
Abstract
Chromosome rearrangements involving telomeres have been established as one of the major causes of idiopathic mental retardation/developmental delay. This case of 7p partial trisomy syndrome in a 3-year-old female child presenting with developmental delay emphasizes the clinical relevance of cytogenetic diagnosis in the better management of genetic disorders. Application of subtelomeric FISH technique revealed the presence of interstitial telomeres and led to the ascertainment of partial trisomy for the distal 7p segment localized on the telomeric end of the short arm of chromosome 19. Whole-genome cytogenetic microarray-based analysis showed a mosaic 3.5 Mb gain at Xq21.1 besides the approximately 24.5 Mb gain corresponding to 7p15.3- > pter. The possible mechanisms of origin of the chromosomal rearrangement and the clinical relevance of trisomy for the genes lying in the critical regions are discussed.
Similar content being viewed by others


Introduction
Unbalanced rearrangements leading to chromosomal and thereby genomic imbalance have been traditionally associated with abnormal phenotypes. While rearrangements involving sex chromosomes may not be linked to severe motor impairment, those involving autosomes may result in dysmorphic facies, malformations and mental retardation (MR). Further, the detection of chromosome ends in majority of the translocations has emphasized the role of telomeric rearrangements as the cause of several genetic diseases. The telomeric regions of human chromosomes are enriched for CpG islands and have been reported to have the highest gene density in the entire genome [1].
Telomeres composed of the hexameric TG-rich repeat (TTAGGG)n sequences and ranging from 2 to 15 kb in length, cap the ends of mammalian chromosomes. They are very essential for the stability and integrity of the chromosomes by preventing end to end fusion and exonucleolytic degradation. Additionally, they also play a role in cell longevity by actively participating in the complete replication of DNA [2]. Telomeres are synthesized by telomerase, a reverse transcriptase-like enzyme that is composed of RNA and protein catalytic subunits [3]. The telomeric sequences serve as binding sites for a specialized set of proteins that give rise to a unique chromatin structure [4] and assist in chromosome attachment to the nuclear matrix [5] and in segregation of chromosomes at meiosis [6].
Interstitial telomeric sequences could result from i) termino-terminal rearrangements with fusion of the telomeres of two chromosomes, ii) rearrangements in which an acentric fragment of one chromosome fuses to the telomere of another chromosome and iii) telomere-centromere rearrangements in which telomeric sequences of one chromosome fuse with the centromere of another chromosome [7]. There is increasing evidence that interstitial telomeres may be hot-spots for chromosomal instability leading to an increased rate of spontaneous and induced chromosomal rearrangements such as translocations, ring chromosomes, and unstable jumping translocations seen in tumor cells [8, 9]. These sequences are also present in certain intrachromosomal sites e.g., within the band 2q13 and have been proposed to have originated as a result of telomeric fusions in the course of genome evolution [10]. In some cases, interstitial telomeric sequences can be considered as non-functional elements analogous to the inactivated centromeres in dicentric chromosomes [11, 7]. Another case of 7p partial trisomy syndrome associated with interstitial telomeres is added to the existing literature through this report stressing the fact that rearrangements involving telomeres or telomere dysfunction can handicap a cell severely and might lead to genetic disease.
Case report
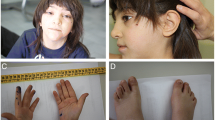
The proband was a 3-year-old female child born to non-consanguineous parents. Her mother was 29 years old and her father was aged 32 years at her birth. She weighed 2.25 kg at birth and was delivered normally after an uneventful pregnancy. The child was referred for cytogenetic analysis with a complaint of severe developmental delay and dysmorphic facies. Clinical examination revealed open anterior fontanelle, hypertelorism, low set ears, squint eyes and clinodactyly of both little fingers. CT scan indicated mild cerebral atrophy. She was afflicted with bronchopneumonia and did not exhibit any organomegaly. T4 and T3 levels were within normal limits. Pedigree analysis did not indicate any genetic disorder or congenital defects in any of the family members.
