
Abstract
Background
The R6K replicon is one of the best studied bacterial plasmid replicons. Replication of the R6K plasmid and derivatives harboring its γ origin of replication (oriR6Kγ) is dependent on the pir gene-encoded π protein. Originally encoded by R6K, this protein is usually provided in trans in hosts engineered to support replication of plasmids harboring oriR6Kγ. In Escherichia coli this is commonly achieved by chromosomal integration of pir either via lysogenization with a λpir phage or homologous recombination at a pre-determined locus.
Findings
Current methods for construction of host strains for oriR6Kγ-containing plasmids involve procedures that do not allow selection for presence of the pir gene and require cumbersome and time-consuming screening steps. In this study, we established a mini-Tn7-based method for rapid and reliable construction of pir+ host strains. Using a curable mini-Tn7 delivery plasmid, pir expressing derivatives of several commonly used E. coli cloning and mobilizer strains were isolated using both the wild-type pir+ gene as well as the copy-up pir-116 allele. In addition, we isolated pir+ and pir-116 expressing derivatives of a clinical isolate of Salmonella enterica serovar Typhimurium. In both E. coli and S. enterica serovar Typhimurium, the presence of the pir+ wild-type or pir-116 alleles allowed the replication of oriR6Kγ-containing plasmids.
Conclusions
A mini-Tn7 system was employed for rapid and reliable engineering of E. coli and S. enterica serovar Typhimurium host strains for plasmids containing oriR6Kγ. Since mini-Tn7 elements transpose in most, if not all, Gram negative bacteria, we anticipate that with relatively minor modifications this newly established method will for the first time allow engineering of other bacterial species to enable replication of plasmids with oriR6Kγ.
Similar content being viewed by others

Background
The γ origin of replication of the broad-host-range plasmid R6K (oriR6Kγ) has been used to construct conditionally replicative cloning and transposon delivery vectors too numerous to cite them all, with some of the most well-known vectors described in the late 1980s and early 1990s [1–6]. Replication of these vectors requires the π protein encoded by the pir gene which on R6K is located next to the γ origin of replication [7, 8]. For maintenance of conditionally replicative plasmids that contain oriR6Kγ but lack pir, the π protein is expressed in trans from pir located on a compatible plasmid or, most frequently, on a λ phage or a gene inserted into the chromosome via homologous recombination at a predetermined locus [9]. In cells harboring a wild-type pir+ gene, oriR6Kγ containing plasmids are maintained at 15 copies or less depending on size of the oriR6Kγ plasmid and pir gene source. A number of pir mutations have been identified that alter plasmid copy number, for example the pir-116 allele [10]. In cells harboring this allele integrated into the chromosome, oriR6Kγ plasmids are maintained at a copy number of about 250 per cell which compares to 15 copies per cell when the pir+ allele is integrated at the same chromosomal locus [9]. The host range for oriR6Kγ-containing plasmids is limited because construction of strains supporting their replication involves methods that do not allow selection for presence of the pir gene and require cumbersome and time-consuming screening steps. To allow expansion of plasmid host range to customized genetic strain backgrounds we therefore developed a mini-Tn7-based method for rapid and reliable construction of enterobacterial pir+ host strains.
Results and discussion
Development of a mini- Tn7 based oriR6Kγ chromosomal insertion system
We sought to employ the mini-Tn7 method described by McKenzie and Craig [11] for chromosomal insertion of pir alleles in the absence of selection. For this purpose, the pir+ and pir-116 genes were cloned into the mini-Tn7 delivery vector pGRG36 (Figure 1) and published procedures [11] followed in an attempt to transpose the cloned pir genes into the chromosomes of various E. coli strains. However, in some strains, despite repetition and exhaustive PCR screening, this method proved ineffective for this purpose as the majority of colonies obtained after completion of the procedure did not contain the desired mini-Tn7 insertions or did not result in any insertions, for unexplained reasons. We therefore designed a method that allows positive selection of strains containing chromosomally inserted pir alleles (Figure 2). The rationale for this method is to establish the delivery vector with a temperature-sensitive replicon (ts), here pSC101orits, at permissive temperature (30°C), then introduce an oriR6Kγ reporter plasmid at 37°C, creating conditions at which the mini-Tn7 delivery vector is cured and replication of the reporter plasmid is dependent on the presence of a chromosomally-integrated pir gene. After verification of the desired mini-Tn7-pir insertions the reporter plasmid is then cured using sucrose counter-selection.

