
Abstract
Background
The majority of Marfan syndrome (MFS) cases is caused by mutations in the fibrillin-1 gene (FBN1), mapped to chromosome 15q21.1. Only few reports on deletions including the whole FBN1 gene, detected by molecular cytogenetic techniques, were found in literature.
Results
We report here on a female patient with clinical symptoms of the MFS spectrum plus craniostenosis, hypothyroidism and intellectual deficiency who presents a 1.9 Mb deletion, including the FBN1 gene and a complex rearrangement with eight breakpoints involving chromosomes 6, 12 and 15.
Discussion
This is the first report of MFS with a complex chromosome rearrangement involving a deletion of FBN1 and contiguous genes. In addition to the typical clinical findings of the Marfan syndrome due to FBN1 gene haploinsufficiency, the patient presents features which may be due to the other gene deletions and possibly to the complex chromosome rearrangement.
Similar content being viewed by others


Background
Marfan syndrome (MFS) is a dominant disorder, mainly caused by mutations in the fibrillin-1 gene (FBN1) located on chromosome 15q21.1. The estimated prevalence of MFS is about 1 in 10000. Approximately 25% of MFS patients are sporadic cases due to new mutations [1, 2]. Different tissues and organs can be affected, especially the cardiovascular, skeletal, and ocular systems. Diagnostic criteria are well established and known as the Ghent criteria [3]. However, the inter- and intra-familial variability of the phenotype limits the establishment of genotype-phenotype correlations. To date, more than 1329 FBN1 mutations have been published (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=FBN1), but only a few are recurring mutations. Missense mutations substituting or creating a cysteine molecule in one of the calcium-binding EGF domains are the most prevalent. The others are frameshift, splice-site, nonsense mutations and in-frame deletions and insertions. Heterozygous mutations in the genes coding for transforming growth factor beta receptors I (TGFBR1) and II (TGFBR2) have also been reported in patients with MFS and MFS-related disorders, indicating genetic heterogeneity [4–6]. Interstitial deletions involving the 15q21.1 band and the FBN1 gene are very rare. To our best knowledge, there are only six reports in the literature describing deletions of the whole FBN1 gene detected by molecular techniques, and only in five of them this gene deletion was associated with classical MFS [2, 7–11]. The patients described by Adès et al. [8] and Hutchinson et al. [7] had clinical features of the MFS spectrum and mental retardation, but the size of their deletions was not determined. The patient described by Faivre et al. [10] had a 2.97 Mb deletion and some features of MFS but no mental retardation. Hilhorst-Hofstee et al. [2] described 10 cases (five of the same family) with a complete FBN1 gene deletion, screened by Multiplex Ligation-dependent Probe Amplification (MLPA) analysis of 300 patients presenting from mild MFS features to the classical MFS or an MFS-related phenotype. Recently, Furtado et al. [11] studied 14 patients from 11 unrelated families with aortic aneurysm. Three patients of the one family who met clinical diagnostic criteria for Marfan syndrome had a 542 Kb deletion in chromosome 15 including the whole FBN1 gene; the study of the region was refined by MLPA and array analysis. Cases with chromosomal alterations including a genomic deletion of the whole FBN1 gene are rare, but their number has increased after the adoption of routine screening by molecular techniques (MLPA, array), especially in patients with MFS. To date, there is no description in the literature of a patient with MFS and a complex chromosome rearrangement (CCR). We report here on a 16-year-old female patient displaying features of the MFS spectrum and mental retardation, who was found to present a 1.9 Mb deletion at 15q21.1 (refined by array) including the FBN1 gene, and a novel CCR among chromosomes 6, 12 and 15.
Case presentation
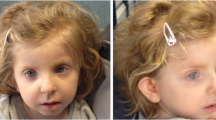
The proband (Figure 1) is the first child of healthy parents. She was born at term, with a length of 50 cm and a weight of 3240 g. She walked at 18 months, and spoke at four years of age. She was however able to attend a regular school, but showed hyperactivity and difficulty to focus. She had a seizure at the age of 3 years, and a cardiac examination evidenced mitral insufficiency with a dystrophic valve. An evolutive scoliosis was noted at the age of 6 years, with progression. At age 11, hypothyroidism was detected. She was referred at 13 years of age for a suspicion of MFS, showing: positive thumb and wrist sign, scoliosis, joint hyperlaxity, high-arched palate with dental crowding, dysmorphism and aortic root dilatation with dystrophic mitral valve. Ophthalmological examination revealed myopia and astigmatism but not ectopia lentis. A neuropsychological assessment showed global intellectual impairment (IQ 50) according to the Brazilian values of the Wechsler Intelligence Scale for Children-III (WISC-III), with major deficits in attention and executive skills. Thus, she met the Ghent criteria for Marfan syndrome, but also presented craniostenosis, hypothyroidism and intellectual deficiency.

