
Abstract
Background
Identification of the Mycobacterium tuberculosis complex organisms to the species level is important for diagnostic, therapeutic and epidemiologic perspectives. Indeed, isolates are routinely identified as belonging to the M. tuberculosis complex without further discrimination in agreement with the high genomic similarity of the M. tuberculosis complex members and the resulting complex available identification tools.
Findings
We herein develop a pyrosequencing assay analyzing polymorphisms within glpK, pykA and gyrB genes to identify members of the M. tuberculosis complex at the species level. The assay was evaluated with 22 M. tuberculosis, 21 M. bovis, 3 M. caprae, 3 M. microti, 2 M. bovis BCG, 2 M. pinnipedii, 1 M. canettii and 1 M. africanum type I isolates. The resulted pyrograms were consistent with conventional DNA sequencing data and successfully identified all isolates. Additionally, 127 clinical M. tuberculosis complex isolates were analyzed and were unambiguously identified as M. tuberculosis.
Conclusion
We proposed a pyrosequencing-based scheme for the rapid identification of M. tuberculosis complex isolates at the species level. The assay is robust, specific, rapid and can be easily introduced in the routine activity.
Similar content being viewed by others


Background
The Mycobacterium tuberculosis complex (MTC) includes M. tuberculosis, M. bovis and BCG-derived clones, M. africanum, M. canettii, M. microti, M. caprae and M. pinnipedii[1]. Recently, an eight member named Mycobacterium mungi was identified in banded mongooses (Mungos mungo) in Botswana [2]. While there is variable host-specificity among the different MTC members, every species, but M. mungi, has been implicated in human tuberculosis with M. tuberculosis being the most common pathogen [3–6]. The exact contribution of each species in human disease may be underestimated due to the limited capacity of laboratories to identify MTC isolates at the species level in routine practice. Indeed, phenotypic methods are time-consuming and the results are difficult to interpret. Molecular assays are hampered by the high genetic similarity reflected by the complete conservation of the 16S rRNA and rpoB genes of the MTC members [7, 8]. This situation indeed, led to propose that the various MTC species would be more accurately described as ecotypes [9]. Despite the high degree of nucleotide sequence homology, some genomic markers such as pncA, mpt40, hupB, gyrB and wbbl1 genes in addition to the regions of difference (RD) have been successfully used for the differentiation of MTC members through direct analysis such as in spoligotyping or after enzymatic digestion of PCR products [10–16]. Nevertheless, most of these techniques allow only partial discrimination between the MTC species and are too fastidious to be implemented in the routine practice of microbiology laboratories. The Genotype MTBC (Hain Lifescience, GmbH, Nehren, Germany) is a commercial DNA-strip based assay identifying MTC species through the detection of gyrB gene single nucleotide polymorphisms (SNPs) and RD1 [17]. The test is easy to use, but failed to identify M. canettii and M. pinnipeddii[17].
Pyrosequencing is a real-time non-electrophoretic DNA sequencing technique that can be easily adapted for routine use in the clinical microbiology laboratory. This technique was successfully used for the identification of non-tuberculous mycobacteria, identification of the Beijing family of M. tuberculosis and detection of rifampin-resistance among MTC members [18–20]. We here developed a pyrosequencing assay for the rapid identification of the MTC species based on polymorphisms within the glpK, pykA and gyrB genes.
Methods
MTC isolates
A set of 8 reference MTC isolates and 47 M. africanum type 2 and human and animal clinical isolates representative of all MTC species (excluding M. mungi and M. africanum type II) after molecular identification as previously described [21] was used in this study [15, 16, 20]. Maria Laura Boschiroli (ANSES, Maisons-Alfort, France) graciously provided DNA extracts of MTC organisms isolated from different animal species (cattle, badger, cat, dog, sea lion, wild boar, wild deer, marmoset and chimpanzee). In addition, 127 clinical MTC organisms identified using ITS-real time PCR were analyzed (Additional file 1). These strains were isolated at the microbiology laboratory of Farhat Hached Hospital, Sousse, Tunisia and the mycobacteria reference laboratory of the Institut Hospitalier Universitaire POLMIT, Marseille, France. This study involves mycobacteria isolated from animals and from patients as a part of the routine diagnostic activity of laboratories and does not involve animals or patients themselves. No informed consent was obtained from individuals in agreement with French law, as the study concerns only microbiota and not the individuals themselves.