Materials and methods
Cytogenetic analysis
Chromosomal preparations were obtained from PHA-stimulated lymphocytes of the proband, her parents and her elder brother following the protocol of Hungerford (1965) [12] and were GTG-banded [13]. High resolution banding was performed by using ethidium bromide [14]. This study was approved by the Institutional Ethics Committee. Twenty-five metaphases were analyzed from each individual. About five well-banded metaphases were documented and karyotyped using Applied Spectral Imaging Systems karyotyping software (BandView version 5.5). Chromosomal anomalies were designated using standard nomenclature (ISCN 2009).
M-FISH analysis
M-FISH was performed on unbanded chromosome preparations from proband using 24Xcyte Human M-FISH probe kit from MetaSystems, Gmbh, Germany, following the manufacturer's specifications. Images were captured under respective filters using Zeiss Axioscope microscope (Zeiss, Jena, Germany). Image processing and analysis were done with the ISIS imaging system (MetaSystems, Gmbh, Altlussheim, Germany).
FISH analysis using WCP probes
Targeted FISH procedures were performed using whole chromosome painting probes for chromosome 7 (WCP7, FITC, MetaSystems, Gmbh, Germany) and chromosome 19 (WCP 19, Spectrum Green, Vysis Inc., USA) according to the manufacturer's instructions.
FISH analysis using subtelomere probes
FISH experiments were performed using ToTelVysion™ subtelomere multicolour DNA probe Mixture 7 (TelVysion 7p Spectrum Green, 7q Spectrum Orange, TelVysion 14q Spectrum Orange and Spectrum Green) and Mixture 14 (TelVysion 19p Spectrum Green, 19q Spectrum Orange, LSI 19p13 Spectrum Aqua) (Vysis Inc., USA) to confirm cryptic subtelomeric rearrangements. About twenty-five metaphases were captured under Zeiss Axioscope microscope and analysed using ISIS imaging system (MetaSystems).
Whole-genome cytogenetic microarray-based hybridization analysis
DNA was extracted using the Nucleon BACC2 DNA extraction kit (Amersham Pharmacia Biotech, Piscataway, NJ) following the manufacturer's protocol. Microarray analysis was performed using the Affymetrix® Whole-Genome Cytogenetic 2.7 M Array which detects both known and novel chromosome aberrations across the entire genome. Detailed analysis was done using Affymetrix® Chromosome Analysis Suite (ChAS) software ver. 1.1. (Affymetrix INC. Santa Clara, CA, USA) and UCSC Genome Browser Assembly was used for localization of gains and losses in the copy number analysis.
Results
Cytogenetic analysis
Chromosomal analysis revealed an abnormal chromosome 19 with no evidence of mosaicism in the proband who was found to have the karyotype 46, XX, der(19)t(19;?)(p13;?) (Figure 1). The origin of the terminal dark band could not be determined as the chromosomal constitution of her parents was normal. Her brother also exhibited a normal karyotype.

GTG-banded karyogram of the proposita showing an abnormal chromosome 19.
FISH analysis
M-FISH allowed the precise identification of the extra material on the derivative chromosome 19 to be a segment of chromosome 7 (Figure 2) and this was confirmed employing WCP7 and WCP19 probes (Figure 3a and 3b). Although M-FISH and FISH with WCPs did not reveal the arm of chromosome 7 involved in the rearrangement, FISH using subtelomere probes showed the presence of three sets of subtelomeric sequences within the abnormal chromosome i.e., subtel 19p, 19q and 7p (Figure 4a-c), substantiating the partial trisomy of chromosome 7p. The karyotype was consequently determined to be 46, XX, der(19)t(7;19)(p15;p13.3).

mFISH reveals the derivative chromosome 19 to contain material from chromosome 7.

FISH using whole chromosome paints - a) WCP7 (green) and b) WCP19 (green) probes confirms that the translocated segment on derivative chromosome 19 (indicated by arrow) was derived from chromosome 7.

FISH using subtelomeric probes showing signals on - a) 7p (green), seen also on derivative chromosome 19 (arrow) and 7q (red); b) 19p (green) and 19q (red); and c) interstitial 19p signal near the breakpoint junction (green, indicated with an arrow), 19q (red) and 7p (green) on the der(19)t(7;19) chromosome.