Patient at age 13 years.
Genetic Study
Classical Cytogenetic Findings
Conventional chromosome analysis was performed on phytohemagglutinin-stimulated lymphocytes from peripheral blood cultures, using GTG banding according to standard protocols. Cell images were captured using the Ikaros Digital Imaging System (Metasystem, Altlussheim, Germany). G-banded analysis with a resolution of 550 bands per haploid karyotype revealed a female karyotype with a de novo balanced translocation involving chromosomes 6, 12 and 15, with breakpoints apparently at 6q22, 12q24 and 15q21 (Figure 2A).

Cytogenetic and molecular data from the patient studied. A) GTG-banded chromosomes showing the translocation involving chromosomes 6, 12 and 15. B) Array result for chromosome 15 showing the 1.9 Mb deletion (red) at 15q21.1 including the FBN1 gene (arrow). C) FISH with WCP probes of chromosomes 6, 12 and 15 in different color combinations showing a complex chromosomal rearrangement. D) FISH with probes RP11-631P6 (6q13) in red and RP11-46N22 (6q14.3) in green, showing signals next to each other on the normal chromosome 6 and separate signals on the der (6) chromosome. E) FISH with probes RPRP11-627A9 (15q23) in green and RP11-793M16 (12q24.13) in red, showing signals of both on the derivative chromosome 12. F) Ideogram of the derivative chromosomes involved in the patient's complex chromosome rearrangement, showing the probes used to define the breakpoints and the 15q21.1 band deletion (arrow).
Molecular Findings
Genomic DNA was isolated from peripheral blood using a Gentra Puregene kit (Qiagen Sciences, Inc., Germantown, MD). Array study was performed with Affymetrix Cytogenetics Array 2.7 (Affymetrix Inc., Santa Clara, CA, USA) according to the manufacturer's instructions, using the Affymetrix Chromosome Analysis Suite software. Copy number state indicated an apparently contiguous interstitial deletion (~1.9 Mb) on chromosome 15q21.1 in the region 45,466,733-47,335,104 bp (NCBI36/hg18) (Figure 2B). Parental array analysis showed normal results.
Molecular Cytogenetic Findings
In order to better characterize the complex chromosome rearrangement, FISH was performed with region-specific BAC probes for chromosomes 6q, 12q and 15q. Clones were selected from the BACPAC Resource Center at the Children's Hospital Oakland Research Institute (Oakland, CA, USA) and prepared according to Guilherme et al. [12]. FISH was performed with whole chromosome painting (WCP) probes for chromosomes 6, 12 and 15 (Cytocell, Cambrigde, UK). Individual and combined WCP probes were used in order to clarify the complex chromosomal rearrangement. Cell images were captured using the Isis Digital Imaging System (Metasystem, Altlussheim, Germany). FISH analysis revealed a quite complex rearrangement with eight breakpoints, as follows:
46, XX, t(6;12;15)(6pter→6q14::15q15.1→15q21.1::15q21.1→15q22.3::6q14→6q21::12q24.1→12qter;12pter→12q24.1::6q21→6q22.2::15q22.3→15qter;15pter→15q15.1::6q22.2→6qter)dn.arr 15q21.1(45,466,733-47,335,104)×1 (Figure 2C, D and 2E).
