
Abstract
Background
The striped bass and its relatives (genus Morone) are important fisheries and aquaculture species native to estuaries and rivers of the Atlantic coast and Gulf of Mexico in North America. To open avenues of gene expression research on reproduction and breeding of striped bass, we generated a collection of expressed sequence tags (ESTs) from a complementary DNA (cDNA) library representative of their ovarian transcriptome.
Results
Sequences of a total of 230,151 ESTs (51,259,448 bp) were acquired by Roche 454 pyrosequencing of cDNA pooled from ovarian tissues obtained at all stages of oocyte growth, at ovulation (eggs), and during preovulatory atresia. Quality filtering of ESTs allowed assembly of 11,208 high-quality contigs ≥ 100 bp, including 2,984 contigs 500 bp or longer (average length 895 bp). Blastx comparisons revealed 5,482 gene orthologues (E-value < 10-3), of which 4,120 (36.7% of total contigs) were annotated with Gene Ontology terms (E-value < 10-6). There were 5,726 remaining unknown unique sequences (51.1% of total contigs). All of the high-quality EST sequences are available in the National Center for Biotechnology Information (NCBI) Short Read Archive (GenBank: SRX007394). Informative contigs were considered to be abundant if they were assembled from groups of ESTs comprising ≥ 0.15% of the total short read sequences (≥ 345 reads/contig). Approximately 52.5% of these abundant contigs were predicted to have predominant ovary expression through digital differential display in silico comparisons to zebrafish (Danio rerio) UniGene orthologues. Over 1,300 Gene Ontology terms from Biological Process classes of Reproduction, Reproductive process, and Developmental process were assigned to this collection of annotated contigs.
Conclusions
This first large reference sequence database available for the ecologically and economically important temperate basses (genus Morone) provides a foundation for gene expression studies in these species. The predicted predominance of ovary gene expression and assignment of directly relevant Gene Ontology classes suggests a powerful utility of this dataset for analysis of ovarian gene expression related to fundamental questions of oogenesis. Additionally, a high definition Agilent 60-mer oligo ovary 'UniClone' microarray with 8 × 15,000 probe format has been designed based on this striped bass transcriptome (eArray Group: Striper Group, Design ID: 029004).
Similar content being viewed by others

Background
The striped bass and its relatives in the genus Morone (the temperate basses) are ecologically and economically important aquaculture and fisheries species native to estuaries and rivers of the Atlantic coast and Gulf of Mexico in North America [1, 2]. Although the striped bass and its hybrids have been reared as commercial aquaculture products in the United States since the late 1980s, little genetic information is available for these species in public databases at the National Center for Biotechnology Information (NCBI) or elsewhere, consisting only of microsatellite DNA markers [3, 4], the mitochondrial genome (GenBank: HM447585), and a medium density genetic linkage map [5]. A major factor contributing to restricted growth of hybrid striped bass farming nationwide is reproductive dysfunction of female striped bass, resulting in non-viable eggs, embryos, and larvae [6]. These reproductive failures hamper selective breeding efforts required for species domestication and improvement. The exact cause(s) of poor egg quality and embryonic mortality in farmed fishes, however, still remain to be discovered, making appropriate and timely corrective measures difficult to achieve [review: [7, 8]].
Functional genomics has emerged as a major research field and gene expression (transcriptomics) and proteomics studies are promising approaches to gain new insights into reproductive molecular biology [7, 9–12]. Marked advancement in striped bass reproductive technology based on such "Omic" analyses is, however, currently restricted due to the lack of an available, comprehensive sequence database for this species or for other members of the genus Morone that are important in aquaculture (e.g. hybrid striped bass) or as research models (e.g. white perch, M. americana). Transcriptome resources are currently available for other commercially important fishes, including rainbow trout (Oncorhynchus mykiss) [13–16], coho salmon (Oncorhynchus kisutch) [17], tilapia (Oreochromis mossambicus) [18], Atlantic halibut (Hippoglossus hippoglossus) [19], Senegalese sole (Solea senegalensis) [20], Atlantic salmon (Salmo salar) [21], and cod (Gadus morhua) [22].
The emergence of pyrosequencing and later generation DNA sequencing technologies has made acquisition of significant genomic resources accessible and affordable for non-model organisms [23–25]. Vast numbers of expressed sequence tags (ESTs) can readily be generated using these methods, providing direct evidence of gene transcription, and collections of such EST sequences are presently the most important resources used for transcriptome exploration [26]. Depending on the number of ESTs sequenced, resulting databases can represent a high proportion of the total number of gene transcripts expressed by a given tissue (i.e. transcriptome), making downstream procedures for transcriptome profiling, such as oligo microarray or real-time quantitative reverse transcription PCR, tractable without the need for an entire genome sequence.
When sequencing depth is limited, organ specific EST collections permit more efficient gene expression analyses using 'UniClone' microarrays, which are comprised of probe sequences isolated from a single organ type [27–30]. UniClone arrays represent a larger proportion of a target organ transcriptome and have reduced redundancy when compared to arrays comprised of ESTs derived from several different tissue types. Additionally, to realize the full benefits of proteomic analyses based on mass spectrometry, species-specific ESTs are required, since algorithms used for spectral analyses (e.g. SEQUEST, Proteome Discoverer Software, Thermo Scientific, West Palm Beach, FL) require a homologous reference sequence database. For non-model organisms, sequence information from even closely related species can be insufficient for the accurate identification of peptides, since these algorithms tend to be conservative and heterospecific amino acid substitutions can result in peptide misidentification or an inability to detect orthologues [31].