Whole-genome microarray cytogenetic hybridization analysis
Array-based comparative genomic hybridization was used for high-resolution mapping of the breakpoints that were identified using conventional and fluorescence techniques. A copy number gain ('partial trisomy') of 24490 kb was observed for the short arm of chromosome 7 involving the bands 7p15.3-pter (chr7: 141029-24632081, hg18) (Figure 5a). Further, an apparent mosaicism for a gain of 3500 kb corresponding to Xq21.1 (chrX: 77294144-80794852, hg18) was also noted (Figure 5b).

Whole-genome microarray analysis with respective segment report showing a) copy number gain "partial trisomy" of 24490 kb corresponding to cytobands 7p15.3- > pter b) mosaic gain of 3500 kb on Xq21.1.
Discussion
Loss of telomere or telomere function can lead to breakage-fusion-bridge events that involve repeated fusion and breakage of chromosomes thereby leading to instability, rearrangements including unbalanced translocations and terminal deletions, gene amplification and subsequently cancer [15, 16]. Generally chromosomes harbouring a break at their ends stabilize themselves through telomere capture which is telomerase-independent by acquisition of a pre-existing telomere through recombination or via telomere healing which is telomerase-dependent by de novo synthesis of a new telomere [17, 18]. Another possibility is that the telomere cap structure might be disrupted without chromosome breakage, through the unfolding of a putative hairpin loop or the transient deficiency of telomere-binding proteins. This would permit the chromosome end to be joined to a broken segment of another chromosome and thus would give rise to a non-reciprocal exchange [11, 9].
Lack of whole chromosome paint probe WCP19 signal on chromosome 7 confirmed the absence of a balanced translocation in the proposita. Therefore, chromosome healing through telomere capture appears to be the most probable mechanism of genesis of the derivative chromosome 19 where the acentric fragment of chromosome 7 fuses to the telomere of chromosome 19. The presence of subtelomeric 19p probe signal on the derivative chromosome indicates that the break had not occurred in the subtelomere but in the distal part removing all or some of the telomeric consensus sequence although this could not be validated. Telomere capture always occur de novo in contrast to unbalanced cryptic translocations which are mostly inherited from balanced carrier parents [9]. The rearrangement observed in the proband was also de novo in this study. The derivative chromosome could have been the outcome of a de novo event arising during gametogenesis from either of the parent. Baird et al. (2006) [19] have elucidated the association between telomere truncation events and the male germ cell line. Such events may be influenced by paternal age at conception [20] and have the potential to generate chromosomal rearrangements and to limit replicative potential [19]. However, this was not a case of advanced paternal age.
The deleted chromosome in the proposita could have been repaired by acquiring the end of chromosome 7p, while the distal end of the broken chromosome 19p and the now broken donor chromosome 7 remains unrepaired. Thus an unbalanced segregation of a parental nonreciprocal translocation at meiosis is the most common mechanism leading to partial trisomy. This process involves recombination-dependent DNA replication termed break-induced replication (BIR) [21]. This mechanism has been observed in yeast and is widely hypothesized to be the etiology behind telomere associations in mammalian cells too. Although such healing events potentially give rise to chromosomes carrying terminal deletions and a new telomere, it allows the truncated chromosome to replicate and segregate normally [22].
The abnormal phenotype observed in the proposita might be partly attributed to telomeric position effect (TPE), a silencing mechanism involving telomere architecture and classical heterochromatin features, and partly due to trisomy 7p15-pter. Repositioning of active genes near telomeres or subtelomeric sequences as a result of chromosome rearrangements may pave way to TPE consequently playing a direct role in human genetic diseases including mental retardation. The subtelomeres are usually prone to extensive recombination and may either buffer or facilitate the spreading of silencing that originates from the telomere [23, 24].