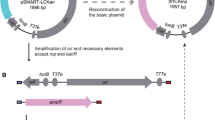
Maps of mini-Tn 7 delivery vector and ori R6Kγ reporter plasmid. A) Map of pir+ delivery vector pGRG36pir. Other versions include pGRG36pir-116 which contains pir-116 in place of pir+ and pGRG36pir-116- FKm contains a FRT-flanked Kmr marker adjacent to pir-116 as illustrated in the inset. B) Map of the oriR6Kγ reporter plasmid pR6KT2. Abbreviations: aacC1, aminoglycoside acetyltransferase conferring gentamicin resistance; araC, encoding the AraC regulator of expression from the E. coli arabinose operon promoter P BAD ; B1 and B2, attB1 and attB2 Gateway recombination sites; bla, β-lactamase gene conferring ampicillin resistance; FRT, Flp recombinase target site; L and R, Tn7 left and right ends; nptII, neomycin phosphotransferase gene conferring kanamycin resistance; oriR6Kγ, R6K plasmid γ replication origin; oriT, RP4 conjugal transfer origin; pSC101ori ts, temperature-sensitive pSC101 replication origin; sacB, levansucrase-encoding gene; tnsABCD, genes encoding the Tn7 site-specific transposition pathway; uidA, glucuronidase-encoding gene.

Overview of steps involved in chromosomal mini-Tn 7 - pir insertion. A) Introduce the mini-Tn7-pir+ delivery vector by conjugation. Select ampicillin resistant (Ampr) colonies at 30°C to establish the delivery vector. B) Grow Ampr cells at 30°C in presence of arabinose to induce the genes encoding the Tn7 site-specific transposition pathway. C) Introduce the oriR6Kγ reporter pR6KT2 by conjugation and grow at 37°C in the presence of gentamicin (Gm) to cure the mini-Tn7-pir+ delivery vector and report integrants based on pir-dependent replication of pR6KT2. Establish Gmr and Amp susceptible (Amps) phenotype. D) Cure pR6KT2 reporter plasmid by plating on sucrose-containing medium. Verify Gms and Amps phenotype, and confirm pir+ integrants by PCR (using primer pair 2372 and 2373 for E. coli or 2374 and 2375 for S. enterica serovar Typhimurium). It must be noted that PCR-based insertion site verification strategies are limited to bacteria for which sequence information about the glmS flanking sequences is available. Methods for identifying Tn7 insertion sites in bacteria for which genome sequences are unknown have been described [12].
Mini-Tn7 insertion of pir genes in E. coli and S. enterica serovar Typhimurium
Following the procedure outlined in Figure 2, we readily obtained mini-Tn7-pir insertions in several commonly used E. coli laboratory cloning and mobilizer strains--DH5α, JM108, MC4100, SM10, RHO3--and a clinical S. enterica serovar Typhimurium isolate. Exconjugants examined by PCR contained the desired insertion, either mini-Tn7-pir+ or mini-Tn7-pir-116 (Figure 3A). The plasmid copy number of pR6KT2 was greatly elevated in pir-116-containing E. coli DH5α and S. enterica serovar Typhimurium 14028S host strains when compared to the same strains containing chromosomally inserted wild-type pir+ (Figure 3B). An alternative to employing an oriR6Kγ reporter plasmid is to use mini-Tn7-pir elements that contain a Kmr selection marker that after verification of desired inserts can be removed using Saccharomyces cerevisiae Flp recombinase-mediated site-specific excision, followed by curing of the Flp recombinase expression plasmid. We have successfully used this strategy in E. coli. Both strategies require equal time and effort.