Therefore, the goal of the present study was to provide an ovary transcriptome database representative of all stages of oogenesis and atresia in striped bass, one that could provide the requisite foundation for functional genomics and proteomics investigations of reproduction and egg quality in this species and that would support similar studies in the other temperate basses.
Results
A total of 230,151 EST short read sequences with a combined length of 51,259,448 bp (average length 224 bp) were generated from cDNA pooled from ovarian tissues and eggs encompassing the various stages of ovary growth, maturation and atresia. A total of 11,208 high-quality contigs with a length of at least 100 bp were assembled and these included 2,984 contigs that were 500 bp or longer (average length 895 bp; total length 5,068,343 bp) (Additional File 1). Blastx comparisons revealed 5,482 orthologues, of which 4,120 (36.7%) were annotated with Gene Ontology (GO) terms. The number of unknown, unique sequences was 5,726 (51.1%). The breakdown of GO annotation classes within the three categories of GO terms for all annotated sequences is shown in Figure 1: Biological Process (2nd level) and Molecular Function and Cellular Component (3rd level). A complete list, in FASTA format, of the contig assemblies identified by their annotations are included as Additional File 2 and a list of the assemblies and their GO terms are included as Additional File 3.

Gene ontology graph of A. Cellular Component (3rd level GO terms), B. Molecular Function (3rd level GO terms), and C. Biological Process (2nd level GO terms) of annotated genes in the striped bass ovary transcriptome. The number of GOs in each class is shown and sections that contained 50-150 entities are represented in black, 151-500 by dark gray, 500 and up by light gray, and the predominant class is indicated in white.
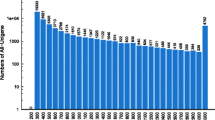
There were 66 contigs that were each assembled from groups of ESTs that comprised ≥ 0.15% of the total 230,151 reads (i.e. ≥ 345 reads per contig) and these contigs were considered to have abundant ovary expression. These contigs were identified by NCBI UniGene cluster and compared to zebrafish, Danio rerio, orthologues evaluated by Digital Differential Display (DDD) (Table 1). Twenty-two striped bass genes from this list (33.3% of the total listed) either had no blastx returns (i.e. were novel), or were identified as being unnamed gene products, or had gene names but no zebrafish UniGene orthologues. These were excluded from further evaluation. Of the remaining informative 44 genes, 23 (52.5%) are predicted to have predominant ovary expression based on DDD of zebrafish orthologues, 11 (25.0%) would be expected to have no difference in expression between ovary and other tissues of the body based on the DDD results, and 10 (22.7%) would likely have predominant expression in other tissues of the body based on the DDD comparison. Overall, the estimated 66 most abundantly expressed striped bass ovary genes were assembled from ~1/6 of the total number of short read sequences (Table 1).
All of the high-quality ESTs have been deposited in the NCBI Short Read Archive (GenBank: SRX007394) and annotated contigs are posted under "Resources" on the National Animal Genome Research Program Aquaculture Genome Projects website (http://www.animalgenome.org/aquaculture/database/) [32]. These contigs also have been submitted to Agilent Technologies eArray (Santa Clara, CA) for ovary UniClone microarray design (http://www.chem.agilent.com/). We designed a high definition 60-mer SurePrint oligo array with 8 × 15,000 probe format comprised of 11,145 UniGene probes from the transcriptome, plus an additional 3,854 probes printed in duplicate or selected from Morone cDNAs available from NCBI or from our own unpublished results (B.J. Reading and C.V. Sullivan, unpublished data) and datasets (eArray Group: Striper Group, Design ID: 029004).
Discussion
This collection of ESTs represents the first contribution of a large reference sequence database for species of the genus Morone and provides a basis for future gene expression studies in these temperate basses. Availability of characterized ovarian transcriptomes from fishes other than zebrafish is limited. Partial transcriptomes have been reported for tilapia (474 EST assemblies) [18] and for cod (1,361 EST assemblies) [22]. Several thousand ovarian ESTs have been reported for salmonid fishes [[13, 15, 33] and references therein], but to our knowledge these have not been assembled into a comprehensive ovarian transcriptome. Numbers of total ESTs currently available in the NCBI EST database for some other commercially important finfishes are as follows: rainbow trout (287,967), coho salmon (4,942), tilapia (Genus Oreochromis, 121,346), Atlantic halibut (20,836), Senegalese sole (10,631), Atlantic salmon (498,212), and cod (229,094). Therefore, the 230,151 ESTs reported herein represent a comparatively valuable transcriptome resource for striped bass.
If the 11,208 contigs are considered to be UniGenes, this represents a substantial proportion of the estimated total protein-coding gene transcripts expressed by the striped bass ovary (i.e. transcriptome) as the average number of mRNA transcripts expressed by a single tissue type is estimated to be between 10,000-15,000 [34], but can be as low as 8,200 [35]. Since over 1,300 GOs from Biological Process classes of Reproduction (121), Reproductive process (55), and Developmental process (1,188) were assigned to the annotated contigs (Figure 1), this sequence collection should prove to be a powerful tool for analysis of ovarian gene expression related to fundamental questions of oogenesis.
