
Abstract
Background
Interest in the detailed lignin and polysaccharide composition of plant cell walls has surged within the past decade partly as a result of biotechnology research aimed at converting biomass to biofuels. High-resolution, solution-state 2D 1H–13C HSQC NMR spectroscopy has proven to be an effective tool for rapid and reproducible fingerprinting of the numerous polysaccharides and lignin components in unfractionated plant cell wall materials, and is therefore a powerful tool for cell wall profiling based on our ability to simultaneously identify and comparatively quantify numerous components within spectra generated in a relatively short time. However, assigning peaks in new spectra, integrating them to provide relative component distributions, and producing color-assigned spectra, are all current bottlenecks to the routine use of such NMR profiling methods.
Results
We have assembled a high-throughput software platform for plant cell wall profiling that uses spectral deconvolution by Fast Maximum Likelihood Reconstruction (FMLR) to construct a mathematical model of the signals present in a set of related NMR spectra. Combined with a simple region of interest (ROI) table that maps spectral regions to NMR chemical shift assignments of chemical entities, the reconstructions can provide rapid and reproducible fingerprinting of numerous polysaccharide and lignin components in unfractionated cell wall material, including derivation of lignin monomer unit (S:G:H) ratios or the so-called SGH profile. Evidence is presented that ROI-based amplitudes derived from FMLR provide a robust feature set for subsequent multivariate analysis. The utility of this approach is demonstrated on a large transgenic study of Arabidopsis requiring concerted analysis of 91 ROIs (including both assigned and unassigned regions) in the lignin and polysaccharide regions of almost 100 related 2D 1H–13C HSQC spectra.
Conclusions
We show that when a suitable number of replicates are obtained per sample group, the correlated patterns of enriched and depleted cell wall components can be reliably and objectively detected even prior to multivariate analysis. The analysis methodology has been implemented in a publicly-available, cross-platform (Windows/Mac/Linux), web-enabled software application that enables researchers to view and publish detailed annotated spectra in addition to summary reports in simple spreadsheet data formats. The analysis methodology is not limited to studies of plant cell walls but is amenable to any NMR study where ROI segmentation techniques generate meaningful results.
Please see Research Article: http://www.biotechnologyforbiofuels.com/content/6/1/46/.
Similar content being viewed by others
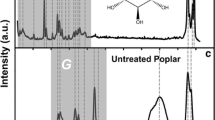


Background
Interest in the detailed lignin and polysaccharide composition of plant cell walls has surged within the past decade partly as a result of biotechnology research aimed at converting biomass to biofuels [1, 2]. Numerous studies have established the link between the relative amount of lignin and cellulose in vascular tissues and the accessibility of plant cell walls to chemical, enzymatic, and microbial digestion [2–4]. Comparisons of different species [5], and transgenic studies in which synthesis of cell wall components is genetically modified [3, 4, 6], are particularly useful in identifying these linkages.
High-resolution, solution-state 2D 1H–13C HSQC NMR spectroscopy has proven to be an effective tool for rapid and reproducible fingerprinting of the numerous polysaccharides and lignin components in unfractionated plant cell wall materials [7–11]. Recent advances in “ball-milled” sample preparations dissolved or swelled in organic solvents have enabled unfractionated material to be profiled without the need for component isolation [12, 13]. The heterogeneous and highly polymeric nature of the ball-milled cell wall material, in which polymers are of significantly lower degree of polymerization (DP) than in the intact cell wall (where DP of cellulose is ~7000-15000) [13], results in spectra with broad linewidths and considerable complexity. However, the dispersion provided by the two-dimensional correlation of protons to their attached 13C nuclei, at natural abundance, enables resolution and assignment of numerous lignin, cellulose, and hemicellulosic components. The 2D 1H–13C HSQC experiment is thus a powerful tool for cell wall profiling based on our ability to simultaneously identify and comparatively quantify numerous components within spectra generated with relatively short acquisition times (15–20 min/sample, but up to 5 h if excellent signal-to-noise and the ability to detect minor components is desirable).
As sample preparation and data acquisition methods have improved [10, 11], the task of spectral analysis has become a bottleneck in large studies. NMR-based chemometrics is one data analysis approach recently applied to investigate structural/compositional differences between wood samples from Populus[14]. Chemometrics is a multivariate approach with an extensive history in metabonomics [15, 16]. General strengths of a multivariate approach that simultaneously examines features from different sample groups include the ability to detect subtle patterns among features across sample groups, albeit sometimes with confusion by artifacts [12], and assess the relative importance of each feature for group discrimination [14].
