
Abstract
Background
Small supernumerary marker chromosomes (sSMC) occur in 0.075% of unselected prenatal and in 0.044% of consecutively studied postnatal cases. Individuals with sSMC present with varying phenotype, ranging from normal to extremely mild or severe depending on the chromosomal region involved, the euchromatic content present and degree of mosaicism. Except for chromosomes 15 and 22, the number of reported cases of sSMC is extremely small to provide us with a good genotype-phenotype correlation. Analphoid sSMC are even rarer. To our knowledge only eight cases of analphoid inversion-duplication 3q sSMC are reported so far.
Results
We describe here a one month old female child with several dysmorphic features and with a de novo analphoid supernumerary marker chromosome only in cultured skin fibroblast cells and not in lymphocytes. The marker was characterized as analphoid inversion-duplication 3q25.33-qter by oligo array comparative genomic hybridization (aCGH) and fluorescence in situ hybridization (FISH) studies. The final skin fibroblast karyotype was interpreted as 47,XX,+der(3).ish inv dup(3)(qter-q25.33::q25.33-qter)(subtel 3q+,subtel 3q+) de novo.
Conclusion
In addition to the eight reported cases of analphoid inversion-duplication 3q supernumerary marker in the literature, this is yet another case of 3q sSMC with a new breakpoint at 3q25.33 and with varying phenotype as described in the case report. Identification of more and more similar cases of analphoid inversion-duplication 3q marker will help in establishing a better genotype-phenotype correlation. The study further demonstrates that aCGH in conjunction with routine cytogenetics and FISH is very useful in precisely identifying and characterizing a marker chromosome, and more importantly help in providing with an accurate genetic diagnosis and better counseling to the family.
Similar content being viewed by others


Background
Small supernumerary marker chromosomes occur in 0.075% of unselected prenatal cases and in 0.044% of consecutively studied postnatal cases, and majority of them are de novo in origin [1–4]. Phenotype of individuals with de novo sSMC vary from normal to extremely mild or severe, depending on the chromosomal region involved and the euchromatic content present [5–7]. Although a number of reports describe the occurrence of a variety of sSMC for nearly all the chromosomes, the number for each type is not large enough to suggest a good genotype-phenotype correlation for a given sSMC, except for inv dup(15) and inv dup(22) where the phenotypic consequences are well described [6, 8–10]. We describe here the phenotype and corresponding molecular cytogenetic results of a child with dysmorphic features. This is yet another case of analphoid 3q supernumerary marker chromosome involving a new break point at 3q25.33. The marker is characterized as inversion-duplication 3q25.33-qter by oligo aCGH and FISH studies and it also reveals tissue specific mosaicism.
Case presentation
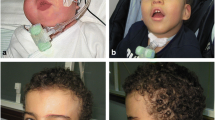
A one month old female child presenting with several dysmorphic features was referred to us for cytogenetic studies. She was the second child of unrelated parents. The first child was normal. The pregnancy and delivery at term were normal. Birth weight was 2.2 Kg, length 45 cm and head circumference was 32 cm. At birth she was noted to have several dysmorphic features including: prominent hairy forehead with hair extending up to the cheeks, upslanting palpebral fissures, depressed nasal bridge with short nose and very smooth philtrum, thin upper lip which was turning downward, low set ears, micrognathia, chubby cheeks, contracture of the fingers with postaxial polydactyly of left hand, widely separated toes which were overlapping, streaky pigmentation on the inner aspect of both fore arms distributed along the lines of Blaschko. Opthalmological exam was normal. Echocardiography showed sub-aortic ventricular septal defect (VSD), pulmonary hypertention and moderate valvular pulmonary stenosis. Skeletal survey showed a tiny projection of the tip of coccyx, a tail-like sacrococcygeal appendage. Computed tomography scan of the lumbosacral spine showed prominence of coccyx and outward projection. Magnetic resonance imaging of the brain showed partial hypoplasia of the corpus callosum and slight hypoplasia of the cerebellum.