PCR verification of mini-Tn 7 - pir insertion and plasmid copy number. A) PCR verification of pir+ and pir-116 mini-Tn7 insertions into att Tn7 of E. coli DH5α. Colony PCR with primers 2372 and 2373 (see Figure 2 for relative priming site locations) was conducted to confirm presence or absence of the mini-Tn7-pir+ and mini-Tn7-pir-116 insertions. The PCR reactions were analyzed by agarose gel electrophoresis. Expected fragment sizes are 678 bp for DH5α without a mini-Tn7 insertion and 2,539 bp for derivatives containing mini-Tn7-pir+ or mini-Tn7-pir-116 insertions. Lane M, Hi-Lo molecular size ladder from Minnesota Molecular (Minneapolis, MN) with the sizes of selected fragments indicated; Lane -, DH5α negative control (no insertion). B) Demonstration of pR6KT2 copy number in pir+ and pir-116 containing E. coli and S. enterica serovar Typhimurium strains. Plasmid DNA was purified from overnight cultures of E. coli DH5α and S. enterica serovar Typhiumurium 14028S containing either mini-Tn7-pir+ or mini-Tn7-pir-116. A 20 μl aliquot of each preparation was digested with Hin dIII to linearize the 5,953-bp plasmid and the samples were analyzed by agarose gel electrophoresis. Lane M contains the same molecular size ladder as shown in panel A and the sizes of selected fragments are indicated.
Conclusions
We have developed simple and effective strategies for engineering of pir expressing strains of Enterobacteriaceae. These strategies allow extension of the host range of oriR6Kγ containing plasmids to virtually any enterobacterial strain, something that was, to date, only possible using relatively cumbersome and time-consuming methods, e.g. isolation of λpir lysogens or chromosomal insertion of cloned pir alleles via site-specific recombination at a pre-determined locus [9]. Mini-Tn7 elements insert at naturally evolved att Tn7 sites that are usually located in intergenic regions downstream of conserved glmS genes [12–16]. This alleviates the need for selecting potential insertion sites not affecting bacterial fitness when choosing recombinant DNA strategies for pir allele insertion into a bacterial genome. Since mini-Tn7 elements transpose in most, if not all, Gram negative bacteria, we anticipate that with relatively minor modifications this newly established method will for the first time allow engineering of other bacterial species to enable replication of plasmids with oriR6Kγ. As described, the procedure relies on availability of ts replicons which may limit its applicability to bacteria that can tolerate the non-permissive temperatures needed for plasmid curing. The methods described here were developed for Enterobacteriaceae which, like most bacteria, can tolerate 37°C, a temperature at which most plasmids with ts replicons, including pSC101orits, are readily cured. For bacteria with growth temperature optima less than 37°C, the described strategy will not work and require appropriate modifications, i.e. inclusion of different counter-selection markers, for example sacB[17]. Lastly, though many manipulations described in this paper use conjugations as means for introduction of plasmids into cells, some them could also be done by plasmid transformation which would alleviate the need for counter-selection strategies required for bacterial matings. We, however, consistently find that conjugations are equally convenient and more efficient means of plasmid transfer than transformation.