NMR-based chemometrics is characterized by a sequence of steps involving: i) NMR data processing, including baseline correction if necessary; ii) generation of a feature set usually by selecting intensity values on each peak or summing over segmented regions (spectral binning); iii) production of a data table in which each sample represents a row and the features are columns; iv) normalization (row-based) and scaling (column-based) of the data; and v) multivariate statistical modeling. The greatest pitfalls lie in feature selection (step ii). Originally developed as a rapid and consistent method to generate data sets automatically and handle problems of peak “drift”, spectral binning unfortunately reduces spectral resolution and can generate artifacts in crowded spectra where the boundary of a bin may lie at the center of a signal. Even when the full resolution spectrum is used without binning, the common technique of analyzing 2D data by generating a 1D row vector from the 2D grid results in a loss of correlation information between the 1H and 13C intensity values during the analysis process, although this may be retained by indexing the 1D data so that 2D spectra can be recreated, including after, for example, principal component analysis [14].
An alternative to peak-based or bin-based feature selection is to mathematically model the data and use the modeled parameters as features for subsequent analysis. If the model can efficiently represent the relevant features of the data, the modeling step dramatically reduces the number of columns in the data matrix (data reduction) without loss of relevant information or generation of artifacts. Recently, spectral deconvolution using fast maximum-likelihood reconstruction (FMLR) was shown to accurately quantify metabolites in 2D 1H–13C HSQC spectra [17, 18]. FMLR constructs the simplest time-domain model (e.g., the model with the fewest number of signals and parameters) whose frequency spectrum matches the visible regions of the spectrum obtained from identical Fourier processing of the data [19, 20].
Spectral analysis of 2D 1H–13C HSQC NMR data by FMLR would appear to be an attractive approach for high-throughput plant cell wall profiling in the following respects:
-
i.
FMLR has already been shown to accurately model the characteristics of complex 2D 1H–13C HSQC solution spectra [17], and can be performed with minimal input information and operator intervention (moderately high throughput).
-
ii.
Because of the high spectral dispersion inherent in 2D 1H–13C NMR data, the detailed but localized amplitude and frequency information derived from FMLR should be easily combinable with assigned region-of-interest tables to generate the relative concentration of cell wall components in each sample (cell wall component profiles). Previous work has shown the utility of region of interest (ROI)-segmentation in quantitative 2D 1H–13C NMR studies [21, 22].
-
iii.
ROIs that correspond to a resolved peak or peak cluster can be defined even when the NMR assignment is tentative or unknown. The cell wall component profiles are thus suitable for both untargeted and targeted profiling.
-
iv.
Simple visual inspection of the cell wall component profiles might suffice to identify patterns of enrichment and depletion of various components between sample groups.
-
v.
The cell wall component profiles are also a robust feature set for input into multivariate analysis.
We apply here the spectral analysis methodology of FMLR with ROI-based segmentation to a large (98 samples) 2D 1H–13C NMR study of Arabidopsis lignin mutants and controls involving 20 sample groups (10 consolidated groups). Our focus here is not on biological conclusions to be drawn from the study (this is published concomitantly) [23], but on the methodology and software implementation of data analysis for powerful cell wall profiling by NMR.
Materials & methods
Biological sources
For ten genes involved in lignin biosynthesis [24], two Arabidopsis thaliana mutant alleles were analyzed (see Table 1). The 20 sample groups were consolidated into 10 effective sample groups based on statistically similar lignin composition. These samples were drawn from an overall pool of forty biological replicates of each homozygous mutant and 32 biological replicates for wild-type type were grown simultaneously in a random block design, spread over different trays, in the same environment. Plants were grown first under short-day conditions (8 h light, 21°C, humidity 55%) during 6 weeks, and then transferred to the greenhouse. For all of the biological repeats, the main stem was harvested just above the rosette when the plant was completely senesced. Once harvested, axillary inflorescences, siliques and seeds, as well as the bottom 1 cm of the main stem, were removed. The rest of the inflorescence stem was cut into 2 mm pieces and biological repeats were pooled per 8 stems to obtain 5 biological replicates for the mutant alleles and 4 repeats for the wild-type, except for c4h-2, ccr1-3, and ccr1-6. In order to have enough biomass for NMR analyses, the senesced inflorescence stems of c4h-2 were pooled in one single pool, for ccr1-3 the stems were pooled in 3 pools, and for ccr1-6 in 4 pools.