Results
Cytogenetic study by Giemsa banding (G-banding) showed normal 46,XX chromosomes in all the 50 lymphocyte cells and 47,XX,+mar in all the 70 skin fibroblast cells studied (Fig. 1a). The marker chromosome was constitutive heterochromatin band (C-band) and nucleolar organizer region (NOR) staining negative. Fluorescence in situ hybridization studies using Pan-centromeric alpha-satellite FISH probe (Cambio) showed no hybridization onto the marker chromosome (Fig. 1b), confirming it to be analphoid. Immuno-FISH studies for centromere specific proteins could not be undertaken to confirm the formation of a neocentromere. However, the fact that the marker was confirmed as analphoid by FISH and was also found to be highly stable in skin fibroblast culture, strongly suggest of a possible neocentromere formation. Peripheral blood chromosomes of the parents were normal by G-banding and by all chromosome specific sub-telomeric FISH analysis. Array-CGH studies on cultured skin fibroblast cells of the patient showed amplification of oligo-probes from probe FLJ14153 at 3q25.33 (160.03 Mb) to 3qter (199.29 Mb) (Fig. 2) confirming the origin and breakpoint of the marker chromosome. The observation was further validated by FISH studies using chromosome 3 specific subtelomere FISH probe (Vysis), which revealed the marker to be inversion-duplication 3q25.33-qter (Fig. 1c).

a) Partial G-banded metaphase from skin fibroblast cell showing the marker chromosome (arrow). b) Supernumerary marker chromosome showing no hybridization with human alpha-satellite Pan-centromeric probe (Cambio). (c) Chromosome 3 specific subtelomeric FISH (Mix 3 – Vysis) showing one normal chromosome 3 (3ptel green/3qtel red) and the marker chromosome with two red signals for 3q subtelomere, confirming the marker to be inversion-duplication 3q25.33-qter.

Oligo aCGH study using Agilent 44 K oligo array. Labeling and hybridization are as described in methods. Figure shows a partial cytogenetic profile of chromosome 3q and amplification of chromosomal region 3q25.33-qter.
Discussion
Supernumerary marker chromosomes are a cause of great concern and pose huge challenge in routine medical practice, particularly in genetic diagnosis and counseling. Precise identification and complete characterization of the marker chromosome is very important to understand the underlying cause of the disease and to establish a good genotype-phenotype correlation.
Analphoid markers are rare observations, in which the marker chromosome lacks centromeric alphoid DNA sequences, that are otherwise typically present in normal functional centromeres. Survival and stability of such an analhpoid marker chromosome depends on the formation of a neocentromere [11]. At present 73 cases of neocentromeric sSMC originating from different chromosomes are reported in the literature, of which 13q, 15q and 3q are the most frequent observations [1]. To our knowledge only 9 cases of analphoid 3q inversion-duplication marker (including the present case) are reported so far, involving chromosomal break points 3q22.3, 3q25.33, 3q26.2, 3q27.1, 3q27.2 and 3q28 [12–19]. A brief summary of the clinical features, chromosomal breakpoints and degree of mosaicism of the reported cases with 3q inversion-duplication supernumerary marker chromosome is presented in Table 1. As can be seen, the most frequent break point is at 3q26.2 (4 out of 9 cases) suggesting that this is possibly the most common site of neocentromere formation in a 3q marker. However, findings of other break points between 3q22 to 3q28 strongly suggest that this region probably has several potential hotspots of necentromere formation.
Although all of the above cases with an analphoid 3q marker chromosome share some common clinical features, they still show widely varying phenotypes, probably due to the presence of varying euchromatic content as well as varying degree of mosiaicism (Table 1). Except for the case described by Portnoi et al [17] where the patient presented only with lines of Blaschko but otherwise healthy and not dysmorphic, all the other reported cases presented with developmental delay, severe dysmorphic features and multiple congenital anomalies involving several organs and systems. Our present case with break point at 3q25.33 presented with strikingly distinct phenotype as described in the case report. The typical facial appearance and the prominent hairy forehead are distinctive features, in addition to the cardiac and skeletal abnormalities that are similar to the cases described by Gimelli et al and Cockwell et al[12, 13].
Mosaicism is reported in 59% of cases where the sSMC are found in association with a normal cell line [4]. Five of the nine cases discussed here showed tissue specific mosaicism where the 3q marker was present only in fibroblasts and not in lymphocytes (case number 1–3 and 5–6 of Table 1), other two cases showed varying degree of mosaicism in lymphocytes as well as in fibroblasts (case number 4 and 7 of Table 1), and the remaining two cases had the marker chromosome in 71% and 100% lymphocytes respectively (case number 8 and 9 of Table 1). Keeping in mind the specific clinical features of the patient and with the application of modern molecular cytogenetic detection methods such as aCGH and FISH, more and more similar cases are likely to be identified that would further help in obtaining a better genotype-phenotype correlation, and in providing with an accurate genetic diagnosis and better informed counseling, which is highly valuable to the patients.