Methods
Bacterial strains, media and growth conditions
Bacterial strains used in this study are listed in Table 1. Bacteria were routinely grown at 37°C in Luria Bertani broth Lennox (LB) [18] or on LB agar purchased from MO BIO Laboratories, Carlsbad, CA. The sacB-containing oriR6Kγ reporter pR6KT2 was cured by plating plasmid-containing cells on yeast extract-tryptone (YT) sucrose medium containing 10 g/l yeast extract (Difco, Detroit, MI), 16 g/l tryptone (Fisher Scientific, Fairlawn, NJ), 16 g/l Bacto agar (Becton, Dickinson and Company, Sparks, MD) and 15% sucrose (w/v)[19]. Strains containing temperature-sensitive (TS) plasmid derivatives were grown at 30°C (permissive temperature) for plasmid maintenance and 37°C or 42°C (non-permissive temperature) for plasmid curing. Antibiotics were added at the following concentrations: 100 μg/ml ampicillin (Amp), 10-15 μg/ml gentamicin (Gm) and 35 μg/ml kanamycin (Km) for E. coli and S. enterica serovar Typhimurium harboring plasmids or for selection of chromosomally-integrated mini-Tn7 elements. Antibiotics were purchased from EMD Biosciences, San Diego, CA (Gm) and Sigma, St. Louis, MO (Amp and Km). For E. coli strain RHO3, media were supplemented with diaminopimelic acid (DAP; LL-, DD-, and meso-isomers; Sigma) which was added at a final concentration of 400 μg/ml for agar plates and 200 μg/ml for broth cultures. 5-bromo-4-chloro-3-indolyl-β-D-glucuronic acid (XGluc; Gold Biotechnology, St. Louis, MO) was added to media at a final concentration of 40 μg/ml. Induction of gene expression from the arabinose operon promoter (P BAD ) was achieved by addition of L-arabinose to media at a final concentration of 0.5% (w/v).
DNA and genetic methods
Isolation of plasmid DNA
Plasmid DNAs were isolated from E. coli and S. enterica serovar Typhimurium by using a Fermentas GeneJET Plasmid MiniPrep Kit (Fermentas, Glen Burnie, MD).
Transposition of mini-Tn7
The respective mini-Tn7 delivery vectors were transformed into E. coli mobilizer strain RHO3 [19]. Conjugation of delivery plasmid into E. coli and S. enterica serovar Typhimurium strains was achieved by biparental mating using previously described methods [19] with some minor modifications. Briefly, RHO3 donor and E. coli and S. enterica serovar Typhimurium recipient cultures were grown overnight at 30°C (pGRG36-based donor strains) or 37°C (recipient strains) in LB medium with the appropriate nutritional (DAP) and antibiotic (Amp) supplements for RHO3 with the mini-Tn7 delivery vector. One ml of donor and recipient were placed into separate 1.7 ml microcentrifuge tubes and harvested by centrifugation in a microcentrifuge for 30 s at 13,400×g and room temperature. Cells were washed twice in 1 ml LB medium and then re-suspended in 200 μl of LB medium. Equal volumes (25 μl) of each cell suspension were transferred to a cellulose acetate membrane (13 mm diameter; 0.45 μM pore size; Sartorius Stedim, Bohemia, NY) sitting on an LB agar plate containing 400 μg/ml DAP and 0.5% arabinose. After overnight incubation at 30°C, the membrane was transferred to a microcentrifuge tube containing 1 ml of LB and cells dislodged by centrifugation in a microfuge for 30 s at 13,400×g and room temperature. After removing the membrane, cells were washed twice in 1 mL LB and then re-suspended in 200 μl of LB medium. The entire sample was placed on an LB-agar plate with 100 μg/ml Amp and 0.5% arabinose, and a portion streaked for single colonies with an inoculating loop. The plates were incubated at 30°C overnight or until single colonies were clearly discernable.
A single purified colony was then used as recipient for the oriR6Kγ reporter pR6KT2. This plasmid was introduced via biparental mating from RHO3 as described above, except that recipient cells were grown in the presence of Amp and arabinose and RHO3/pR6KT2 cultures were grown in the presence of Gm and DAP. After overnight incubation at 30°C mating mixtures were recovered and plated on LB with 15 μg/ml Gm at 37°C to cure the temperature-sensitive mini-Tn7 delivery vector and select for pR6KT2. Purified colonies were patched on LB, LB + Gm and LB + Amp to confirm the loss of the mini-Tn7 delivery vector (Amp susceptibility) and presence of pR6KT2 (Gm resistance). After verification of mini-Tn7 insertions by PCR, pR6KT2 was cured by streaking a single colony on YT medium with sucrose and XGluc, and incubating overnight at 37°C. Single colonies were patched on LB and LB + Gm plates to confirm the loss of the plasmid. The pir gene insertions in the resulting strains were then re-confirmed by PCR and sequencing of the resulting DNA fragments.