Sample preparation and cell wall dissolution
Preparation of whole cell wall samples for NMR was largely as described previously [8, 10]. In brief, pre-ground Arabidopsis stem samples (~200 mg) were extracted with water (3×) and then 80% aqueous ethanol (sonication 3 × 20 min) yielding 70–100 mg of cell wall material. Isolated cell walls (~80 mg) were ball-milled (4 × 30 min milling and 5 min cooling cycles, total time 2 h 20 min) using a Fritsch (Idar-Oberstein, Germany) Planetary Micro Pulverisette 7 ball mill vibrating at 800 rpm with 12 mL ZrO2 vessels containing thirty 5 mm ZrO2 ball bearings. Aliquots of the ball-milled whole cell walls (~60 mg) were transferred into NMR sample tubes, swollen in DMSO-d6:pyridine-d5 (4:1, v/v, 600 μl), and subjected to 2D NMR experiments.
Analysis overview
The process of FMLR reconstruction with ROI segmentation can be viewed as a sequence of steps involving:
-
1.
NMR data acquisition and processing
-
2.
Ensemble matrix formation and importation of grouping information
-
3.
Spectral normalization
-
4.
ROI segmentation
-
5.
Spectral deconvolution by FMLR
-
6.
ROI assignment and generation of a feature matrix
-
7.
ROI normalization of the feature matrix
-
8.
Statistical analysis of the features
NMR data acquisition and processing
NMR spectra were acquired on a Bruker Biospin (Billerica, MA) AVANCE 700 MHz spectrometer fitted with a cryogenically cooled 5-mm TXI gradient probe with inverse geometry (proton coils closest to the sample). Cell wall samples were swollen in 4:1 DMSO-d6:pyridine-d5, 0.5 mL; the central DMSO solvent peak was used as internal reference (δC, 49.5; δH, 3.49 ppm). Adiabatic HSQC experiments (hsqcetgpsisp.2.2) were carried out using the parameters described previously [10].
The initial steps of NMR data processing (conversion from time-domain to frequency domain) were performed using Topspin 3.1-Macintosh (Bruker Biospin, Rheinsteten, Germany). The processing consisted of i) apodization (matched Gaussian in F2, squared cosine-bell in F1), ii) zero-filling, iii) Fourier transformation, and iv) phase correction; no linear prediction was used.
The apodization and zero-filling parameters associated with steps i-iv along each dimension d define a vector operator that can be applied identically to both the acquired FID and the model FID along dimension d. In the FMLR algorithm, the operator converts discrete basis functions in the time domain (see Table 2) to discrete basis functions in the frequency domain.
Ensemble matrix formation
To facilitate concerted analysis of multiple data sets, the 2D absorption spectra (portions remaining after phase correction and discarding of imaginary components) were appended together to form an “ensemble” data set (pseudo-3D matrix). Two of the dimensions correspond to the 1H and 13C spectral frequencies and the remaining dimension is a “pseudo-dimension” that encodes the spectral index (and identity of the sample source).
Spectral normalization
The intensity of each data point in the spectrum was normalized to the sum of all intensity points prior to spectral analysis. This pre-analysis normalization step removes intensity modulation due to varying concentrations of biological material and allows the same intensity thresholds to be applied across all data sets.
ROI segmentation
A region of interest (ROI) as used in this context refers simply to a 2D spectral window or “box” associated with a spectral transition from a molecular entity. Regions of interest were manually defined for 91 ROIs within Newton by drawing boxes overlaid on the spectra (see graphical view in Figure 1A-C). Results from previous cell wall profiling studies [8, 10, 11, 25] and model compounds were used to determine the footprint of the ROIs appearing in the figures and to assign 52/91 ROIs in the various spectral regions. As an ROI is drawn once and can be superimposed onto any spectrum, the time required to define their boundaries is based only on the number of ROIs, rather than the number of spectra.

Annotated high-resolution, solution-state 2D 1H–13C HSQC NMR of a wild-type Arabidopsis spectrum in the A) lignin aromatic, B) polysaccharide anomeric, and C) lignin-polysaccharide regions. The rectangular boxes denote ROIs that correspond to assigned NMR transitions (colored boxes with annotations) or simply resolved regions of the spectrum that have yet to be assigned (gray boxes). The unassigned regions are associated with an ID that is used to identify them in the feature matrix. To avoid crowding the figure, the ID does not appear as a label. The lowest contour in the figure corresponds to an intensity level of 3 SD of rms noise.
For future studies, ROIs defined from earlier studies can be imported and graphically adjusted to align with the local spectra.
Fast maximum-likelihood reconstruction (FMLR)
The detailed theory and equations for applying the maximum-likelihood method to analysis of NMR data have been reported previously [19, 20], and most recently for the analysis of 2D 1H–13C data sets in a metabolomics context [17]. The specific steps for performing spectral deconvolution of the Arabidopsis data in this study consisted of:
-
1.