Methods
Cytogenetic and fish studies
Cytogenetic study was carried out on peripheral blood lymphocytes by G-banding according to the standard procedures [20], and 50 G-banded metaphases were analyzed. FISH study was undertaken using all chromosome specific sub-telomere FISH probes (Vysis). Hybridization and washing was done as per manufacturer's protocol and 100 metaphases were analyzed. Due to the strong abnormal clinical presentation and presence of prominent streaky pigmentation, further studies were undertaken on cultured skin fibroblast cells by routine G-banding and 70 metaphase spreads were analyzed, followed by C-band and NOR staining [20]. To further characterize the marker chromosome, alpha-satellite Pan-centromeric (Cambio) and chromosome 3 specific sub-telomeric (Vysis) FISH studies were performed as per manufacturer's instructions.
Oligo array CGH studies and validation
Oligo aCGH analysis was carried out using Human 44A microarray (Agilent) to determine the origin of the marker chromosome and to precisely identify the breakpoint region involved in the formation of the marker. The array contained in situ synthesized 60-mer oligonucleotides representing a total of 44,290 features. The oligonucleotide probes span the human genome with an average spatial resolution of approximately ~75 Kb, including coding and non-coding sequences, providing sufficient coverage for a genome-wide survey of DNA aberrations. Genomic DNA was extracted from cultured skin fibroblast cells of the patient by routine Proteinase K method and co-hybridized with normal male control DNA (Promega). DNA labelling, hybridization to oligonucleotide-array and post washing was carried out according to the protocol from Agilent Technologies and as described in Murthy et al [21]. Array CGH result was further validated by FISH study using chromosome 3 specific subtelomere FISH probe (Vysis).
Consent
This case report is presented with the consent of the patient's family.
References
Liehr T, Utine GE, Trautmann U, Rauch A, Kuechler A, Pietracz J, Bocian E, Kosyakova N, Mrasek K, Broduroglu K, Weise A, Aktas D: Neocentromeris small supernumerary marker chromosomes (sSMC) – three more cases and review of literature. Cytogenet Genome Res 2007, 118: 31–37. 10.1159/000106438
Buckton KE, Spowart G, Newton MS, Evans HJ: Fortyfour probands with an additional 'marker' chromosome. Hum Genet 1985, 69: 353–370. 10.1007/BF00291656
Crolla JA: FISH and molecular studies of autosomal supernumerary marker chromosomes excluding those derived from chromosome 15. II. A review of literature. Am J Med Genet 1998, 75: 367–381. 10.1002/(SICI)1096-8628(19980203)75:4<367::AID-AJMG5>3.0.CO;2-N
Crolla JA, Youings SA, Ennis S, Jacobs PA: Supernumeray marker chromosomes in man: parental origin, mosaicism and maternal age revisited. Eur J Hum Genet 2005, 13: 154–160. 10.1038/sj.ejhg.5201311
Liehr T, Mrasek K, Weise A, Dufke A, Rodriguez L, Martinez Guardia N, Sanchis A, Vermeesch JR, Ramel C, Polityko A, Haas OA, Anderson J, Claussen U, von Eggeling F, Starke H: Small supernumerary marker chromosomes: progress towards a genotype-phenotype correlation. Cytogenet Genome Res 2006, 112: 23–34. 10.1159/000087510
Dennis NR, Veltman MW, Thompson R, Craig E, Bolton PF, Thomas NS: Clinical findings in 33 subjects with large supernumerary marker(15) chromosomes and 3 subjects with triplication of 15q11-q13. Am J Med Genet 2006, 140(5):434–441. 10.1002/ajmg.a.31091
Starke H, Nietzel A, Weise A, Heller A, Mrasek K, Belitz B, Kelbova C, Volleth M, Albrecht B, Mitulla B, Trappe R, Bartels I, Adolph S, Dufke A, Singer S, Stumm M, Wegner RD, Seidel J, Schmidt A, Kuechler A, Schreyer I, Claussen U, von Eggeling F, Liehr T: Small supernumerary marker chromosomes (SMCs): genotype-phenotype correlation and classification. Hum Genet 2003, 114(1):51–67. 10.1007/s00439-003-1016-3
Crolla JA, Harvey JF, Sitch FL, Dennis NR: Supernumerary marker 15 chromosomes: a clinical, molecular and FISH approach to diagnosis and prognosis. Hum Genet 1995, 95: 161–70. 10.1007/BF00209395
Crolla JA, Howard P, Mitchell C, Long FL, Dennis NR: Am J Med Genet. 1997, 72: 440–7. 10.1002/(SICI)1096-8628(19971112)72:4<440::AID-AJMG13>3.0.CO;2-R
Warburton D: De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: clinical significance and distribution of breakpoints. Am J Hum Genet 1991, 49: 995–1013.