Approximately 52.5% of the informative contigs considered to have abundant ovary expression (i.e. those with ≥ 345 reads per contig) were also predicted to have predominant expression in striped bass ovary through DDD comparisons to zebrafish orthologues (Table 1). These include cyclin B2 (ccnb2, contig10186), several egg envelope and zona pellucida proteins, histone H2A (h2af1o, contig00030), oogenesis-related gene (org, contig11081), cyclin B1 (ccnb1 contig00092), karyopherin alpha 2 (kpna2, contigs 00126 and 11014), claudin 4 (cldnd, contig00036), securin (LOC566690, contig 09337), cell division cycle 20 homolog (cdc20, contig10986), cyclin K (LOC100331304, contig11174), ribonucleotide reductase M2 polypeptide (rrm2, contig10438), ribosomal protein S20 (rps20, contig11198), cytochrome C oxidase copper chaperone (cox17, contig00771), and epididymal secretory protein E1 (npc2, contig00231). Many of these are well-characterized ovary transcripts and several recent and informative papers have been published detailing the functions of these genes and their protein products in fish oocytes and embryos [see: [7, 8, 13–20, 27, 28, 36–38]]; others are briefly detailed below.
The remaining 47.5% of abundant striped bass ovary genes that were compared to zebrafish orthologues in the DDD were predicted to have indifferent or predominant expression levels in other tissues of the body relative to the ovary. These may represent constitutively expressed genes or those expressed at high levels in the ovary albeit comparatively lower than in other tissues of the body, respectively. Examples of potential genes with constitutive expression include NADH quinone 1 (nqo1, contig00146), tubulin (zgc:123194, contig08878 and MGC171407, contig10107), fatty acid binding proteins (fabp1b, contig10472 and fabp11a, contig11147), H1 histone member oocyte-specific (h1m, contig06635), nucleoside diphosphate kinase B (nme2b, contig00127), geminin DNA replication inhibitor (gmnn, contig09321), superoxide dismutase (sod1, contig09354), ubiquitin B (ubb, contig10048), and cyclin A2 (ccna2, contig10083). Of these, fatty acid-binding protein heart (fabp11a) has been shown to be up-regulated in ovary of rainbow trout females that mature precociously [13] and an orthologue of h1m (H1foo) is generally considered to be an oocyte specific histone in mouse (Mus musculus) [39, 40], contrary to the DDD prediction. The UniGene EST Profile of zebrafish h1m (Dr. 75735) indicates that it is predominantly expressed in skin, however the second most abundant site of expression is the reproductive system.
The following genes expressed in striped bass ovary are also expressed in zebrafish ovary, however the DDD indicates that they are predominantly expressed in other tissues of the body (Table 1): histone (h3f3c, contig09329), beta-actin (bactin2, contig10058), ADP/ATP translocase (slc25a5, contig11098), 60S acidic ribosomal protein P0 (rplp0, contig10309), elongation factor 1 alpha (ef1a, contig10120), peptidylprolyl isomerase A (ppia, contig10741), eukaryotic translation elongation factor 1 gamma (eef1g, contig10746), stathmin 1 oncoprotein 18 variant 8 (stmn1b, contig00216), and heat-shock proteins 8 (hspa8, contig09917) and 90 (hsp90ab1, contig10015). Ovarian representation of gene transcripts that show predominant expression in other tissues of the body is not surprising given the heterogeneous complexity of the ovary, which is comprised of vasculature, blood and other connective tissues, the somatic follicle, and germ cells. Furthermore, most of these genes, for example ef1a and bactin2, are considered to have constitutively high expression in most tissues, and this is supported by the corresponding zebrafish UniGene EST Profiles (Dr. 31797 and Dr.75125, respectively). There were, however, three exceptional genes whose expression, although considered to be lower in comparison to other tissues of the body by DDD, have been shown to be highly expressed in ovary. Stathmin (stmn) is expressed in oocytes and pre-implantation embryos of mice [41] and in cod ovary [22], and Stmn proteins have been detected in zebrafish ovary [36]. Contig00216 encodes a full-length, 147 amino acid Stmn and has been putatively identified as stmn1b, however it is highly similar to two zebrafish stmn isoforms (95% and 94% amino acid identity with stmn1b and stmn1a, respectively). Although stmn1b has body predominant expression in zebrafish by DDD (Table 1), zebrafish stmn1a (UniGene Dr.52664) shows ovary predominant expression and, therefore, contig00216 may actually be orthologous to stmn1a. Given the high similarity of this sequence to both zebrafish stmn1 isoforms, it is not possible to definitively assign identity without comparison to the other striped bass stmn isoform, which is unavailable. Recently, hsp8 and hsp90 (corresponding to striped bass hspa8 and hsp90ab1, respectively) have been characterized as some of the most abundant genes expressed in mouse and fish eggs at both the transcript and protein levels [36, 37, 42].