Prototype Signal Generation: An isolated signal was graphically selected by the operator as an archetypal signal. The signal giving rise to the peak was fitted using a model whose basis functions and model parameters are specified in Table 2. The decay rate (linewidths) obtained from this optimization were used as initial values for further modeling. For the Arabidopsis study, the prototype linewidth was 80 Hz along both the 1H and 13C dimensions.
-
2.
Constraint Specification: The FMLR algorithm uses constraints on linewidth to assist in convergence of the fitting algorithm in crowded spectral areas. Linewidth constraints are specified as a multiple of the prototype linewidth along each dimension. For the study reported here, the linewidth was constrained to be a factor of 1/2 to 2 relative to the prototype linewidth, i.e., 40–160 Hz.
-
3.
Choosing Noise Thresholds: During spectral deconvolution (see below), signals are added incrementally in a series of iterations. Initially the pick threshold is set to the maximum peak height and is then reduced geometrically by a factor of at the conclusion of each iteration. The analysis algorithm is terminated when the pick threshold reaches a minimum value specified as a multiple of signal-to-noise. The S/N threshold for this study was 4.0.
-
4.
Spectral Deconvolution: To avoid modeling extraneous features of the spectrum, only those peaks in a spectrum contained within at least one ROI were modeled by spectral deconvolution. Spectral deconvolution was initiated after steps 1–3 above and continued without operator intervention for a series of 10 iterations that yielded 22,389 signals (5 × 22,389 = 111,945 total parameters) across the 98 data sets. The total duration time of the analysis was 28 minutes on an off-the-shelf Pentium laptop [AMD Phenom II N870 Triple-Core Processor 2.3 GHz, 6.0 GB RAM, Windows 7 SP 1 2009 64 bit OS, Java 1.6.0_25_b06 with Java Hot Spot (TM) 64 bit server virtual machine].
ROI assignment and feature matrix generation
A signal was assigned to a target ROI if its peak center existed within the boundaries of that ROI. When a source peak is contained within more than one target ROI (i.e., two or more target ROIs overlap), the Newton assignment algorithm assigns the source peak to the target ROI with the greatest “gravity metric” (product of source peak and target peak intensities divided by the spectral distance between the source and target peak summed over all target peaks).
The amplitude of each ROI was calculated as the simple sum of all signal amplitudes (obtained from spectral deconvolution) assigned to that ROI. From this information, a “feature matrix” can be constructed of a 2D n r × n s matrix where n r is the number of regions of interest and n s is the number of spectra.
ROI normalization
After generation of the feature matrix, which can be imported into any standard spreadsheet program (csv file format), the value of each ROI amplitude (i.e., the sum of amplitudes of all signals located within the region of interest) was normalized by a value L representing lignin content in the spectrum. The value L is the weighted sum of integrals of the following ROI amplitudes:
Where [S 2/6], [S'2/6], [G 2], [G'2], [H 2/6] represent the ROI amplitudes in regions corresponding to the S (syringyl), G (guaiacyl), and H (p-hydroxyphenyl) lignin types [See also Figure 1A]. The coefficients are derived from the relative ratio of proton/carbon pairs assigned to the spectral regions. This normalization step produces a meaningful metric (i.e., as a fraction of lignin content in the sample) for reporting the amplitudes of cell wall components. The normalization operation was performed within a spreadsheet program (Microsoft Excel).
For spectra in which an internal standard (e.g., DSS or formate) is present at a fixed concentration (not shown here), the software also supports normalization by the intensity of the ROI associated with the internal standard.
Statistical data analysis
Differences in ROI amplitudes between Arabidopsis mutant lines and a wild type were analyzed with analysis of variance using the glm procedure of the SAS/STAT software, Version 9.3 of the SAS System for windows. Copyright © 2011, SAS Institute Inc., Cary, NC, USA. P-values were adjusted for multiple testing using the Dunnett approach. All reported significant differences are at the overall α level of 0.05.
Data visualization
All of the contour plots contained in the figures here were rendered by Newton and exported in the vector-based format of encapsulated postscript (EPS). Annotations were added using Adobe Illustrator. Bar charts and similar graphics comparing ROI amplitudes were produced by Microsoft Excel and SAS.
Software availability
The software application can be downloaded and run from instructions found at http://newton.nmrfam.wisc.edu/. The host machine must have an installed version of the Java Runtime Environment (JRE) v1.6+ to run the application; Microsoft Windows, Apple MacOS, and various Linux implementations are all supported.
