PCR conditions were as described in the previous section. The ITS2 chromatograms of five individuals comprised double peaks: obtaining the haplotypes of three of them was trivial as each of these individuals had only one double peak, and we used Clark’s method [42] to determine the haplotypes of the two remaining individuals that had two double peaks each. Although other authors have reported intra-individual mitogenome heterogeneity in L. pertusa using next-generation sequencing [44], we did not observe double peaks in the chromatograms obtained for any of the mitochondrial markers sequenced. The complete mitogenomes of individuals #302 and #362 were deposited in public databases [GenBank: KC875348-KC875349], as well as the CR and ITS sequences obtained in this study [GenBank: KC875351-KC875375]. Haplotype networks were built using the median-joining approach [63] implemented in Network 4.1 (available online at http://www.fluxus-engineering.com/); the ITS2 haplonet was turned into a haploweb by adding connections between alleles found co-occurring in heterozygous individuals [39].
References
Shearer TL, van Oppen MJH, Romano SL, Wörheide G: Slow mitochondrial DNA sequence evolution in the Anthozoa (Cnidaria). Mol Ecol. 2002, 11: 2475-2487. 10.1046/j.1365-294X.2002.01652.x.
Huang D, Meier R, Todd P, Chou L: Slow mitochondrial COI sequence evolution at the base of the metazoan tree and its implications for DNA barcoding. J Mol Evol. 2008, 66: 167-174. 10.1007/s00239-008-9069-5.
Medina M, Collins AG, Takaoka TL, Kuehl JV, Boore JL: Naked corals: skeleton loss in Scleractinia. Proc Natl Acad Sci USA. 2006, 103: 9096-9100. 10.1073/pnas.0602444103.
Chen C, Chiou C-Y, Dai C-F, Chen CA: Unique mitogenomic features in the scleractinian family Pocilloporidae (Scleractinia: Astrocoeniina). Marine Biotechnol. 2008, 10: 538-553. 10.1007/s10126-008-9093-x.
Chen C, Dai C-F, Plathong S, Chiou C-Y, Chen CA: The complete mitochondrial genomes of needle corals, Seriatopora spp. (Scleractinia: Pocilloporidae): an idiosyncratic atp8, duplicated trnW gene, and hypervariable regions used to determine species phylogenies and recently diverged populations. Mol Phylogenet Evol. 2008, 46: 19-33. 10.1016/j.ympev.2007.09.013.
Flot J-F, Licuanan W, Nakano Y, Payri C, Cruaud C, Tillier S: Mitochondrial sequences of Seriatopora corals show little agreement with morphology and reveal the duplication of a tRNA gene near the control region. Coral Reefs. 2008, 27: 789-794. 10.1007/s00338-008-0407-2.
Fukami H, Chen C, Chiou C-Y, Knowlton N: Novel group I introns encoding a putative homing endonuclease in the mitochondrial cox1 gene of scleractinian corals. J Mol Evol. 2007, 64: 591-600. 10.1007/s00239-006-0279-4.
Flot J-F, Tillier S: The mitochondrial genome of Pocillopora (Cnidaria: Scleractinia) contains two variable regions: the putative D-loop and a novel ORF of unknown function. Gene. 2007, 401: 80-87. 10.1016/j.gene.2007.07.006.
Linnaeus C: Systema naturae per regna tria naturæ secundum classes, ordines, genera, species, cum characteribus, differentilis, synonymis, locis. Edited by: Tomus I. 1758, Stockholm: Laurentius Salvius, Editio decima, reformata
Emblem Å, Karlsen BO, Evertsen J, Johansen SD: Mitogenome rearrangement in the cold-water scleractinian coral Lophelia pertusa (Cnidaria, Anthozoa) involves a long-term evolving group I intron. Mol Phylogenet Evol. 2011, 61: 495-503. 10.1016/j.ympev.2011.07.012.
Rogers AD: The biology of Lophelia pertusa (Linnaeus 1758) and other deep-water reef-forming corals and impacts from human activities. Int Rev Hydrobiol. 1999, 84: 315-406.
Jensen A, Frederiksen R: The fauna associated with the bank-forming deepwater coral Lophelia pertusa (Scleractinia) on the Faroe shelf. Sarsia. 1992, 77: 53-69.