A survey of cases of 7p partial trisomy reported in the literature reveals a well-defined pattern of musculoskeletal, cardiovascular, neurological, genital, and ocular abnormalities in decreasing order of frequency. Further, approximately one-third of the patients were described to have died in infancy and one-half had severe mental retardation [25]. The first case was reported by Willner et al. (1977) [26] who also stressed the potential of the derivative chromosome in regional gene mapping. The most striking effect of this anomaly is found to be a severe failure in cerebral development resulting in gross retardation. The clinical findings observed in the proposita including psychomotor retardation, large anterior fontanelle, hypertelorism and low-set ears were in accordance with these earlier reports (Table 1). However, congenital heart defects and skeletal abnormalities [27, 28] were not recorded in the index case. This difference may be explained by the exact position of the breakpoints at the molecular level, individual gene dosage and also probably TPE.
Normal development is strongly associated with gene dosage and it is established that an imbalance leads to abnormal phenotype. The majority of genes exhibiting dosage sensitivity are regulatory which affect the expression of vital target genes [29, 30]. Rapid progress in mapping of candidate genes has led to the identification of several loci involved in neurological dysfunction and, in particular, mental retardation. Array-based comparative genomic hybridization confirmed partial trisomy 7p (7p15.3-pter) which harbors about 130 protein coding genes and five of these genes found to encode transcription factors could be crucial in the neuronal pathway (Table 2). Two genes P2PRY10 and BRWD3 of 11 genes on the cytoband Xq21.1, found to show copy number variation in mosaic form, could also have a possible role in the causation of mental retardation. Triple dosage of the TWIST gene on 7p21.1-p21.2 has been attributed to abnormal skull development due to delayed closure of fontanelles [31, 32].
Neuronal migration disorders, in which newly born neurons fail to migrate correctly from the ventricular zone to their final neocortical positions, are associated with neurological dysfunction [33]. Mental retardation could reflect an abnormal dosage effect of NPY gene during brain development. The neuropeptide Y, a neurotransmitter found in the CNS, functions through G protein-coupled receptors (GPCR) and influences many physiological processes including cortical excitability [34, 35]. The P2Y purinoceptor 10 protein, encoded by the P2PRY10 gene localized on Xq21.1, is itself is a GPCR and is also known to have a role in neurotransmission. Both the genes activate MAPK/MEK signaling pathway.
Another gene mapping in the partial 7p trisomic region is RADIL (RA [Ras association] and DIL domains) and its product is known to play a critical role in cell adhesion and migration [36]. Knockdown studies of radil in the zebrafish model presented multiple defects in neuronal cell (NC)-derived lineages such as cartilage, pigment cells and enteric neurons [37]. RADIL, a downstream effector of RAP, a member of RAS family also activates MAPK/MEK signaling pathway. Murine models indicate a key-role for RAS signaling in memory, learning and cognitive abilities [35, 38]. Human constitutional defects associated with NC function account for up to 30% of all congenital birth defects [39] which often include craniofacial abnormalities.
Mental retardation frequently occurs in patients with disorders of the RAS/MAPK signaling cascade [40]. MAPK/MEK activation thus seems to be a crucial event in neuronal adhesion and synaptic functions. The association of the three genes NPY, P2PRY10 and RADIL with this critical pathway and the probable cross-talk between them (Figure 6) identifies them as the prime candidates for the observed phenotype.

A hypothetical schematic representation of the interaction of genes NPY and RADIL on 7p15.3- > pter and P2RY10 on Xq21.1.
In conclusion, application of molecular cytogenetic techniques has not only resolved the origin of the abnormality but also has revealed the presence of interstitial telomeric sequences on the derivative chromosome. Further, segmental imbalances thus detected prove significant in phenotype-genotype correlation and in the identification of probable candidate genes.