Results and discussion
Region of interest specification
After processing the spectra and creating the ensemble, a set of 91 ROIs were specified as 2D rectangles along the 1H and 13C axis as shown in the lignin, lignin-polysaccharide, and polysaccharide-anomeric regions of Figure 1A-C. The spectral regions shown in each figure were obtained from a selected spectrum from the wild-type sample group of Arabidopsis. The boundaries were graphically drawn to segment the spectrum into clusters of signals that are resolved from one another (although the signals within a cluster may be only partially resolved). Assignments of plant cell wall components from previous studies [10, 11] using model compounds were used to assign 52 of the 91 ROIs (see Figure 1A-C). Once specified for a given study, a ROI table can be exported and imported into other studies with minimal adjustment.
Spectral deconvolution by FMLR
A mathematical model of all signals present in the spectral ensemble was obtained by spectral deconvolution using fast maximum likelihood reconstruction (see FMLR section of methods for details). Signals present in an ROI were modeled if the height of the residual peak was at least 4.0 standard deviations (SD) above the measured root-mean-square (rms) noise of the ensemble. Peaks outside of any ROI were ignored. Each signal was modeled with five parameters: a scalar amplitude, a frequency along each dimension, and a decay rate (linewidth) along each dimension. The final statistics associated with the deconvolution are summarized in Table 3.
The data, model, and residual of spectra from the complex lignin-side-chain plus polysaccharide region of a wild-type sample are shown in Figure 2. Each marker in the figure denotes the center of a signal obtained from spectral deconvolution. Evidence for the suitability of the model to account for major features of the data is that a minimal number of observed signals yields a reconstructed model with a small associated residual (difference between the data and the model). As evident from the figure plotted at a threshold intensity of 3.0 SD, there are few signals in the residual with a peak threshold greater than 3.0 SD (SD of rms noise).

Processed spectrum (data), FMLR reconstruction (model), and residual of the aromatic (A) and polysaccharide (B) region of the 2D 1H–13C HSQC for a wild-type sample of Arabidopsis . The color of a contour is assigned to the color of the ROI associated with the dominant signal in that region. As can be seen in the figure, a minimal number of reconstructed signals is required to yield a model with an associated residual that is less than the noise floor (noise floor = 3.0 SD). The set of contours near (3.6, 76) ppm and (4.7, 63) ppm in (B) are not reflective of poor modeling but are a consequence of the fact that no ROI was defined near those positions. Signals in that region of the spectrum were simply not modeled.
Feature set of ROI amplitudes
The generation of a meaningful “feature set” of ROI-based amplitudes from FMLR is straightforward. Each peak was automatically assigned to an ROI based on whether its peak center was located within a given ROI (see ROI Assignment section of methods). The amplitude of an ROI was calculated as the simple sum of all signal amplitudes assigned to that ROI. To provide a more meaningful comparison of ROI amplitudes between sample groups, each ROI amplitude was normalized by total lignin content (see ROI Normalization section of Methods). This normalized ROI amplitudes per spectrum results in a feature matrix of 91 ROI amplitudes × 98 spectra (available from Additional Information).
SGH lignin composition
The relative composition of S (syringyl), G (guaiacyl), and H (p-hydroxyphenyl) lignin units is an important element of plant cell wall profiling. The spectral data associated with the SGH ROIs for the sample groups in the study (averaged over all spectra per mutant sample group) is shown as a series of contour plots in Figure 3. In discerning whether relative percentages of SGH lignin are modulated across the sample groups, the bar chart of Figure 4 provides a graphical view of the normalized profiles obtained from the SGH portion of the ROI feature matrix. Differences in S, G, and H percentages between the Arabidopsis mutant lines and the wild-type together with Dunnett adjusted p-values are given in Table 3. The overall pattern of enrichment and depletion in the mutant sample groups compared to the wild-types is displayed in the bar chart of Figure 5 where 3 patterns are evident: i) increase of H and S relative to G (c4h, 4cl1, ccoaomt1); ii) increase of H relative to S (ccr1), and iii) depletion of S relative to G (f5h1 and comt). These results are confirmed by thioacidolysis on the same set of Arabidopsis lignin mutants and are published concomitantly [23].

Contour plots of 2D 1H–13C HSQC spectral regions associated with signals assigned to the S′2/6, S2/6, G′2, G2, G5/6, and H2/6 transitions. The data shown represent the mean spectra of all samples belonging to each sample group (number of spectra for each sample group shown in parentheses). The color of each contour is assigned based on the FMLR reconstructions, i.e., the dominant signal associated with each grid point is used to assign a color to that pixel (and related contour). The contour plots show the ability of the reconstructions to discriminate between assigned (colored) and unassigned (black) signals that partially overlap.