Costello M, McCrea M, Freiwald A, Lundälv T, Jonsson L, Bett BJ, van Weering TCE, de Haas H, Roberts JM, Allen D: Role of cold-water Lophelia pertusa coral reefs as fish habitat in the NE Atlantic. Cold-Water Corals and Ecosystems. Edited by: Freiwald A. 2005, Springer: Berlin Heidelberg, 771-805.
Lessard-Pilon SA, Podowski EL, Cordes EE, Fisher CR: Megafauna community composition associated with Lophelia pertusa colonies in the Gulf of Mexico. Deep-Sea Res II Top Stud Oceanogr. 2010, 57: 1882-1890. 10.1016/j.dsr2.2010.05.013.
Fosså JH, Mortensen PB, Furevik DM: The deep-water coral Lophelia pertusa in Norwegian waters: distribution and fishery impacts. Hydrobiologia. 2002, 471: 1-12. 10.1023/A:1016504430684.
Hall-Spencer J, Allain V, Fosså JH: Trawling damage to Northeast Atlantic ancient coral reefs. Proc R Soc Lond B Biol Sci. 2002, 269: 507-511. 10.1098/rspb.2001.1910.
Guinotte JM, Orr J, Cairns S, Freiwald A, Morgan L, George R: Will human-induced changes in seawater chemistry alter the distribution of deep-sea scleractinian corals?. Front Ecol Environ. 2006, 4: 141-146. 10.1890/1540-9295(2006)004[0141:WHCISC]2.0.CO;2.
Turley C, Roberts J, Guinotte J: Corals in deep-water: will the unseen hand of ocean acidification destroy cold-water ecosystems?. Coral Reefs. 2007, 26: 445-448. 10.1007/s00338-007-0247-5.
Le Goff-Vitry MC, Rogers AD, Baglow D: A deep-sea slant on the molecular phylogeny of the Scleractinia. Mol Phylogenet Evol. 2004, 30: 167-177. 10.1016/S1055-7903(03)00162-3.
Le Goff-Vitry M-C: Molecular ecology of the deep-sea coral Lophelia pertusa in the North East Atlantic. 2003, University of Southampton, School of Ocean and Earth Science: PhD thesis
Le Goff-Vitry MC, Pybus OG, Rogers AD: Genetic structure of the deep-sea coral Lophelia pertusa in the northeast Atlantic revealed by microsatellites and internal transcribed spacer sequences. Mol Ecol. 2004, 13: 537-549. 10.1046/j.1365-294X.2004.2079.x.
Morrison C, Ross S, Nizinski M, Brooke S, Järnegren J, Waller R, Johnson R, King T: Genetic discontinuity among regional populations of Lophelia pertusa in the North Atlantic Ocean. Conserv Genet. 2011, 12: 713-729. 10.1007/s10592-010-0178-5.
Dahl MP, Pereyra RT, Lundälv T, André C: Fine-scale spatial genetic structure and clonal distribution of the cold-water coral Lophelia pertusa. Coral Reefs. 2012, 31: 1135-1148. 10.1007/s00338-012-0937-5.
U.S. Department of Commerce NOaAA: 2-minute Gridded Global Relief Data (ETOPO2v2). 2006, : ,http://www.ngdc.noaa.gov/mgg/fliers/06mgg01.html,
Jakobsson M, Macnab R, Mayer L, Anderson R, Edwards M, Hatzky J, Schenke HW, Johnson P: An improved bathymetric portrayal of the Arctic Ocean: implications for ocean modeling and geological, geophysical and oceanographic analyses. Geophys Res Lett. 2008, 35: L07602
Zhang Z, Li J, Zhao X-Q, Wang J, Wong GK-S, Yu J: KaKs_Calculator: calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinformatics. 2006, 4: 259-263. 10.1016/S1672-0229(07)60007-2.
Nei M, Kumar S: Molecular Evolution and Phylogenetics. 2000, Oxford: Oxford University Press
Nei M, Gojobori T: Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986, 3: 418-426.
Li W-H, Wu C-I, Luo C-C: A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol Biol Evol. 1985, 2: 150-174.
Tzeng Y-H, Pan R, Li W-H: Comparison of three methods for estimating rates of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 2004, 21: 2290-2298. 10.1093/molbev/msh242.
Li W-H: Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J Mol Evol. 1993, 36: 96-99. 10.1007/BF02407308.