Discussion
We report here on a girl with clinical features of the MFS spectrum and a 15q21.1 deletion including the entire FBN1 gene. This is the seventh study in the literature in which the deletion of FBN1 is confirmed by molecular techniques. Thus, to this date 18 patients with deletions including the whole FBN1 gene were reported, five of them belonging to the same family. Interestingly, only 12 of these patients [2,7,11, present case] present the typical MFS phenotype according to the Ghent criteria. Clinical variability in patients with different point mutations has been described, with patients with a nonsense mutation presenting milder phenotypes than patients with a missense mutation [10]. Cases with a deletion can be compared to patients with a nonsense mutation, since the truncated mRNA in the latter cases is believed to be reduced, due to the nonsense-mediated decay (NMD) mechanism that prevents the expression of the truncated mRNA [2, 10, 13]. Thus, in cases of deletion and nonsense mutation, the MFS phenotype can result from FBN1 gene haploinsufficiency and, especially in cases of missense mutations, from a dominant negative effect. Hutchinson et al [7] suggested that the clinical variability in MFS could be also due to variable FBN1 expression of the normal allele. The size of the deletions reported varies from small (less than 300 kb in 5 members of the same family) [2], including only the FBN1 gene, to large (up to 17.7 Mb) [9]. As found in our patient, when the deletion involves other genes besides FBN1, other unusual features can be found, such as those described here (craniostenosis, hypothyroidism and intellectual deficit). The patient described by Hiraki et al [9] presented no sign of the MFS syndrome, probably due to her young age and severe clinical phenotype. Our patient showed most of the skeletal features of MFS and an aortic root dilatation, but no ectopia lentis. Concerning the ocular system, she presented myopia and astigmatism. Her deletion comprises 19 genes and predicted genes including FBN1, besides SEMA6D and COPS2 that may have contributed to the intellectual deficit and hypothyroidism, respectively. Of the 18 patients with a complete deletion of the FBN1 gene described so far, only seven were karyotyped: two presented normal karyotypes [2, 10], three had visible 15q deletions [7–9], and one had a de novo translocation between the long arms of chromosomes 12 and 15 and a 4.9 Mb interstitial deletion at the translocation breakpoint of the long arm of chromosome 15 between the bands q21.1 and q21.2 [2]. Hilhorst-Hofstee et al [2] performed karyotype analysis only in two patients, as part of the mental retardation screening. So, our patient is the first MFS case described presenting a complex chromosome rearrangement among chromosomes 6, 12 and 15. In the literature, up to 30-50% of the patients with a chromosomal rearrangement, both complex and reciprocal translocations, show an imbalance on the chromosomal or molecular level as an explanation for their phenotype [14]. The greater the number of breakpoints involved in a CCR, the greater the likelihood of genomic imbalances or position effect. Disruption of a gene could unmask a recessive mutation on the homologue allele, suggesting a greater chance for an abnormal phenotypic outcome [15].
Conclusion
We emphasize the importance of using a combination of different molecular cytogenetic techniques in cases of chromosomal and/or genomic rearrangements involving the FBN1 gene, in order to better understand the extent of the molecular etiology of the Marfan syndrome and also to elucidate the genetic constitution of CCRs associated with diseases.
Consent
Written informed consent was obtained from the patient's parents for the publication of this case report and accompanying images. A copy of the consent form is available for review by the Editor-in-Chief of this journal.
References
Turner CL, Emery H, Collins AL, Howarth RJ, Yearwood CM, Cross E, Duncan PJ, Bunyan DJ, Harvey JF, Foulds NC: Detection of 53 FBN1 mutations (41 novel and 12 recurrent) and genotype-phenotype correlations in 113 unrelated probands referred with Marfan syndrome, or a related fibrillinopathy. American journal of medical genetics 2009, 149A(2):161–170. 10.1002/ajmg.a.32593
Hilhorst-Hofstee Y, Hamel BC, Verheij JB, Rijlaarsdam ME, Mancini GM, Cobben JM, Giroth C, Ruivenkamp CA, Hansson KB, Timmermans J, Moll HA, Breuning MH, Pals G: The clinical spectrum of complete FBN1 allele deletions. European journal of human genetics 2011, 19(3):247–252. 10.1038/ejhg.2010.174
Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, Pyeritz RE, Sponseller PD, Wordsworth P, De Paepe AM: The revised Ghent nosology for the Marfan syndrome. Journal of medical genetics 2010, 47(7):476–85. 10.1136/jmg.2009.072785
Mizuguchi T, Collod-Beroud G, Akiyama T, Abifadel M, Harada N, Morisaki T, Allard D, Varret M, Claustres M, Morisaki H, Ihara M, Kinoshita A, Yoshiura K, Junien C, Kajii T, Jondeau G, Ohta T, Kishino T, Furukawa Y, Nakamura Y, Niikawa N, Boileau C, Matsumoto N: Heterozygous TGFBR2 mutations in Marfan syndrome. Nature genetics 2004, 36(8):855–60. 10.1038/ng1392
Mátyás G, Arnold E, Carrel T, Baumgartner D, Boileau C, Berger W, Steinmann B: Identification and in silico analyses of novel TGFBR1 and TGFBR2 mutations in Marfan syndrome-related disorders. Human mutation 2006, 27(8):760–9. 10.1002/humu.20353
Chung BH, Lam ST, Tong TM, Li SY, Lun KS, Chan DH, Fok SF, Or JS, Smith DK, Yang W, Lau YL: Identification of novel FBN1 and TGFBR2 mutations in 65 probands with Marfan syndrome or Marfan-like phenotypes. American journal of medical genetics 2009, 149A(7):1452–1459. 10.1002/ajmg.a.32918
Hutchinson S, Furger A, Halliday D, Judge DP, Jefferson A, Dietz HC, Firth H, Handford PA: Allelic variation in normal human FBN1 expression in a family with Marfan syndrome: a potential modifier of phenotype? Human molecular genetics 2003, 12(18):2269–2276. 10.1093/hmg/ddg241
Ades LC, Sullivan K, Biggin A, Haan EA, Brett M, Holman KJ, Dixon J, Robertson S, Holmes AD, Rogers J, Bennetts B: FBN1, TGFBR1, and the Marfan-craniosynostosis/mental retardation disorders revisited. American journal of medical genetics 2006, 140(10):1047–1058.