glpK and pykA gene analyses
The sequence of the glpK and pykA genes of M. tuberculosis H37Rv (Genes ID: 885280, 885501), H37Ra (Genes ID: 5214476, 5215168), F11(Genes ID: 5224418, 5222312), KZN1435 (Genes ID: 8164550, 8160651) and CDC1551(Genes ID: 922690, 924212), M. bovis AF2122 (Genes ID: 1093713, 1092578), M. bovis BCG Tokyo (Genes ID: 7563752, 7560567) and M. bovis BCG Pasteur (Genes ID: 4698362, 4695734) were downloaded from GenBank and aligned using the Clustal software (http://www.clustal.org). In addition, specific primers were designed using the NCBI Primer BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) for amplification and conventional sequencing of both genes in M. tuberculosis (N = 10), M. bovis (N = 10), M. caprae (N = 3), M. microti (N = 3), M. bovis BCG (N = 2), M. pinnipedii (N = 2), M. canettii (N = 1), and M. africanum type I (N = 1) isolates (Table 1). PCR reaction was performed in a final volume of 50 μl comprising 22.75 μl of water, 10 μl of 5x Q-buffer (Qiagen, Courtaboeuf, France), 5 μl of 10x buffer, 200 mM of each dDNTP, 1 μl of forward and reverse primers (10 pmol/μl), 2.5 U of hotstar Taq polymearse (Qiagen) and 5 μl of template DNA. Negative controls consisting of reaction mix without DNA were added in each PCR run. The PCR conditions consisted of enzyme activation at 95°C for 15 min, 40 cycles of 30 s at 94°C, 2.5 min at 60°C, 1 min at 72°C and a final elongation at 72°C for 5 min. Sequencing was performed using the BigDye Terminator v1.1 Cycle Sequencing kit (Applied Biosystems, Courtaboeuf, France) and the 3100 genetic analyser (Applied Biosystems) [21]. Sequences were assembled and analysed with the Seqscape software (Applied Biosystems).
Pyrosequencing analysis
After analysis of the glpK and pykA gene sequences, MTC species-specific polymorphic regions were targeted for pyrosequencing [15]. To allow discrimination between all MTC members, single nucleotide polymorphisms (SNPs) within the gyrB gene were also used [13]. Specific primers were designed using the PSQ assay (Qiagen) (Table1). In a first step, simplex PCR reactions were performed as described above with 1-min of elongation instead of 2.5 min. PCR products were the n subjected to simplex pyrosequencing analysis using the PyroMark Q96 ID System (Qiagen) as previously described [18]. SNP analyses were performed using the SNP program of the PyroMark Q96 ID software. Sequencing analysis was realized using the sequencing program with a 6(GATC) dispension order. Secondly, we performed multiplex PCR and pyrosequencing reactions with the same conditions as for simplex analysis. The choice of the gene fragments to be simultaneously analyzed was dictated by the PyroMark Q96 ID software so that polymorphic regions did not overlap. For multiplex SNP analysis, two sequencing primers were hybridized either to a single or two PCR amplified loci.
Specificity testing
A set of clinical isolates of non tuberculous mycobacteria (Mycobacterium smegmatis, Mycobacterium immunogenum, Mycobacterium bolletii, Mycobacterium chelonae, Mycobacterium abscessus, Mycobacterium massillense, Mycobacterium avium and Mycobacterium intracellulare) and other bacteria (Escherichia coli, Klebsiella pneumoniae, Enterococcus feacalis, Salmonella enterica, Proteus mirabilis, Proteus vulgaris, Pseudomonas aeruginosa, Citrobacter freundii and Stenotrophomonas maltophilia) were tested on the pyrosequencing analysis to assess the specificity of the assay. Isolates were identified by sequencing the 16S rRNA or the rpoB gene and/or using matrix-assisted laser desorption ionization time-of-flight mass spectrometry (Bruker Daltonics, Wissembourg, France) [22].
Results and discussion
The pykA (encoding for the pyruvate kinase) and glpK (encoding for the glycerol kinase) genes were previously analyzed to disclose the genetic basis of difference in the glycerol metabolism between M. tuberculosis and M. bovis in comparison with other MTC species [23, 24]. The authors reported a specific gene structure for M. tuberculosis, M. bovis and M. bovis BCG but this particularity has not been further used for identifying MTC organisms. In our study, we extended this previous observation to all MTC species and a larger collection of clinical isolates in the perspective of MTC member's identification.