Bar charts of the mean normalized percentages of S (syringyl), G (guaiacyl), and H ( p -hydroxyphenyl) lignin units with their standard errors and number of observations (in parentheses). The values are derived from the ROI feature matrix in which each ROI amplitude is the sum of the amplitude of all modeled signals assigned to that ROI (derived from FMLR, see text for details).

Bar chart showing pattern of enrichment and depletion of S (syringyl), G (guaiacyl), and H ( p -hydroxyphenyl) lignin levels (normalized percentages) per sample group. The pal and cad6 mutants (not shown) showed no significant difference to wild-type. The displayed levels represent the mean predicted difference between each sample group and the effective wild-type sample group.
When comparing %S, %G, and %H changes between the mutant groups and wild-type groups, the corresponding p-values are all < 0.0001 (Table 3) for any change greater than 4% (Table 3). The differences are in general larger in magnitude for patterns detected with FMLR reconstruction (Table 3A) versus ROI integration (Table 3B).
Correlation of ROI changes to SGH modulation
To assess which ROIs might be correlated with the SGH patterns, Pearson correlations were calculated between all ROI amplitudes and the lignin compounds G2, G′2, S2/6, S′2/6, and H2/6. LA-Sβ was highly positively correlated to S2/6 (r = 0.94, p < 0.0001) and S′2/6 (r = 0.94, p < 0.0001) and highly negatively correlated to G2 (r = -0.88, p < 0.0001). LA-Sβ is assigned specifically to β-syringyl ethers and therefore relates to the S-G distribution, being obviously lower when the S-content is lower. LB α is highly positively correlated to G2 (r = 0.82, p < 0.0001). The LB α region is assigned to phenylcoumaran (β–5) units in lignins. Such units arise from coupling of a monolignol (at its β-position) with a guaiacyl G (or H) unit (at its 5-position), but not a syringyl unit (which has the 5-position blocked with a methoxyl group); thus levels are higher when relative syringyl levels are lower (S/G is lower). The correlations are visualized in Figure 6. Such correlations or associations can be powerful aids in enhancing our assignment capabilities in these complex cell wall samples. For example, the profile of two of the unassigned regions (ROI55 and ROI66) in the lignin region of the spectrum (Figure 1A) are highly positively correlated with H2/6 (r = 0.93, p < 0.0001 for both).

Bar charts reflecting the correlations between the ROIs and the Arabidopsis mutant lines.
Conclusions
The spectral dispersion inherent in 2D 1H–13C HSQC renders ROI segmentation methods useful for semi-quantitative studies of complex biological systems [21, 22]. The profile of any single cross peak in the spectrum is linearly proportional to the concentration of the underlying species giving rise to the resonance. The term “semi-quantitative” is used here because the amplitude of different cross peaks in the 2D 1H–13C HSQC spectrum is not strictly comparable due to a range of factors relating to NMR methods themselves, and to the properties of the various polymers. For example the finite RF power available on the carbon channel in proton-carbon correlation experiments leads to non-uniform excitation of carbon resonances across the spectrum, although this is somewhat ameliorated by using adiabatic-pulse experiments [26]. If the experiment permits longer acquisition times, a range of quantitative 2D HSQC experiments [27, 28] have been developed to mitigate this artifact.
We provide evidence here using a sizeable mutant study that FMLR reconstruction is useful both for rapid profiling of plant cell wall material and in improving the accuracy of conventional ROI segmentation methods for analysis of NMR spectra. The approach of generating a frequency domain spectrum from Fourier processing of a model time domain signal was used to reconstruct a model spectrum with close agreement to the processed data (Figure 2) using a small number of signals (degrees of freedom). An analysis of variance (ANOVA) in the SGH regions of the ROI feature matrix between pairs of mutant and wild-type sample groups yielded differences larger in magnitude using ROI segmentation coupled with FMLR reconstruction than with simple ROI integration alone. The difference between fixed-window integration techniques and spectral deconvolution is expected to be more pronounced in heterogeneous systems that display broad line widths such as in ball-milled preparations of plant cell wall material.
Even more significant is that assignment of ROIs to a mathematical model of the data rather than the data itself makes subsequent quantification less sensitive to changes in ROI definition. When modeled mathematically, the entire amplitude of a signal is assigned to an ROI as long as the peak center associated with the signal is encapsulated by the ROI. With direct integration of the spectrum itself, however, the ROI amplitude values are always modulated by changing the size or position of the ROI. This is an important consideration for general profiling using ROI segmentation because ROIs can be reused between studies with a minimal amount of adjustment (e.g., a constant ppm shift applied across all ROIs).