Consent
Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
References
Saccone S, de Sario A, Valle GD, Bernardi G: The highest gene concentrations in the human genome are in telomeric bands of metaphase chromosomes. Proc Nati Acad Sci USA 1992, 89: 4913–4917. 10.1073/pnas.89.11.4913
Knight JLS, Flint J: Perfect endings: a review of subtelomeric probes and their use in clinical diagnosis. J Med Genet 2000, 37: 401–409. 10.1136/jmg.37.6.401
Masutomi K, Yu EY, Khurts S, Ben-Porath I, Currier JL, Metz GB, Brooks MW, Kaneko S, Murakami S, DeCaprio JA, Weinberg RA, Stewar SA, Hahn WC: Telomerase maintains telomere structure in normal human cells. Cell 2003, 114: 241–253. 10.1016/S0092-8674(03)00550-6
Tommerup H, Dousmanis A, de Lange T: Unusual chromatin in human telomeres. Molecular and Cellular Biology 1994, 14: 5777–5785.
Ludérus MEE, van Steensel B, Chong L, Sibon OCM, Cremers FFM, de Lange T: Structure, subnuclear distribution, and nuclear matrix association of the mammalian telomeric complex. The Journal of Cell Biology 1996, 135: 867–881. 10.1083/jcb.135.4.867
Conrad NM, Dominguez AM, Dresser ME: Ndj1p, a meiotic telomere protein required for normal chromosome synapsis and segregation in yeast. Science 1997, 276: 1252–1255. 10.1126/science.276.5316.1252
Rossi E, Floridia G, Casali M, Danesino C, Chiumello G, Bernardi F, Magnani I, Papi L, Mura M, Zuffardi O: Types, stability, and phenotypic consequences of chromosome rearrangements leading to interstitial telomeric sequences. J Med Genet 1993, 30: 926–931. 10.1136/jmg.30.11.926
Kilburn EA, Shea MJ, Sargent RG, Wilson JH: Insertion of a telomere repeat sequence into a mammalian gene causes chromosome instability. Mol Cellular Biology 2001, 21: 126–135. 10.1128/MCB.21.1.126-135.2001
Fortin F, Bergeron MB, Fetni R, Lemieux N: Frequency of chromosome healing and interstitial telomeres in 40 cases of constitutional abnormalities. Cytogenet Genome Res 2009, 125: 176–185. 10.1159/000230002
IJdo JW, Baldini A, Ward DC, Reeders ST, Wells RA: Origin of human chromosome 2: An ancestral telomere-telomere fusion. Proc Natl Acad Sci USA 1991, 88: 9051–9055. 10.1073/pnas.88.20.9051
Park MV, Gustashaw KM, Wathen TM: The presence of interstitial telomeric sequences in constitutional chromosome abnormalities. Am J Hum Genet 1992, 50: 914–923.
Hungerford DA: Leucocytes cultured from small inocula of whole blood and the preparation of metaphase chromosomes by treatment with hypotonic KCl. Stain Technology 1965, 40: 333–338.
Seabright M: A rapid banding technique for human chromosomes. Lancet 1971, ii: 971–972.
Ikeuchi T, Sasaki M: Accumulation of early mitotic cells in ethidium bromide treated human lymphocyte cultures. Proc Jpn Acad 1979, 55: 15–18.
Hackett AJ, Feldser DM, Greider CW: Telomere dysfunction increases mutation rate and genomic instability. Cell 2001, 106: 275–286. 10.1016/S0092-8674(01)00457-3
Lo WIA, Sabatier L, Fouladi B, Pottier G, Ricoul M, Murnane JP: DNA amplification by Breakage/Fusion/Bridge cycles initiated by spontaneous telomere loss in a human cancer cell line. Neoplasia 2002, 4: 531–538. 10.1038/sj.neo.7900267
Flint J, Craddock CF, Villegas A, Bentley DP, Williams HJ, Galanello R, Cao A, Wood WG, Ayyub H, Higgs DR: Healing of broken human chromosomes by the addition of telomeric repeats. Am J Hum Genet 1994, 55: 505–512.
Daniel A, St Heaps L: Chromosome loops arising from intrachromosomal tethering of telomeres occur at high frequency in G1 (non-cycling) mitotic cells: Implications for telomere capture. Cell & Chromosome 2004, 3: 3. [http://www.cellandchromosome.com/content/3/1/3] 10.1186/1475-9268-3-3
Baird MD, Britt-Compton B, Rowson J, Amso NN, Gregory L, Kipling D: Telomere instability in the male germline. Hum Mol Genet 2006, 15: 1 45–151.