When using the mini-Tn7-pir-FKm vectors, the protocol for conjugation was as described above for mini-Tn7 delivery without antibiotic selection. Exconjugants were grown overnight in LB + DAP and arabinose at 30°C, and Kmr transposon insertions were selected by plating conjugation mixtures on LB plates with 35 μg/ml Km followed by incubation at 42°C to cure the delivery plasmid. The Kmr marker can optionally be deleted from the strain with the mini-Tn7-pir-FKm insertion by transformation with pFLP2 (or any other Flp recombinase-expressing plasmid such as pCP20 [25]), testing transformants for Km susceptibility and then curing pFLP2 by plating on sucrose-containing media following previously described protocols [17].
Confirmation of mini-Tn7-pir insertions
Insertions of mini-Tn7-pir at att Tn7 in E. coli and S. enterica serovar Typhimurium were performed by colony PCR using DNA in boiling preparation lysates as templates. These lysates were obtained by transferring separate colonies to individual sterile microcentrifuge tubes containing 30 μl of sterile water and boiling for 10 min. Using 6 μl of these boiling preparations and Taq DNA polymerase from New England Biolabs, PCR reactions were performed in a total volume of 50 μl. Primer pairs 2372 (5'-GATGCTGGTGGCGAAGCTGTC) & 2373 (5'-GATGACGGTTTGTCACATGGAG) and 2374 (5'-CAGCAACGACAACATGCACA) & 2375 (5'-AAACCATCAGCGCGGAACAA) were used for E. coli and S. enterica serovar Typhimurium, respectively. In each case, the entire PCR reaction was analyzed on a 1% agarose gel. Expected PCR fragment sizes are 678 bp for E. coli without a mini-Tn7 insertion and 2,539 bp for derivatives containing mini-Tn7-pir+ and mini-Tn7-pir-116. Fragment sizes for S. enterica serovar Typhimurium without and with mini-Tn7-pir insertions are 485 bp and 2,345 bp, respectively. When utilizing mini-Tn7-pir-FKm, the fragment sizes obtained with strains containing pir+ or pir-116 insertions change by +1,470 bp when the Kmr marker is present or by +145 bp after its Flp-mediated excision.
Plasmid construction
Plasmids used in this study are listed in Table 2. The Gateway-compatible mini-Tn7 delivery vector pGRG36GW was constructed by cloning of a 1,770-bp Stu I-Xho I fragment from pUC18-mini-Tn7T-Gm-GW [16] between the Sma I and Xho I sites of pGRG36 [11], followed by transformation into the gyrA462 strain DB3.1.
The pir+ and pir-116 Gateway donor clones were constructed as follows. First, the pir+ and pir-116 genes were PCR-amplified from E. coli strain PIR2 (pir+) and PIR1 (pir-116) chromosomal DNA templates using HiFi Taq polymerase (Invitrogen) and primers 1333 (5'-TGAGCGTCGCAGAACATTACA) & 1334 (5'-ACCTGGGTGGACGATATCAC). The resulting 1,264-bp PCR fragments were cloned into the TA cloning vector pCR2.1 (Invitrogen) to create pCR2.1pir and pCR2.1pir-116. Next, attB sequences were attached to the respective pir gene segments using PCR with primers 1558 (5'-GGGGACAAGTTTGTACAAAAAAGCAGGCT TGAGCGTCGCAGAACATTACA; the attB1 sequence is underlined) & 1559 (5'-GGGGACCACTTTGTACAAGAAAGCTGGGT ACCTGGGTGGACGATATCAC; the attB2 sequence is underlined), and pCR2.1-pir and pCR2.1-pir-116 DNA as templates. This generated 1,323-bp DNA fragments that were recombined into pDONR221 using Gateway BP clonase reactions which created pDONR221pir and pDONR221pir-116, respectively. Finally, the inserts from pDONR221pir and pDONR221pir-116 were transferred to pGRG36GW using LR Gateway recombination to yield pGRG36pir and pGRG36pir-116. The pGRG36pir-116-FKm plasmid was constructed by cloning a 1,470-bp Sal I fragment from pFKM4 containing a Kmr gene flanked by Flp recombinase target (FRT) sites into the Xho I site of pGRG36pir-116.