A strength of ROI segmentation methods is that prior information about spectral assignments can be used but is not required for profiling. In plant cell wall profiling, for example, the assignment of the lignin components is important not only in calculating SGH composition but also as a means of normalizing cross peaks from other regions of the spectrum. Even if a cluster of peaks is not assigned, the cluster may be associated with a region of interest and profiled across sample groups.
Conventional approaches create a feature set using spectral binning and then apply multivariate techniques to detect patterns among features across sample groups. The feature set of such an analysis is large and must eventually be related to a molecular species for targeted studies. This study provides an example of detecting patterns of enriched and depleted cell wall components using simple one-way ANOVA techniques directly on a meaningful feature set.
The analysis methodology has been implemented in a publicly-available, cross-platform (Windows/Mac/Linux), web-enabled software application (http://newton.nmrfam.wisc.edu) that enables researchers to view and publish detailed annotated spectra in addition to summary reports in standard csv formats. The csv format of the ROI feature matrix, for example, can be directly imported into dedicated software packages for metabolomic data processing and statistical analysis such as MetaboAnalyst 2.0 (http://www.metaboanalyst.ca) [29], as well as general statistical packages such as R (http://www.r-project.org/) and Matlab (http://www.mathworks.com/products/matlab/).
Abbreviations
- 1D:
-
1-dimensional
- 2D:
-
2-dimensional
- 3D:
-
3-dimensional
- 4CL:
-
4-coumarate: CoA ligase
- 5-OH-G:
-
5-hydroxy-guaiacyl
- ANOVA:
-
Analysis of variance
- C3H:
-
p-coumarate 3-hydroxylase
- C4H:
-
Cinnamate 4-hydroxylase
- CAD:
-
Cinnamyl alcohol dehydrogenase
- CCoAOMT:
-
Caffeoyl-CoA O-methyltransferase
- COMT:
-
Caffeic acid O-methyltransferase
- CCR:
-
Cinnamoyl-CoA reductase
- DMSO:
-
Dimethyl-sulfoxide (-d6)
- DOE:
-
(US) Department of energy
- DP:
-
Degree of polymerization
- DSS:
-
4,4-dimethyl-4-silapentane-1-sulfonic acid (NMR standard)
- EPS:
-
Encapsulated postscript
- F5H:
-
Ferulate 5-hydroxylase
- FID:
-
Free induction decay
- FMLR:
-
Fast maximum likelihood reconstruction
- G:
-
Guaiacyl
- H:
-
p-hydroxyphenyl
- HCT:
-
p-hydroxycinnamoyl-CoAquinate/shikimate p:-hydroxycinnamoyltransferase
- HSQC:
-
Heteronuclear single-quantum coherence (spectroscopy)
- NMR:
-
Nuclear magnetic resonance (spectrometry)
- PAL:
-
Phenylalanine ammonia lyase
- Rms:
-
Root-mean-square
- ROI:
-
Region of interest
- ROIs:
-
Regions of interest
- S:
-
Syringyl
- SD:
-
Standard deviation.
References
Carroll A, Somerville C: Cellulosic Biofuels. Annu Rev Plant Biol 2009, 60: 165-182. 10.1146/annurev.arplant.043008.092125
Ragauskas AJ, Williams CK, Davison BH, Britovsek G, Cairney J, Eckert CA, Frederick WJ, Hallett JP, Leak DJ, Liotta CL: The Path Forward for Biofuels and Biomaterials. Science 2006, 311: 484-489. 10.1126/science.1114736
Boerjan W, Ralph J, Baucher M: Lignin biosynthesis. Annu Rev Plant Biol 2003, 54: 519-546. 10.1146/annurev.arplant.54.031902.134938
Chen F, Dixon RA: Lignin modification improves fermentable sugar yields for biofuel production. Nat Biotechnol 2007, 25: 759-761. 10.1038/nbt1316
Davison BH, Drescher SR, Tuskan GA, Davis MF, Nghiem NP: Variation of S/G ratio and lignin content in a Populus family influences the release of xylose by dilute acid hydrolysis. Appl Biochem Biotechnol 2006, 130: 427-435. 10.1385/ABAB:130:1:427
Vanholme R, Morreel K, Ralph J, Boerjan W: Lignin biosynthesis and structure. Plant Physiol 2010, 153: 895-905. 10.1104/pp.110.155119
Lu F, Ralph J: Non-degradative dissolution and acetylation of ball-milled plant cell walls: high-resolution solution-state NMR. Plant J 2003, 35: 535-544. 10.1046/j.1365-313X.2003.01817.x
Kim H, Ralph J, Akiyama T: Solution-state 2D NMR of ball-milled plant cell wall gels in DMSO-d 6 . BioEnergy Research 2008, 1: 56-66. 10.1007/s12155-008-9004-z
Ralph J, Lu F: Cryoprobe 3D NMR of acetylated ball-milled pine cell walls. Org Biomol Chem 2004, 2: 2714-2715. 10.1039/b412633e
Kim H, Ralph J: Solution-state 2D NMR of ball-milled plant cell wall gels in DMSO-d 6 /pyridine-d 5 . Org Biomol Chem 2010, 8: 576-591. 10.1039/b916070a
Mansfield SD, Kim H, Lu F, Ralph J: Whole plant cell wall characterization using solution-state 2D NMR. Nat Protocols 2012, 7: 1579-1589. 10.1038/nprot.2012.064
Craig A, Cloarec O, Holmes E, Nicholson JK, Lindon JC: Scaling and normalization effects in NMR spectroscopic metabonomic data sets. Anal Chem 2006, 78: 2262-2267. 10.1021/ac0519312
Fengel D, Wegener G: Wood: chemistry, ultrastructure, reactions. Berlin-New York: Walter De Gruyter; 2003.