Pamilo P, Bianchi NO: Evolution of the Zfx and Zfy genes: rates and interdependence between the genes. Mol Biol Evol. 1993, 10: 271-281.
Yang Z, Nielsen R: Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol. 2000, 17: 32-43. 10.1093/oxfordjournals.molbev.a026236.
Zhang Z, Li J, Yu J: Computing Ka and Ks with a consideration of unequal transitional substitutions. BMC Evol Biol. 2006, 6: 44-10.1186/1471-2148-6-44.
Goldman N, Yang Z: A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol. 1994, 11: 725-736.
Muse SV, Gaut BS: A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol Biol Evol. 1994, 11: 715-724.
Flot J-F, Magalon H, Cruaud C, Couloux A, Tillier S: Patterns of genetic structure among Hawaiian corals of the genus Pocillopora yield clusters of individuals that are compatible with morphology. Comptes Rendus Biologies. 2008, 331: 239-247. 10.1016/j.crvi.2007.12.003.
Forsman Z, Barshis D, Hunter C, Toonen R: Shape-shifting corals: molecular markers show morphology is evolutionarily plastic in Porites. BMC Evol Biol. 2009, 9: 45-10.1186/1471-2148-9-45.
Flot J-F, Couloux A, Tillier S: Haplowebs as a graphical tool for delimiting species: a revival of Doyle’s “field for recombination” approach and its application to the coral genus Pocillopora in Clipperton. BMC Evol Biol. 2010, 10: 372-10.1186/1471-2148-10-372.
Flot J-F, Blanchot J, Charpy L, Cruaud C, Licuanan W, Nakano Y, Payri C, Tillier S: Incongruence between morphotypes and genetically delimited species in the coral genus Stylophora: phenotypic plasticity, morphological convergence, morphological stasis or interspecific hybridization?. BMC Ecol. 2011, 11: 22-10.1186/1472-6785-11-22.
Stefani F, Benzoni F, Yang SY, Pichon M, Galli P, Chen C: Comparison of morphological and genetic analyses reveals cryptic divergence and morphological plasticity in Stylophora (Cnidaria, Scleractinia). Coral Reefs. 2011, 30: 1033-1049. 10.1007/s00338-011-0797-4.
Clark A: Inference of haplotypes from PCR-amplified samples of diploid populations. Mol Biol Evol. 1990, 7: 111-122.
Doyle JJ: The irrelevance of allele tree topologies for species delimitation, and a non-topological alternative. Syst Bot. 1995, 20: 574-588. 10.2307/2419811.
Emblem Å, Karlsen BO, Evertsen J, Miller DJ, Moum T, Johansen SD: Mitogenome polymorphism in a single branch sample revealed by SOLiD deep sequencing of the Lophelia pertusa coral genome. Gene. 2012, 506: 344-349. 10.1016/j.gene.2012.06.040.
Hellberg M: No variation and low synonymous substitution rates in coral mtDNA despite high nuclear variation. BMC Evol Biol. 2006, 6: 24-10.1186/1471-2148-6-24.
Fukami H, Knowlton N: Analysis of complete mitochondrial DNA sequences of three members of the Montastraea annularis coral species complex (Cnidaria, Anthozoa, Scleractinia). Coral Reefs. 2005, 24: 410-417. 10.1007/s00338-005-0023-3.
De Mol B, Van Rensbergen P, Pillen S, Van Herreweghe K, Van Rooij D, McDonnell A, Huvenne V, Ivanov M, Swennen R, Henriet JP: Large deep-water coral banks in the Porcupine Basin, southwest of Ireland. Mar Geol. 2002, 188: 193-231. 10.1016/S0025-3227(02)00281-5.
De Mol B, Henriet J-P, Canals M: Development of coral banks in Porcupine Seabight: do they have Mediterranean ancestors?. Cold-Water Corals and Ecosystems. Edited by: Freiwald A, Roberts JM. 2005, Springer: Berlin Heidelberg, 515-533.
McCulloch M, Taviani M, Montagna P, López Correa M, Remia A, Mortimer G: Proliferation and demise of deep-sea corals in the Mediterranean during the Younger Dryas. Earth Planet Sci Lett. 2010, 298: 143-152. 10.1016/j.epsl.2010.07.036.