Choo KHA: Centromere DNA dynamics: latent centromeres and neocentromere formation. Am J Hum Genet 1997, 61: 1225–33. 10.1086/301657
Gimelli G, Giorda R, Beri S, Gimelli S, Zuffardi O: A large analphoid inv dup(3q22.3) marker chromosome characterized by array-CGH in a child with malformations, mental retardation, ambiguous genitalia and Blaschko's lines. Eur J Med Genet 2007, 50(4):264–273. 10.1016/j.ejmg.2007.04.003
Cockwell A, Gibbons B, Moore I, Crolla JA: An analphoid supernumerary marker chromosome derived from chromosome 3 ascertained in a fetus with multiple malformations. J Med Genet 2000, 37: 807–809. 10.1136/jmg.37.10.807
Teshima T, Bawle M, Weksberg R, Shuman C, Van Dyke DL, Schwartz S: Analphoid 3qter markers. Am J Med Genet 2000, 94(2):113–119. 10.1002/1096-8628(20000911)94:2<113::AID-AJMG3>3.0.CO;2-Q
Yu J, Qi Z, Thompson K, Modaff P, Wells W, Meisner L, Pauli R: Characterization of a rare neocentric marker chromosome using chromosome microdissection. 54th annual meeting of the American Society of Human Genetics 2004, (Abstract # 982):192.
Sullivan CM, Mountford ST, Emmerson JM, Ellis RJ, Turmbull C, Waters KS: A mosaic karyotype with an additional inv dup(3)(qter-q26.2::q26.2-qter), containing a neocentromere, detected in a skin biopsy from a girl with skeletal abnormalities, abnormal skin pigmentation and developmental delay. J Med Genet 2005, 42(Suppl 1):S71. (Abstract # 2.23)
Portnoi MF, Boutchnei S, Bouscarat F, Morlier G, Nizard S, Dersarkissian H, Crickx B, Nouchy M, Taillemite JL, Belaich S: Skin pigmentary anomalies and mosaicism for an acentric marker chromosome originating from 3q. J Med Genet 1999, 36: 246–250.
Barbi G, Spaich C, Adolph S, Kehrer-Sawatzki H: Analphoid de novo marker chromosome inv dup(3)(q28qter) with neocentromere in a dysmorphic and developmentally retarded girl. J Med Genet 2003, 40(3):e27. 10.1136/jmg.40.3.e27
Liehr T: Small supernumerary marker chromosome (sSMC).[http://www.med.uni-jena.de/fish/sSMC/03.htm]
Rooney DE: Chromosome staining and banding techniques. In Human Cytogenetics: constitutional analysis. A practical approach. Edited by: Rooney DE. Oxford university press; 2001.
Murthy SK, Nygren AOH, El Shankiry HM, Schouten JP, Al Khayat AI, Ridha A, Al Ali MT: Detection of a novel familial deletion of four genes between BP1 and BP2 of the Prader-Willi/Angelman syndrome critical region by oligo-array CGH in a child with neurological disorder and speech impairment. Cytogenet Genome Res 2007, 116: 135–140. 10.1159/000097433
Acknowledgements
We acknowledge the Department of Health and Medical Services (DOHMS), Dubai and Center for Arab Genomic studies (CAGS), Dubai for supporting the work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SKM conceived the study and drafted the manuscript. LG referred the patient, and RN and LG contributed clinical information. AKM, EEMR, SM, RP performed banding and cytogenetic analysis; SN and SP did FISH and PSJ did aCGH studies. MTA provided valuable support. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Murthy, S.K., Malhotra, A.K., Jacob, P.S. et al. Analphoid supernumerary marker chromosome characterized by aCGH and FISH as inv dup(3)(q25.33qter) de novo in a child with dysmorphic features and streaky pigmentation: case report. Mol Cytogenet 1, 19 (2008). https://doi.org/10.1186/1755-8166-1-19
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8166-1-19