De Meyer T, Rietzschel ER, De Buyzere ML, De Bacquer D, Van Criekinge W, De Backer GG, Gillebert TC, Van Oostveldt P, Bekaert S, on behalf of the Asklepios investigators: Paternal age at birth is an important determinant of offspring telomere length. Hum Mol Genet 2007, 16: 24 3097–3102.
Bosco G, Haber JE: Chromosome break-induced DNA replication leads to nonreciprocal translocations and telomere capture. Genetics 1998, 150: 1037–1047.
Varley H, Di S, Scherer SW, Royle NJ: Characterization of terminal deletions at 7q32 and 22q13.3 healed by de novo telomere addition. Am J Hum Genet 2000, 67: 610–622. 10.1086/303050
Mefford HC, Trask BJ: The complex structure and dynamic evolution of human subtelomeres. Nature Reviews (Genet.) 2002, 3: 91–102.
Ottaviani A, Gilson E, Magdinier F: Telomeric position effect: From the yeast paradigm to human pathologies? Biochimie 2008, 90: 93–107. 10.1016/j.biochi.2007.07.022
Kozma C, Haddad BR, Meck JM: Trisomy 7p resulting from 7p15;9p24 translocation: Report of a new case and review of associated medical complications. Amer J Med Genet 2000, 91: 286–290. 10.1002/(SICI)1096-8628(20000410)91:4<286::AID-AJMG9>3.0.CO;2-2
Willner JP, Paciuc S, Criatian S, Hsu LYF, Murphy R, Hirschhorn K: The 7p partial trisomy syndrome. Pediatr Res 1977, 11: 530.
Larson LM, Wasdahl WA, Jalal SM: Partial trisomy 7p associated with familial 7p;22q translocation. J Med Genet 1977, 14: 258–261. 10.1136/jmg.14.4.258
Berry AC, Honeycombe J, Macoun SJR: Two children with partial trisomy for 7p. J Med Genet 1979, 16: 320–321. 10.1136/jmg.16.4.320
Birchler JA, Riddle NC, Auger DL, Veitia RA: Dosage balance in gene regulation:biological implications. Trends in Genetics 2005, 21: 219–226. 10.1016/j.tig.2005.02.010
FitzPatrick DA: Transcriptional consequences of autosomal trisomy: primary gene dosage with complex downstream effects. Trends in Genetics 2005, 21: 249–253. 10.1016/j.tig.2005.02.012
Mégarbané A, Le Lorc'h M, Elghezal H, Joly G, Gosset P, Souraty N, Samaras L, Prieur M, Vekemans M, Turleau C, Romana SP: Pure partial 7p trisomy including the TWIST, HOXA, and GLI3genes. J Med Genet 2001, 38: 178–182. 10.1136/jmg.38.3.178
Stankiewicz P, Thiele H, Baldermann C, Krüger A, Giannakudis I, Dörr S, Werner N, Kunz J, Rappold GA, Hansmann I: Phenotypic findings due to trisomy 7p15.3-pter including the TWIST locus. Am J Med Genet 2001, 103: 56–62. 10.1002/ajmg.1512
Pentney RJ, Mullan BA, Felong AM, Dlugos CA: The total numbers of cerebellar granule neurons in young and aged Fischer 344 and Wistar-Kyoto rats do not change as a result of lengthy ethanol treatment. Cerebellum 2002, 1: 79–89. 10.1080/147342202753203113
Sun L, Philipson LH, Miller RJ: Regulation of K+ and Ca++ Channels by a Family of Neuropeptide Y Receptors. J Pharmacol Exp Therapeut 1998, 284: 625–632.
Pellieux C, Sauthier T, Domenighetti A, Marsh DJ, Palmiter RD, Brunner HR, Pedrazzini T: Neuropeptide Y (NPY) potentiates phenylephrine-induced mitogen-activated protein kinase activation in primary cardiomyocytes via NPY Y5 receptors. Proc Nati Acad Sci USA 2000, 97: 1595–1600. 10.1073/pnas.030533197
Smolen GA, Schott BJ, Stewart RA, Diederichs S, Muir B, Provencher HL, Look AT, Sgroi DC, Peterson RT, Haber DA: A Rap GTPase interactor, RADIL, mediates migration of neural crest precursors Genes Dev. 2007, 21: 2131–2136.