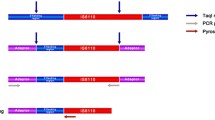
In PCR-based experiments all negative controls remained negative and no PCR products were obtained when testing non-tuberculous mycobacteria and other bacteria confirming the specificity of the primer sets used. After alignment of the glpK and pykA gene sequences herein achieved and those downloaded from GenBank, we observed MTC species-specific signatures within the glpK gene. Four silent SNPs were found at nucleotide (nt) position 279 (A→G), 336 (T→C), 435 (T→A) and 1,497 (T→C) specifically within M. canettii. Insertion of one and three guanine at nt 573 was identified in M. microti and M. bovis BCG, respectively. M. bovis reference isolates also shared the same frameshift as M. microti at nucleotide 573 in addition to a G→C SNP at nt 779, but these polymorphisms were not found in the human and animal clinical M. bovis isolates (N = 10), in agreement with a previous study [23]. Thus, these SNPs further were not considered as M. bovis specific feature for subsequent analysis. M. pinnipedii specifically exhibits a G→A SNP at nt 845 and M. tuberculosis a C→T SNP at nt 1,397. Excluding M. bovis reference strains, no specific feature in the glpK gene was observed within M. bovis and M. caprae isolates. As for the pykA gene, a unique G→T SNP at nt 660 was commonly detected in M. bovis, M. caprae, M. microti and M. pinnipedii. This SNP substitutes glutamic acid with aspartic acid at codon 220, resulting in an inactive pyruvate kinase and the inability of M. bovis to use glycerol as a carbone source [23, 24]. By analyzing a larger collection of isolates, our results confirmed the presence of this SNP within M. bovis and provide the novel evidence for its occurrence also in M. caprae and M. pinnipedii which were not included in the Keating's study [23]. Accordingly, M. bovis, M. caprae and M. pinnipedii are all characterized by the inability to use glycerol and require pyruvate for growth [24]. We were not able to sequence glpK and pykA genes in M. africanum, nevertheless positive results were obtained by pyrosequencing analysis (see below). Polymorphic regions within the glpK and pykA genes were targeted for MTC species identification using a pyrosequencing approach. Since no specific signature was obtained for M. bovis and M. caprae, we combined previously described SNP of the gyrB gene to allow complete MTC discrimination [15]. In a first step, the assay was evaluated with the well characterized MTC species (N = 55). The pyrograms obtained from simplex and multiplex data were consistent with the results obtained by conventional DNA sequencing and correctly identifies MTC isolates including 22 M tuberculosis, 21 M. bovis, 2 M. bovis BCG, 3 M. caprae, 3 M. microti, 2 M. pinnipeddii, 1 M. canettii and 1 M. africanum type I. Accordingly, we propose a generalized diagram for the rapid identification of the MTC species (Figure 1). In a first step, multiplex amplification and pyrosequencing analysis of the glpK 1379 and pyk A660 nt positions in one reaction tube provide four different pyrograms: T/T and G/G specific for M. tuberculosis, T/T and T/T specific for M. africanum type I, C/C and G/G specific for M. bovis BCG and M. canettii (group 1) and C/C and T/T specific for M. bovis, M. caprae, M. microti and M. pinnipedii (group 2). If pyrogram of group 1 is obtained, sequencing of the glpK 573 may discriminate between M. bovis BCG and M. canettii. In the case where pyrogram of group 2 is obtained, analysis of the gyrB 6406 SNP may specifically identifies M. bovis (genotype C/C) from the remaining group 2 species (genotype T/T). M. pinnipedii can be identified by SNP analysis of the glpK 845 (genotype A/A), M. caprae by SNP analysis of the gyrB6307 and M. microti by sequencing the glpK 573. These steps can be done in parallel in order to minimize the delay for final identification of any MTC species. A total of 127 MTC clinical isolates were tested following this scheme and were unambiguously identified as M. tuberculosis.

A proposed scheme for Mycobacterium tuberculosis species identification by pyrosequencing. 1 Genes analysed by multiplex SNP program. 2Gene analysed by sequencing program. 3Genes analysed by simplex SNP program.
Our proposed scheme allows in one step the specific identification of M. tuberculosis, the leading cause of human tuberculosis worldwide and M. africanum type I, an important cause of human tuberculosis in west Africa [6, 25]. M. bovis, an important zoonotic tuberculosis agent and M. bovis BCG a safe vaccine exceptionally responsible for disseminated disease in immuno-compromised vaccinated neonates and bladder cancer-patients, could be unambiguously identified in a second reaction after excluding M. tuberculosis and M. africanum type I [26, 27]. The remaining MTC species are less common in human disease and their identification using specific genomic marker described here could be directed by epidemiologic data.