Hiraki Y, Moriuchi M, Okamoto N, Ishikawa N, Sugimoto Y, Eguchi K, Sakai H, Saitsu H, Mizuguchi T, Harada N, Matsumoto N: Craniosynostosis in a patient with a de novo 15q15-q22 deletion. American journal of medical genetics 2008, 146A(11):1462–1465. 10.1002/ajmg.a.32339
Faivre L, Khau Van Kien P, Callier P, Ruiz-Pallares N, Baudoin C, Plancke A, Wolf JE, Thauvin-Robinet C, Durand E, Minot D, Dulieu V, Metaizeau JD, Leheup B, Coron F, Bidot S, Huet F, Jondeau G, Boileau C, Claustres M, Mugneret F: De novo 15q21.1q21.2 deletion identified through FBN1 MLPA and refined by 244K array-CGH in a female teenager with incomplete Marfan syndrome. European journal of medical genetics 2010, 53(4):208–212. 10.1016/j.ejmg.2010.05.002
Furtado LV, Wooderchak-Donahue W, Rope AF, Yetman AT, Lewis T, Plant P, Bayrak-Toydemir P: Characterization of large genomic deletions in the FBN1 gene using multiplex ligation-dependent probe amplification. BMC medical genetics 2011, 12(1):119. 10.1186/1471-2350-12-119
Guilherme RS, de Freitas Ayres Meloni V, Sodre CP, Christofolini DM, Pellegrino R, de Mello CB, Conlin LK, Hutchinson AL, Spinner NB, Brunoni D, Kulikowski LD, Melaragno MI: Cytogenetic and molecular evaluation and 20-year follow-up of a patient with ring chromosome 14. American journal of medical genetics 2010, 152A(11):2865–2869. 10.1002/ajmg.a.33689
Matyas G, Alonso S, Patrignani A, Marti M, Arnold E, Magyar I, Henggeler C, Carrel T, Steinmann B, Berger W: Large genomic fibrillin-1 (FBN1) gene deletions provide evidence for true haploinsufficiency in Marfan syndrome. Human genetics 2007, 122(1):23–32. 10.1007/s00439-007-0371-x
Papadopoulou E, Sismani C, Christodoulou C, Ioannides M, Kalmanti M, Patsalis P: Phenotype-genotype correlation of a patient with a "balanced" translocation 9;15 and cryptic 9q34 duplication and 15q21q25 deletion. American journal of medical genetics 2010, 152A(6):1515–1522.
de Vree PJ, Simon ME, van Dooren MF, Stoevelaar GH, Hilkmann JT, Rongen MA, Huijbregts GC, Verkerk AJ, Poddighe PJ: Application of molecular cytogenetic techniques to clarify apparently balanced complex chromosomal rearrangements in two patients with an abnormal phenotype: case report. Molecular cytogenetics 2009, 2: 15. 10.1186/1755-8166-2-15
Acknowledgements
This work was supported by FAPESP, Brazil (grant to M.I.M. #09/54261-4).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MESC performed the molecular karyotyping and data analysis and wrote the manuscript; ABAP made the clinical evaluation of the patient; SST, TIM, ARND and RSG did the molecular analysis (FISH, WCP and SNP array); CBM performed a neuropsychological evaluation of the patient; LRJS made the cytogenetic analysis; and ABAP and MIM coordinated the study. All the authors have read and approved the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Colovati, M.E., da Silva, L.R., Takeno, S.S. et al. Marfan syndrome with a complex chromosomal rearrangement including deletion of the FBN1 gene. Mol Cytogenet 5, 5 (2012). https://doi.org/10.1186/1755-8166-5-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8166-5-5