This inconsistent result may relate to the inherent weaknesses of DDD, since only highly expressed genes are adequately represented in the EST libraries used to conduct the in silico comparisons and the Fisher's exact test (FET) is conservative [43]. Although this method does not offer quantitation, ranking of the striped bass contigs by number of short reads used in assembly paired with comparisons to zebrafish orthologues evaluated by DDD proved to be a useful tool for estimating relative ovarian abundance of the striped bass gene transcripts. Reservation must be taken when considering such interspecific DDD comparisons for the purpose of excluding genes that are predicted to have less predominant expression in one tissue compared to another, since they may be highly expressed in both. This is a promising approach for characterization of novel gene transcripts from EST libraries and has recently been used to identify ovary specific genes in zebrafish [44] and rainbow trout [15], however such results should be further validated using an experimental evaluation of gene expression.
The growing oocyte is considered to be largely transcriptionally inactive, acting as a storehouse of specific maternal RNAs, proteins, and other molecules required for competency for fertilization, initiation of zygotic development, and transition to embryonic gene expression [review: [37, 38]]. These maternal factors may be stored in oocytes for extended periods of time until use (e.g. months to years). Therefore, a system of regulatory proteins and RNAs must mediate the oocyte cell cycle during growth, ovarian maturation (OM), and zygotic development from fertilization until activation of the embryonic genome at the mid-blastula transition [45]. A number of known cell-cycle regulators and proteins critical for these processes have been identified as predominantly expressed in striped bass ovary (Table 1). Examples include cyclins B1 and B2 (ccnb1, ccnb2) [46–49], cyclin K (ccnk) [50], securin [51], cdc20[27], kpna2[22, 52], gmnn[53], h2af1o[54] and org[44]. Transcripts encoding several different cell division and cell cycle regulatory proteins were similarly reported in the ovaries of cod [22] and rainbow trout [13].
Solute carrier protein (SLC) family members are selected to illustrate representation of sequences in the striped bass ovary transcriptome encoding proteins from a large gene series. The SLCs are a diverse group of eukaryotic membrane proteins that control cellular influx and efflux of solutes, including ions, fatty acids, amino acids, sugars, drugs, and vitamins [55, 56]. The Human Genome Gene Nomenclature Committee [57] classifies approximately 400 different human SLCs into 47 families. At least one representative protein from 19 (~40.4%) of these families was identified in the striped bass ovary transcriptome (Table 2). Characterization of SLC gene expression in growing oocytes and during OM would be of direct importance to understanding mechanisms of oogenesis and egg quality in light of what is known of oocyte and egg physiology. Due to osmoregulatory requirements imposed by both fresh and marine waters, embryos of egg-laying fishes develop within the confines of an established chorion that becomes osmotically closed after fertilization. Therefore, ovulated eggs must contain all of the water required during embryogenesis as a medium and substrate for biochemical reactions and as a diluent for waste products (e.g. ammonia). Furthermore, water contributes to appropriate egg buoyancy, especially in marine fishes that spawn pelagic eggs. Prior to ovulation, a hyperosmotic solute concentration develops within the oocytes of these species, followed by passive influx of water through aquaporin membrane channels [review: [58, 59]]. Inorganic ions have primarily been implicated in this phenomenon, however the exact mechanisms of their entry have not been verified. Bobe et al. [14] demonstrated up regulation of slc26 (Pendrin) and aqp4 (aquaporin 4) expression in ovary of rainbow trout during OM. Gene transcripts encoding a slc26a6-like protein, along with several other ion transporters (Table 2) and aquaporin 1 (contig08717) were identified in striped bass ovary. This indicates the potential for discovery of previously unknown mechanisms of teleost oocyte hydration by gene expression analyses of these particular SLCs and water transport genes in the striped bass and related species (genus Morone), which can tolerate a wide range of environmental salinities.
Conclusions
In summary, as we continue to advance our understanding of reproduction in temperate basses of the genus Morone, this reference sequence database of ovarian transcripts will provide the requisite foundation for gene expression studies and will open avenues of research related to reproduction and egg quality. Several important candidate genes have already been identified for future study. Furthermore, these sequences have been used to design an ovary UniClone oligo microarray for assessing changes in gene expression during oogenesis and in female striped bass spawning good and poor quality eggs. Our recent deployment of this microarray in a study of striped bass egg quality has allowed us to detect differences in ovarian gene expression explaining and predicting most of the eventual variance in early embryo mortality among good and poor quality spawners.
Methods
Sample collection and preparation
Striped bass were reared in outdoor tanks at the N.C. State University Pamlico Aquaculture Field Laboratory [60]. As the striped bass is a group synchronous, single clutch, iteroparous spawner, ovarian tissues were collected by dissection or through ovarian biopsy [61] from females whose most advanced clutch of oocytes/eggs represented one of several stages (≥ 3 females/stage) of oocyte growth (early primary growth oocytes, diameter 49-81 μm; late primary growth oocytes showing evidence of lipid droplet accumulation, diameter 162-184 μm; vitellogenic growth oocytes, diameter 558-764 μm [see:[62][63]]), oocyte maturation (post-vitellogenic and maturing oocytes, diameter > 900 μm), and atresia [64], and ovulated eggs. All samples were preserved in RNAlater® (Applied Biosystems/Ambion; Austin, TX). Tissues were pooled in equal weight by oocyte/egg stage and total RNA was extracted in TRIzol® Reagent (Invitrogen; Carlsbad, CA). RNA quality was assessed by agarose gel electrophoresis and NanoDrop™ spectrophotometry (Fisher Scientific; Pittsburgh, PA). Dynabeads® (Invitrogen) were used to purify mRNA as described by the manufacturer.