Schönfeld J, Zahn R: Late Glacial to Holocene history of the Mediterranean Outflow. Evidence from benthic foraminiferal assemblages and stable isotopes at the Portuguese margin. Palaeogeogr Palaeoclimatol Palaeoecol. 2000, 159: 85-111. 10.1016/S0031-0182(00)00035-3.
Frank N, Freiwald A, Correa ML, Wienberg C, Eisele M, Hebbeln D, Van Rooij D, Henriet J-P, Colin C, van Weering T: Northeastern Atlantic cold-water coral reefs and climate. Geology. 2011, 39: 743-746. 10.1130/G31825.1.
Tursi A, Mastrototaro F, Matarrese A, Maiorano P, D’Onghia G: Biodiversity of the white coral reefs in the Ionan Sea (Central Mediterranean). Chem Ecol. 2004, 20: S107-S116.
Taviani M, Remia A, Corselli C, Freiwald A, Malinverno E, Mastrototaro F, Savini A, Tursi A: First geo-marine survey of living cold-water Lophelia reefs in the Ionian Sea (Mediterranean basin). Facies. 2005, 50: 409-417. 10.1007/s10347-004-0039-0.
Mastrototaro F, D’Onghia G, Corriero G, Matarrese A, Maiorano P, Panetta P, Gherardi M, Longo C, Rosso A, Sciuto F: Biodiversity of the white coral bank off Cape Santa Maria di Leuca (Mediterranean Sea): An update. Deep-Sea Res II Top Stud Oceanogr. 2010, 57: 412-430. 10.1016/j.dsr2.2009.08.021.
Sargent TD, Jamrich M, Dawid IB: Cell interactions and the control of gene activity during early development of Xenopus laevis. Dev Biol. 1986, 114: 238-246. 10.1016/0012-1606(86)90399-4.
Fukami H, Budd AF, Levitan DR, Jara J, Kersanach R, Knowlton N: Geographic differences in species boundaries among members of the Montastraea annularis complex based on molecular and morphological markers. Evolution. 2004, 38: 324-337.
Rozen S, Skaletsky H: Primer3 on the WWW for general users and for biologist programmers. Bioinformatics Methods and Protocols. Edited by: Misener S, Krawetz SA. 2000, Totowa, NJ: Humana Press, 365-386.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R: DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994, 3: 294-299.
Altschul SF, Gish W, Miller W, Meyers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215: 403-410.
Gray M, Lang B, Cedergren R, Golding G, Lemieux C, Sankoff D, Turmel M, Brossard N, Delage E, Littlejohn T: Genome structure and gene content in protist mitochondrial DNAs. Nucleic Acids Res. 1998, 26: 865-878. 10.1093/nar/26.4.865.
Lowe T, Eddy S: tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25: 955-964.
Benson G: Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999, 27: 573-580. 10.1093/nar/27.2.573.
Bandelt HJ, Forster P, Röhl A: Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999, 16: 37-48. 10.1093/oxfordjournals.molbev.a026036.
Flot J-F, Tillier S: Molecular phylogeny and systematics of the scleractinian coral genus Pocillopora in Hawaii. Proceedings of the 10th International Coral Reef Symposium. 2006, 1: 24-29.
Acknowledgements
Thanks to the crews and researchers aboard the R/V Meteor Cruise 70/1 (Santa Maria di Leuca) and R/V G.O. Sars Cruise No. 2006112 (Korallen). Sampling was supported by HERMES EC contract n°GOCE-CT-2005-511234 funded by the European Commission’s Sixth Framework Programme under the priority ‘Sustainable Development, Global Change and Ecosystems’. Funding was provided by grants from the Oscar and Lili Lamm Foundation and the Linnaeus Centre for Marine Evolutionary Biology at the University of Gothenburg (http://www.cemeb.science.gu.se).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MD initiated the study and obtained the samples, JFF sequenced and analyzed the markers and CA supervised the study. JFF drafted the manuscript that was revised by MD and CA. All authors approved the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Flot, JF., Dahl, M. & André, C. Lophelia pertusa corals from the Ionian and Barents seas share identical nuclear ITS2 and near-identical mitochondrial genome sequences. BMC Res Notes 6, 144 (2013). https://doi.org/10.1186/1756-0500-6-144
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-0500-6-144