Le Douarin NM, Kalcheim C: The Neural Crest. 2nd edition. Cambridge University Press: New York; 1999:445.
Thomas GM, Huganir RL: MAPK cascade signaling and synaptic plasticity. Nat Rev Neurosci 2004, 5: 173–183.
Jones NC, Trainor P: The therapeutic potential of stem cells in the treatment of craniofacial abnormalities. Expert Opin Biol Ther 2004, 4: 645–657. 10.1517/14712598.4.5.645
Cesarini L, Alfieri P, Pantaleoni F, Vasta I, Cerutti M, Petrangeli V, Mariotti P, Leoni C, Ricci D, Vicari S, Selicorni A, Tartaglia M, Mercuri E, Zampino G: Cognitive profile of disorders associated with dysregulation of the RAS/MAPK signaling cascade. Am J Med Genet Part A 2008, 149A: 140–146.
Moore CM, Pfeiffer RA, Craig-Holmes AP, Scott CI, Meisel-Stosiek M: Partial trisomy 7p in two families resulting from different balanced translocations. Clin Genet 1982, 21: 112–121.
Ohdo S, Suzumori K, Madokoro H, Sonoda T, Hayakawa K: Partial trisomy for 7p due to maternal balanced translocation. Jpn J Hum Genet 1983, 28: 297–300. 10.1007/BF01876794
Gabarrón J, Glover G, Jiménez A, Salas P, Pérez-Bryan J, Parra MJ: Chromosomal imbalance in the offspring of translocation carriers involving 7p - Further contribution with three cases to the partial trisomy 7p phenotype. Clin Genet 1988, 33: 211–219.
Caiulo A, Bardoni B, Camerino G, Guioli S, Mineli A, Piantanida M, Crosato F, Fior TD, Maraschio P: Cytogenetic and molecular analysis of an unbalanced translocation (X;7)(q28;p15) in a dysmorphic girl. Hum Genet 1989, 84: 51–54. 10.1007/BF00210670
Pallotta R, Dalpra L, Fusili P, Zuffardi O: Further delineation of 7p trisomy. Case report and review of the literature. Ann Genet 1996, 39: 152–158.
Back E, Jung C, Zeitler S, Schempp W: De novo duplication of 7pter?p21.2 and deletion of 9pter?p23.5: clinical and cytogenetic diagnosis. Clin Genet 1997, 51: 56–60.
Verzi MP, Anderson JP, Dodou E, Kelly KK, Greene SB, North BJ, Cripps RM, Black BL: N-Twist, an Evolutionarily Conserved bHLH Protein Expressed in the Developing CNS, Functions as a Transcriptional Inhibitor. Developmental Biology 2002, 249: 174–190. 10.1006/dbio.2002.0753
Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, Gangadharan U, Hobbs C, Di Marzo V, Doherty P: Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. The Journal of Cell Biology 2003, 163: 463–468. 10.1083/jcb.200305129
Acknowledgements
S.A. and N.C. acknowledge the support of UGC-SAP in the use of Applied Spectral Imaging Systems karyotyping software.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
AS performed the cytogenetic analysis; MJ referred and clinically examined the patient; VOP, RT and KMK participated in laboratory work concerning M-FISH and FISH analyses using WCP & subtelomere probes; SG and DT signed out the Whole-genome cytogenetic microarray-based hybridization analysis results; AS, CN and LRK drafted the manuscript; LS has critically reviewed and approved the manuscript. All authors read and approved the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
S, A., Padmalatha, V.O., G, S. et al. De novo 7p partial trisomy characterized by subtelomeric FISH and whole-genome array in a girl with mental retardation. Mol Cytogenet 4, 21 (2011). https://doi.org/10.1186/1755-8166-4-21
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8166-4-21