cDNA library construction and sequencing
Ovary mRNA was submitted for cDNA synthesis at the N.C. State University Genomic Sciences Laboratory (Raleigh, NC). First and second strand cDNA was synthesized from 2.5 μg of Dnase treated mRNA using the SuperScript™ Double-Stranded cDNA Synthesis Kit (Invitrogen) and oligo (dT)17 according to the manufacturer. Approximately 2 μg of cDNA was prepared for FLX sequencing using standard Roche protocols [65]. Briefly, cDNA was nebulized to generate fragments averaging ~500 bp in length, fragment ends were repaired, and adapters containing PCR and sequencing primer annealing sites were ligated. Fragments were immobilized on beads, clonally amplified and then sequenced on a 1/2 plate using standard FLX platform (Roche; Indianapolis, IN).
Sequence assembly and annotation
Short reads were assembled into contigs using Roche's Newbler software (gsAssembler) with default settings except that the minimum overlap was set to 30 bp. Parameters were set to generate files for large contigs (> 500 bp) and for all contigs > 100 bp. High quality contig assemblies were subjected to BLAST (blastx) [66] of the NCBI database and annotated according to the Gene Ontology Consortium [67] using Blast2GO 2048 M version 12.2.0 [10, 68, 69]. Parameters for blastx were: Expect value 1.0E-3 and HSP Length Cutoff 33. Parameters for the GO annotations were: E-value-hit-filter 1.0E-6, Annotation Cutoff 55, GO Weight 5, and HSP-Hit Coverage Cutoff 0. Combined GO graphs for the annotated sequences (4,120 total) were created using percentages of 2nd level GO terms for Biological Process and 3rd level GO terms for Molecular Function and Cellular Component. Represented GO classes were restricted to those with 50 or more entities (sequence cutoff = 50.0); Sequence Filter = 50, Score alpha = 0.6, Node Score Filter = 10. Parameters for the Combined Graphs, Level Pie Configuration were: Ontology Level = Level 2 or 3 as described above.
Estimation of abundant gene transcripts
Contigs that were assembled from a number of ESTs comprising ≥ 0.15% of the total 230,151 short reads (i.e. those having ≥ 345 reads per contig) were considered to be abundant [see: [38]]. These contigs were ranked by relative abundance and compared to zebrafish orthologues shown to be ovary predominant by NCBI UniGene DDD [70], see: [15, 44]. Zebrafish EST libraries were used to determine relative representation by DDD of orthologous UniGene clusters in ovary (104, 986 ESTs; Lib.IDs 20503, 15519, 20772, 20502, 19214, 15930, 9874, 9767) and body tissues excluding gonads (714, 604 ESTs; Lib.IDs 1520, 1521, 15438, 1028, 17704, 17768, 19753, 1522, 19745, 19746, 20694, 20725, 15518, 21372, 19747, 19748, 4913, 9766, 21371, 19741, 19749, 20771, 19739, 19740, 10504, 19737, 13027, 1029, 17276, 15077, 19752, 15517, 2387, 17282, 17284, 19738, 9968, 9993, 14182, 14249, 19217, 24670, 20072, 20071, 19253, 19219, 19218, 19215, 17283, 17275, 14410, 14409, 13866, 12106, 9706, 4264, 1727). Libraries with sequences derived from embryos, larvae, or whole bodies including gonads were excluded. The Fisher's exact test (FET) was used to determine difference between the number of times sequences from the ovary or body libraries were assigned to a specific UniGene cluster (P ≤ 0.05). Numerical DDD scores of genes with significantly different expression profiles were reported as the fraction of sequences within the EST libraries that mapped to the UniGene cluster.
Availability of supporting data
The data sets supporting the results of this article are available in the National Center for Biotechnology Information repository, Short Read Archive: SRX007394 and the National Animal Genome Research Program Aquaculture Genome Projects repository, http://www.animalgenome.org/aquaculture/database/.
References
Garber AF, Sullivan CV: Selective breeding for the hybrid striped bass (Morone chrysops, Rafinesque x M. saxatilis, Walbaum) industry: status and perspectives. Aquacult Res. 2006, 37: 319-338.
Richards RA, Rago PJ: A case history of effective fishery management: Chesapeake Bay striped bass. N Am J Fish Manage. 1999, 19: 356-375.
Couch CR, Garber AF, Rexroad CE, Abrams JM, Stannard JA, Westerman ME, Sullivan CV: Isolation and characterization of 149 novel microsatellite DNA markers for striped bass, Morone saxatilis, and cross-species amplification in white bass, M. chrysops, and their hybrid. Mol Ecol Notes. 2006, 6: 667-669.
Rexroad CE, Vallejo R, Coulibaly I, Westerman ME, Sullivan CV: Identification and characterization of microsatellites for striped bass from repeat-enriched libraries. Conserv Genet. 2006, 7: 971-982.