The oriR6Kγ reporter pR6KT2 was constructed as follows. First, the oriR6Kγ and oriT regions were PCR-amplified from pUC18R6KT-mini-Tn7 T [16] using Taq polymerase (NEB) and primers 2298 (5'-ATTCCCGGGAGGCCACCACTTCAAGAACTC) & 2299 (5'-TAATCCCGGGCTTCCGCTTCCTCGCTCA). The resulting 824-bp amplicon was cloned into
pCR2.1 to create pCR2.1-R6KoriT. Second, the 901-bp Xba I fragment from non-methylated pPS856 [17] DNA obtained by isolating the plasmid from dam dcm E. coli JM110 [21] was ligated with the 6.5-kb Spe I and Xba I digested backbone of pEXKm5 [19] to construct pEXGm5B. Third, the 915-bp oriR6Kγ and oriT Xba I and Eco 53K1 fragment was released from pCR2.1::R6KoriT and ligated with the 6.2-kb Xba I and Sma I digested pEXGm5B backbone to create pR6KTSCE. Finally, pR6KTSCE was digested with Bmg B1 and Fsp I and the 5.9-kb backbone was re-circularized to construct the reporter vector pR6KT2.
All plasmid constructions were verified by restriction digest and DNA sequence analysis.
References
Miller VL, Mekalanos JJ: A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J Bacteriol. 1988, 170: 2575-2583.
Taylor RK, Manoil C, Mekalanos JJ: Broad-host-range vectors for delivery of TnphoA: Use in genetic analysis of secreted virulence determinants of Vibrio cholerae. J Bacteriol. 1989, 171: 1870-1878.
Herrero M, De Lorenzo V, Timmis KN: Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in Gram-negative bacteria. J Bacteriol. 1990, 172: 6557-6567.
Donnenberg MS, Kaper JB: Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect Immun. 1991, 59: 4310-4317.
Metcalf WW, Wanner BL: Mutational analysis of an Escherichia coli fourteen-gene operon for phosphonate degradation, using TnphoA' elements. J Bacteriol. 1993, 175: 3430-3442.
Skrzypek E, Haddix PL, Plano GV, Straley SC: New suicide vector for gene replacement in Yersinia and other gram-negative bacteria. Plasmid. 1993, 29: 160-163. 10.1006/plas.1993.1019.
Kolter R, Inuzuka M, Helinski DR: Trans-complementation-dependent replication of a low molecular weight origin fragment from plasmid R6K. Cell. 1978, 15: 1199-1208. 10.1016/0092-8674(78)90046-6.
Stalker DM, Kolter R, Helinski DR: Nucleotide sequence of the region of an origin of replication of the antibiotic resistance plasmid R6K. Proc Natl Acad Sci USA. 1979, 76: 1150-1154. 10.1073/pnas.76.3.1150.
Metcalf WW, Jiang W, Wanner BL: Use of the rep technique for allele replacement to construct new Escherichia coli hosts for maintenance of R6Kγ origin plasmids at different copy numbers. Gene. 1994, 138: 1-7. 10.1016/0378-1119(94)90776-5.
Filutowicz M, McEachern MJ, Mukhopadhyay P, Greener A, Yang SL, Helinski DR: DNA and protein interactions in the regulation of plasmid replication. J Cell Science Supplement. 1987, 7: 15-31.