The ability of rapid and accurate identification of MTC members has an important impact both for public health and veterinary facilities. Although a few patients infected with M. bovis have been treated by antibiotic regimens incorporating the pyrazinamide, the natural resistance of these species to pyrazinamide makes pyrazinamide generally not recommended for treating such patients [10]. Moreover, it can help epidemiologists and health care professionals to measure the relative contribution of each MTC species in human and animal disease, to identify specific MTC-specie outbreak and to rapidly intercept the impact of the zoonotic transmission particularly in case of M. bovis infections so that appropriate control measure could be undertaken.
Conclusion
Pyrosequencing analysis of the glpK, pykA and gyrB genes provides a robust and easy tool for the rapid identification of all MTC species that can be easily introduced in the routine laboratory activity, just requiring basics skills in PCR. The assay allows the simultaneous analyses of up to 96 isolates within 10 to 20 min after a 4-hour DNA extraction and PCR amplification rounds comprising of 3 different PCRs, and 7 different pyrosequencing steps which can be run in parallel. Moreover, pyrosequencing has several advantages compared to other molecular methods: it determines the exact sequence thereby providing the same accuracy as conventional sequencing methods, the technique dispenses with the need for labeled nucleotides, labeled primers and electrophoresis.
References
Brosh R, Gordon SV, Marmiesse M, Brodin P, Buchrieser C, Eiglmeier K, Garnier T, Gutierrez C, Hewinson G, Kremer K, Parsons LM, Pym AS, Samper S, van Soolingen D, Col ST: A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci. 2002, 99: 3684-3689.
Alexander KA, Laver PN, Michel AL, Williams M, van Helden PD, Warren RM, Gey van Pittius NC: Novel Mycobacterium tuberculosis complex pathogen, M. mungi. Emerg Infect Dis. 2010, 16: 1296-1299.
Kiers A, Klarenbeek A, Mendelts B, Van Soolingen D, Koëter G: Transmission of Mycobacterium pinnipedii to humans in a zoo with marine mammals. Int J Tuberc Lung Dis. 2008, 12: 1469-1473.
Rodríguez E, Sánchez LP, Pérez S, Herrera L, Jiménez MS, Samper S, Iglesias MJ: Human tuberculosis due to Mycobacterium bovis and M. caprae in Spain, 2004-2007. Int J Tuberc Lung Dis. 2009, 13: 1536-1541.
van Soolingen D, van der Zanden AG, de Haas PE, Noordhoek GT, Kiers A, Foudraine NA, Portaels F, Kolk AH, Kremer K, van Embden JD: Diagnosis of Mycobacterium microti infections among humans by using novel genetic markers. J Clin Microbiol. 1998, 36: 1840-1845.
World Health Organisation: Global tuberculosis control: WHO report 2010. 2010
Frothingham R, Hills HG, Wilson KH: Extensive DNA sequence conservation throughout the Mycobacterium tuberculosis complex. J Clin Microbiol. 1994, 32: 1639-1643.
Adékambi T, Shinnick TM, Raoult D, Drancourt M: Complete rpoB gene sequencing as a suitable supplement to DNA-DNA hybridization for bacterial species and genus delineation. Int J Syst Evol Microbiol. 2008, 58: 1807-1814.
Djelouadji Z, Raoult D, Drancourt M: Palaeogenomics of Mycobacterium tuberculosis: epidemic bursts with a degrading genome. Lancet Infect Dis. 2011, 11: 641-650.
Scorpio A, Collins D, Whipple D, Cave D, Bates J, Zhang Y: Rapid differentiation of bovine and human tubercle bacilli based on a characteristic mutation in the bovine pyrazinamidase gene. J Clin Microbiol. 1997, 35: 106-110.
Liebana E, Aranaz A, Francis B, Cousins D: Assessment of genetic markers for species differentiation within the Mycobacterium tuberculosis complex. J Clin Microbiol. 1996, 34: 933-938.
Prabhakar S, Mishra A, Singhal A, Katoch VM, Thakral SS, Tyagi JS, Prasad HK: Use of the hupB gene encoding a histone-like protein of Mycobacterium tuberculosis as a target for detection and differentiation of M. tuberculosis and M. bovis. J Clin Microbiol. 2004, 42: 2724-2732.
Niemann S, Harmsen D, Rusch-Gerdes S, Richter E: Differentiation of clinical Mycobacterium tuberculosis complex isolates by gyrB DNA sequence polymorphism analysis. J Clin Microbiol. 2000, 38: 3231-3234.