Liu S, Rexroad CE, Couch CR, Cordes J, Reece K, Sullivan CV: A microsatellite linkage map of striped bass (Morone saxatilis) reveals conserved synteny with the three-spined stickleback (Gasterosteus aculeatus). Mar Biotechnol. doi: 10.1007/s10126-011-9407-2,
Zohar Y, Mylonas CC: Endocrine manipulations of spawning in cultured fish: from hormones to genes. Aquaculture. 2001, 197: 99-136.
Cerdà J, Bobe J, Babin PJ, Admon A, Lubzens E: Functional genomics and proteomic approaches for the study of gamete formation and viability in farmed finfish. Rev Fish Sci. 2008, 16 (Suppl 1): 54-70.
Bobe J, Labbé C: Egg and sperm quality in fish. Gen Comp Endocrinol. 2010, 165: 535-548.
Wang Z, Gerstein M, Snyder M: RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009, 10: 57-63.
Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talón M, Dopazo J, Conesa A: High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36: 3420-3435.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B: Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008, 5: 621-628.
Nesvizhskii AI: Protein identification by tandem mass spectrometry and sequence database searching. Methods in Molecular Biology. Edited by: Matthiesen R. 2007, New York: Springer, 87-119.
von Schalburg KR, Rise ML, Brown GD, Davidson WS, Koop BF: A comprehensive survey of the genes involved in maturation and development of the rainbow trout ovary. Biol Reprod. 2005, 72: 687-699.
Bobe J, Montfort J, Nguyen T, Fostier A: Identification of new participants in the rainbow trout (Oncorhynchus mykiss) oocyte maturation and ovulation process using cDNA microarrays. Reprod Biol Endocrinol. 2006, 4: 39-
Bobe J, Nguyen T, Mahé S, Monget P: In silico identification and molecular characterization of genes predominantly expressed in the fish oocyte. BMC Genomics. 2008, 9: 499-
Gohin M, Bobe J, Chesnel F: Comparative transcriptomic analysis of follicle-enclosed oocyte maturational and developmental competence acquisition in two non-mammalian vertebrates. BMC Genomics. 2010, 11: 18-
Luckenbach JA, Iliev DB, Goetz FW, Swanson P: Identification of differentially expressed genes during primary and early secondary oocyte growth in coho salmon. Oncorhynchus kisutch. Reprod Biol Endocrinol. 2008, 6: 2-
Chu S-L, Weng C-F, Hsiao C-D, Hwang P-P, Chen Y-C, Ho J-M, Lee S-J: Profile analysis of expressed sequence tags derived from ovary of tilapia, Oreochromis mossambicus. Aquacult. 2006, 251: 537-548.
Mommens M, Fernandes JMO, Bizuayehu TT, Bolla SL, Johnston IA, Babiak I: Maternal gene expression in Atlantic halibut (Hippoglossus hippoglossus L.) and its relation to egg quality. BMC Res Notes. 2010, 3: 138-
Cerdà J, Mercadé J, Lozano JJ, Manchado M, Tingaud-Sequeira A, Astola A, Infante C, Halm S, Viñas J, Castellana B, Asensio E, Cañavate P, Martínez-Rodríguez G, Piferrer F, Planas JV, Prat F, Yúfera M, Durany O, Subirada F, Rosell E, Maes T: Genomic resources for a commercial flatfish, the Senegalese sole (Solea senegalensis): EST sequencing, oligo microarray design, and development of the Soleamold bioinformatic platform. BMC Genomics. 2008, 9: 508-
Leong JS, Jantzen SG, von Schalburg KR, Cooper GA, Messmer AM, Liao NY, Munro S, Moore R, Holt RA, Jones SJM, Davidson WS, Koop BF: Salmo salar and Esox lucius full-length cDNA sequences reveal changes in evolutionary pressures on a post-tetraploidization genome. BMC Genomics. 2010, 11: 279-
Goetz FW, McCauley L, Goetz GW, Norberg B: Using global genome approaches to address problems in cod mariculture. ICES J Mar Sci. 2006, 63: 393-399.
Hudson ME: Sequencing breakthroughs for genomic ecology and evolutionary biology. Mol Ecol Resour. 2008, 8: 3-17.
Vera JC, Wheat CW, Fescemyer HW, Frilander MJ, Crawford DL, Hanski I, Marden JH: Rapid transcriptome sequencing for a nonmodel organism using 454 pyrosequencing. Mol Ecol. 2008, 17: 1636-1647.
Toth AL, Varala K, Newman TC, Miguez FE, Hutchison SK, Willoughby DA, Simons JF, Egholm M, Hunt JH, Hudson ME, Robinson GE: Wasp brain gene expression supports an evolutionary link between maternal behavior and eusociality. Science. 2007, 318: 441-444.
Nagaraj SH, Gasser RB, Ranganathan S: A hitchhiker's guide to expressed sequence tag (EST) analysis. Brief Bioinform. 2006, 8: 6-21.
Sreenivasan R, Cai M, Bartfai R, Wang X, Christoffels A, Orban L: Transcriptomic analyses reveal novel genes with sexually dimorphic expression in the zebrafish gonad and brain. PLoS ONE. 2008, 3: e1791-
Li Y, Chia JM, Bartfai R, Christoffels A, Yue GH, Ding K, Ho MY, Hill JA, Stupka E, Orban L: Comparative analysis of the testis and ovary transcriptomes in zebrafish by combining experimental and computational tools. Comp Funct Genomics. 2004, 5: 403-418.