McKenzie GJ, Craig NL: Fast, easy and efficient: site-specific insertion of transgenes into enterobacterial chromosomes using Tn7 without need for selection of the insertion event. BMC Microbiol. 2006, 6: 39-10.1186/1471-2180-6-39.
Choi K-H, DeShazer D, Schweizer HP: mini-Tn7 insertion in bacteria with multiple glmS-linked att Tn7 sites: example Burkholderia mallei ATCC 23344. Nat Protocols. 2006, 1: 162-169. 10.1038/nprot.2006.25.
Peters JE, Craig NL: Tn7: smarter than we thought. Nat Rev Mol Cell Biol. 2001, 2: 806-814. 10.1038/35099006.
Choi K-H, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, Schweizer HP: A Tn7-based broad-range bacterial cloning and expression system. Nat Methods. 2005, 2: 443-448. 10.1038/nmeth765.
Choi K-H, Schweizer HP: mini-Tn7 insertion in bacteria with secondary, non-glmS-linked att Tn7 sites: example Proteus mirabilis HI4320. Nat Protocols. 2006, 1: 170-178.
Choi K-H, Schweizer HP: mini-Tn7 insertion in bacteria with single att Tn7 sites: example Pseudomonas aeruginosa. Nat Protocols. 2006, 1: 153-161.
Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP: A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene. 1998, 212: 77-86. 10.1016/S0378-1119(98)00130-9.
Sezonov G, Joseleau-Petit D, D'Ari R: Escherichia coli physiology in Luria-Bertani broth. J Bacteriol. 2007, 189: 8746-8749. 10.1128/JB.01368-07.
Lopez CM, Rholl DA, Trunck LA, Schweizer HP: Versatile dual-technology system for markerless allele replacement in Burkholderia pseudomallei. Appl Environ Microbiol. 2009, 75: 6496-6503. 10.1128/AEM.01669-09.
Liss L: New M13 host: DH5αF' competent cells. Focus. 1987, 9: 13-
Yanisch-Perron C, Vieira J, Messing J: Improved M13 cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985, 33: 103-119. 10.1016/0378-1119(85)90120-9.
Casadaban MJ: Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J Mol Biol. 1976, 104: 541-555. 10.1016/0022-2836(76)90119-4.
Peters JE, Thate DE, Craig NL: Definition of the Escherichia coli MC4100 genome by use of a DNA array. J Bacteriol. 2003, 185: 2017-2021. 10.1128/JB.185.6.2017-2021.2003.
Simon R, Priefer U, Pühler A: A broad-host-range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Bio/Technology. 1983, 1: 784-791. 10.1038/nbt1183-784.
Cherepanov PP, Wackernagel W: Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995, 158: 9-14. 10.1016/0378-1119(95)00193-A.
Choi K-H, Mima T, Casart Y, Rholl D, Kumar A, Beacham IR, Schweizer HP: Genetic tools for select agent compliant manipulation of Burkholderia pseudomallei. Appl Env Microbiol. 2008, 74: 1064-1075. 10.1128/AEM.02430-07.
Acknowledgements
HPS is supported by several NIH grants and this research was made possible by Indirect Cost Recovery funds. The authors acknowledge the contributions of Dr. Takehiko Mima to this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
BK and SB performed most of the experiments, JP, IM, EB, SL, KM and KHC contributed to plasmid construction and performed pilot experiments, BK and RKS designed and supervised experiments, and BK, RKS and HPS drafted the final manuscript. All authors read and approved the final manuscript.
Brian H Kvitko, Steven Bruckbauer contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kvitko, B.H., Bruckbauer, S., Prucha, J. et al. A simple method for construction of pir+ Enterobacterial hosts for maintenance of R6K replicon plasmids. BMC Res Notes 5, 157 (2012). https://doi.org/10.1186/1756-0500-5-157
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-0500-5-157