Reddington K, O'Grady J, Dorai-Raj S, Maher M, van Soolingen D, Barry T: Novel multiplex real-time PCR diagnostic assay for identification and differentiation of Mycobacterium tuberculosis, Mycobacterium canettii, and Mycobacterium tuberculosis complex strains. J Clin Microbiol. 2011, 49: 651-657.
Huard RC, Lazzarini LC, Butler WR, van Soolingen D, Ho JL: PCR-based method to differentiate the subspecies of the Mycobacterium tuberculosis complex on the basis of genomic deletions. J Clin Microbiol. 2003, 41: 1637-1650.
Parsons LM, Brosch R, Cole ST, Somoskövi A, Loder A, Bretzel G, Van Soolingen D, Hale YM, Salfinger M: Rapid and simple approach for identification of Mycobacterium tuberculosis complex isolates by PCR-based genomic deletion analysis. J Clin Microbiol. 2002, 40: 2339-45.
Richter E, Weizenegger M, Fahr AM, Rüsch-Gerdes S: Usefulness of the GenoType MTBC assay for differentiating species of the Mycobacterium tuberculosis complex in cultures obtained from clinical specimens. J Clin Microbiol. 2004, 42: 4303-4306.
Heller LC, Jones M, Widen RH: Comparison of DNA pyrosequencing with alternative methods for identification of mycobacteria. J Clin Microbiol. 2008, 46: 2092-2094.
Djelouadji Z, Henry M, Bachtarzi A, Foselle N, Raoult D, Drancourt M: Pyrosequencing identification of Mycobacterium tuberculosis W-Beijing. BMC Res Notes. 2009, 2: 239-
Halse TA, Edwards J, Cunningham PL, Wolfgang WJ, Dumas NB, Escuyer VE, Musser KA: Combined real-time PCR and rpoB gene pyrosequencing for rapid identification of Mycobacterium tuberculosis and determination of rifampin resistance directly in clinical specimens. J Clin Microbiol. 2010, 48: 1182-1188.
Djelouadji Z, Arnold C, Gharbia S, Raoult D, Drancourt M: Multispacer sequence typing for Mycobacterium tuberculosis genotyping. PLoS ONE. 2008, 3: e2433-
Seng P, Drancourt M, Gouriet F, La Scola B, Fournier PE, Rolain JM, Raoult D: Ongoing revolution in bacteriology: routine identification of bacteria by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Clin Infect Dis. 2009, 49: 543-551.
Keating LA, Wheeler PR, Mansoor H, Inwald JK, Dale J, Hewinson RG, Gordon SV: The pyruvate requirement of some members of the Mycobacterium tuberculosis complex is due to an inactive pyruvate kinase: implication for in vivo growth. Mol Microbiol. 2005, 56: 163-174.
Chavadi S, Wooff E, Coldham NG, Sritharan M, Hewinson RG, Gordon SV, Wheeler PR: Global effects of inactivation of the pyruvate kinase gene in the Mycobacterium tuberculosis complex. J Bacteriol. 2009, 191: 7545-7553.
de Jong BC, Antonio M, Gagneux S: Mycobacterium africanum-review of an important cause of human tuberculosis in West Africa. PLoS Negl Trop Dis. 2010, 4: e744-
Dankner WM, Davis CE: Mycobacterium bovis as a significant cause of tuberculosis in children residing along the United States-Mexico border in the Baja California region. Pediatrics. 2000, 105: e79-
Nadasy KA, Patel RS, Emmett M, Murillo RA, Tribble MA, Black RD, Sutker WL: Four cases of disseminated Mycobacterium bovis infection following intravesical BCG instillation for treatment of bladder carcinoma. South Med J. 2008, 101: 91-95.
Acknowledgements
The authors would like to thank Maria Laura BOSCHIROLI for generously providing DNA from different animal Mycobacterium tuberculosis complex isolates. IBK is financially supported by the Oeuvre Antituberculeuse des Bouches du Rhône, Marseille, France.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
IBK and MH designed and performed analyses; IBK, JB and MD interpreted data and wrote the draft. All authors read and approved the final manuscript.
Electronic supplementary material
13104_2011_1186_MOESM1_ESM.XLS
Additional file 1:List of M. tuberculosis complex isolates used in this study. The table lists from left to right the reference number, the identification at the species level, the source, the geographical origin, and the genotype according to Multispacer Sequence Typing of the isolates used in this work. (XLS 50 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kahla, I.B., Henry, M., Boukadida, J. et al. Pyrosequencing assay for rapid identification of Mycobacterium tuberculosis complex species. BMC Res Notes 4, 423 (2011). https://doi.org/10.1186/1756-0500-4-423
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-0500-4-423