Lo J, Lee SC, Xu M, Liu F, Ruan H, Eun A, He Y, Ma W, Wang W, Wen Z, Peng J: 15000 unique zebrafish EST clusters and their future use in microarray for profiling gene expression patterns during embryogenesis. Genome Res. 2003, 13: 455-466.
Clark MD, Hennig S, Herwig R, Clifton SW, Marra MA, Lehrach H, Johnson SL: WU-GSC EST Group: An oligonucleotide fingerprint normalized and expressed sequence tag characterized zebrafish cDNA library. Genome Res. 2001, 11: 1594-1602.
Habermann B, Oegema J, Sunyaev S, Shevchenko A: The power and the limitations of cross-species protein identification by mass spectrometry-driven sequence similarity searches. Mol Cell Proteomics. 2004, 3: 238-249.
Reading BJ, Sullivan CV, Chapman RW: Striped Bass Transcriptome Sequence Contigs Database. [http://www.animalgenome.org/aquaculture/database/]
Rise ML, von Schalburg KR, Brown GD, Mawer MA, Devlin RH, Kuipers N, Busby M, Beetz-Sargent M, Alberto R, Gibbs AR, Hunt P, Shukin R, Zeznik JA, Nelson C, Jones SR, Smailus DE, Jones SJ, Schein JE, Marra MA, Butterfield YS, Stott JM, Ng SH, Davidson WS, Koop BF: Development and application of a salmonid EST database and cDNA microarray: data mining and interspecific hybridization characteristics. Genome Res. 2004, 14: 478-490.
Hastie ND, Bishop JO: The expression of three abundance classes of messenger RNA in mouse tissues. Cell. 1976, 9: 761-774.
Su AI, Wiltshire T, Batalov B, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB: A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA. 2004, 101: 6062-6067.
Ziv T, Gattegno T, Chapovetsky V, Wolf H, Barnea E, Lubzens E, Admon A: Comparative proteomics of the developing fish (zebrafish and gilthead seabream) oocytes. Comp Biochem Physiol Part D Genomics Proteomics. 2008, 3: 12-35.
Knoll-Gellida A, Babin PJ: Zebrafish ovarian follicle transcriptome. The Fish Oocyte: From Basic Studies to Biotechnological Applications. Edited by: Babin PJ, Cerdà J, Lubzens E. 2007, New York: Springer, 77-97.
Knoll-Gellida A, André M, Gattegno T, Forgue J, Admon A, Babin PJ: Molecular phenotype of zebrafish ovarian follicle by serial analysis of gene expression and proteomic profiling, and comparison with the transcriptomes of other animals. BMC Genomics. 2006, 7: 46-
Hennebold JD: Characterization of the ovarian transcriptome through the use of differential analysis of gene expression methodologies. Hum Reprod Update. 2004, 10: 227-239.
Tanaka M, Hennebold JD, Macfarlane J, Adashi EY: A mammalian oocyte-specific linker histone gene H1oo: homology with the genes for the oocyte-specific cleavage stage histone (CS-H1) of sea urchin and the B4/H1M histone of the frog. Development. 2001, 128: 655-664.
Koppel J, Rehák P, Baran V, Veselá J, Hlinka D, Manceau V, Sobel A: Cellular and subcellular localization of stathmin during oocyte and preimplantation embryo development. Mol Reprod Dev. 1999, 53: 306-317.
Calvert ME, Digilio LC, Herr JC, Coonrod SA: Oolemmal proteomics-identification of highly abundant heat shock proteins and molecular chaperones in the mature mouse egg and their localization on the plasma membrane. Reprod Biol Endocrinol. 2003, 1: 27-
Murray D, Doran P, MacMathuna P, Moss AC: In silico gene expression analysis-an overview. Mol Cancer. 2007, 6: 50-
Dai L, Ma W, Li J, Xu Y, Li W, Zhao Y, Deng F: Cloning and characterization of a novel oocyte-specific gene zorg in zebrafish. Theriogenology. 2009, 71: 441-449.
Stitzel ML, Seydoux G: Regulation of the oocyte-to-zygote transition. Science. 2007, 316: 407-408.
Hirai T, Yamashita M, Yoshikuni M, Lou YH, Nagahama Y: Cyclin B in fish oocytes: its cDNA and amino acid sequences, appearance during maturation, and induction of p34 cdc2 activation. Mol Reprod Dev. 1992, 33: 131-140.
Nagahama Y: 17α,20β-dihydroxy-4-pregnen-3-one, a maturation inducing hormone in fish oocytes: mechanisms of synthesis and action. Steroids. 1997, 62: 190-196.
Taieb F, Thibier C, Jessus C: On cyclins, oocytes, and eggs. Mol Reprod Dev. 1997, 48: 397-411.
Qiu G, Ramachandra RK, Rexroad CE, Yao J: Molecular characterization and expression profiles of cyclin B1, B2 and Cdc2 kinase during oogenesis and spermatogenesis in rainbow trout (Oncorhynchus mykiss). Anim Reprod Sci. 2008, 105: 209-225.
Edwards MC, Wong C, Elledge SJ: Human cyclin K, a novel polymerase II-associated cyclin possessing both carboxy-terminal domain kinase and cdk-activating kinase activity. Mol Cell Biol. 1998, 18: 4291-4300.
Solomon MJ, Burton JL: Securin' M-phase entry. Nat Cell Biol. 2008, 10: 381-383.
Gen K, Yamaguchi S, Okuzawa K, Kagawa H, Alam MS: Novel expression of importin α homologue in marine teleost, Pagrus major. Comp Biochem Physiol B Biochem Mol Biol. 2008, 151: 420-427.
McGarry TJ, Kirschner MW: Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell. 1998, 93: 1043-1053.
Wu N, Yue H, Chen B, Gui JF: Histone H2A has a novel variant in fish oocytes. Biol Reprod. 2009, 81: 275-283.
Schlessinger A, Matsson P, Shima JE, Pieper U, Yee SW, Kelly L, Apeltsin L, Stroud RM, Ferrin TE, Giacomini KM, Sali A: Comparison of human solute carriers. Protein Sci. 2010, 19: 412-428.
Hediger M, Romero MF, Peng J, Rolfs A, Takanaga H, Bruford EA: The ABCs of solute carriers: physiological, pathological and therapeutic implications of human membrane transport proteins. Pflugers Arch. 2004, 447: 465-468.
Povey S, Lovering R, Bruford E, Wright M, Lush M, Wain H: The HUGO Gene Nomenclature Committee (HGNC). Hum Genet. 2001, 109: 678-680.
Fabra M, Raldúa D, Power DM, Deen PM, Cerdà J: Marine fish egg hydration is aquaporin-mediated. Science. 2005, 307: 545-
Reading BJ, Sullivan CV: Vitellogenesis in Fishes. Encyclopedia of Fish Physiology: From Genome to Environment. Edited by: Farrell AP, Stevens ED, Cech JJ Jr, Richards JG. 2011, Maryland Heights: Academic Press, Inc., 257-
Hodson RG, Sullivan CV: Induced maturation and spawning of domestic and wild striped bass Morone saxatilis (Walbaum), broodstock with implanted GnRH analogue and injected hCG. Aquacult Res. 1993, 24: 389-398.
Rees RA, Harrell RM: Artificial spawning and fry production of striped bass and hybrids. Culture and Propagation of Striped Bass and its Hybrids. Edited by: Harrell RM, Kerby JH, Minton RV. 1990, Bethesda: American Fisheries Society, 43-72.
Berlinsky DL, Specker JL: Changes in gonadal hormones during oocyte development in the striped bass, Morone saxatilis. Fish Physiol Biochem. 1991, 9: 51-62.
Tao Y, Hara A, Hodson RG, Woods LC, Sullivan CV: Purification, characterization and immunoassay of striped bass (Morone saxatilis) vitellogenin. Fish Physiol Biochem. 1993, 12: 31-46.
Mylonas CC, Woods LC, Zohar Y: Cyto-histological examination of post-vitellogenesis and final oocyte maturation in captive-reared striped bass. J Fish Biol. 1997, 50: 34-49.
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM: Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005, 437: 376-80.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215: 403-410.
Ashburner M, Ball K, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G: Gene Ontology: tool for the unification of biology. Nat Genet. 2000, 25: 25-29.
Conesa A, Götz S: Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics. 2008, 2008: 619832-
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M: Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005, 21: 3674-3676.
Tokuzawa Y, Maruyama M, Yamanaka S: Utilization of digital differential display to identify novel targets of Oct3/4. Methods Mol Biol. 2006, 329: 223-231.
Acknowledgements
The authors are indebted to Zhanjiang (John) Liu (Auburn University) and Zhiliang Hu (Iowa State University) for organizing the online transcriptome posted to the National Aquaculture Genome Project (NAGP) webpage. We thank Andy S. McGinty and Michael S. Hopper (N.C. State University Pamlico Aquaculture Field Laboratory) for care and maintenance of the striped bass and Marion Beal (The Hollings Marine Laboratory) for posting the short read archive to NCBI. This is an NAGRP Aquaculture Genome (NRSP-8) Project and one of the authors (CVS) is the striped bass NRSP-8 species coordinator. This work was supported by research grants R/AF-46 and MG-XX from the North Carolina Sea Grant Program to C.V.S. and by a special grant NC09211 from the U.S. Department of Agriculture (USDA) National Institute of Food and Agriculture (NIFA) to C.V.S. and three other Co-principal investigators.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
BJR conducted the sample preparation, DDD statistical analyses, and drafted the manuscript. JES performed the FLX pyrosequencing and contig sequence assemblies. RWC performed the GO annotations. RWC, EHS, and CHO participated in design of the study and critical review of the manuscript. CVS conceived the study, participated in its design and coordination, and helped draft the manuscript. All authors read and approved the final manuscript.
Benjamin J Reading, Robert W Chapman contributed equally to this work.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Reading, B.J., Chapman, R.W., Schaff, J.E. et al. An ovary transcriptome for all maturational stages of the striped bass (Morone saxatilis), a highly advanced perciform fish. BMC Res Notes 5, 111 (2012). https://doi.org/10.1186/1756-0500-5-111
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-0500-5-111