Hedenström M, Wiklund-Lindström S, Öman T, Lu F, Gerber L, Schatz P, Sundberg B, Ralph J: Identification of Lignin and Polysaccharide Modifications in Populus Wood by Chemometric Analysis of 2D NMR Spectra from Dissolved Cell Walls. Mol Plant 2009, 2: 933-942. 10.1093/mp/ssp047
Nicholson JK, Lindon JC, Holmes E: ‘Metabonomics’: understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29: 1181-1189. 10.1080/004982599238047
Webb-Robertson BJ, Lowry DF, Jarman KH, Harbo SJ, Meng QR, Fuciarelli AF, Pounds JG, Lee KM: A study of spectral integration and normalization in NMR-based metabonomic analyses. J Pharm Biomed Anal 2005, 39: 830-836. 10.1016/j.jpba.2005.05.012
Chylla RA, Hu K, Ellinger JJ, Markley JL: Deconvolution of two-dimensional NMR spectra by fast maximum likelihood reconstruction: application to quantitative metabolomics. Anal Chem 2011, 83: 4871-4880. 10.1021/ac200536b
Hu K, Ellinger JJ, Chylla RA, Markley JL: Measurement of Absolute Concentrations of Individual Compounds in Metabolite Mixtures by Gradient-Selective Time-Zero 1H-13C HSQC with Two Concentration References and Fast Maximum Likelihood Reconstruction Analysis. Anal Chem 2011, 83: 9352-9360. 10.1021/ac201948f
Chylla RA, Markley JL: Theory and application of the maximum likelihood principle to NMR parameter estimation of multidimensional NMR data. J Biomol NMR 1995, 5: 245-258.
Chylla RA, Volkman BF, Markley JL: Practical model fitting approaches to the direct extraction of NMR parameters simultaneously from all dimensions of multidimensional NMR spectra. J Biomol NMR 1998, 12: 277-297. 10.1023/A:1008254432254
Lewis IA, Schommer SC, Markley JL: rNMR: open source software for identifying and quantifying metabolites in NMR spectra. Magn Reson Chem 2009, 47: s123-s126. 10.1002/mrc.2526
Lewis IA, Schommer SC, Hodis B, Robb KA, Tonelli M, Westler WM, Sussman MR, Markley JL: Method for determining molar concentrations of metabolites in complex solutions from two-dimensional 1H-13C NMR spectra. Anal Chem 2007, 79: 9385-9390. 10.1021/ac071583z
Van Acker R, Vanholme R, Storme V, Mortimer J, Dupree P, Boerjan W: Perturbation of lignin biosynthesis in Arabidopsis thaliana affects secondary cell wall composition and saccharification yield. Biotechnol Bioenergy 2013, 6: 45.
Vanholme R, Storme V, Vanholme B, Sundin L, Christensen JH, Goeminne G, Halpin C, Rohde A, Morreel K, Boerjan W: A systems biology view of responses to lignin biosynthesis perturbations in Arabidopsis . Plant Cell 2012, 24: 3506-3529. 10.1105/tpc.112.102574
Yelle DJ, Ralph J, Frihart CR: Characterization of nonderivatized plant cell walls using high-resolution solution-state NMR spectroscopy. Magn Reson Chem 2008, 46: 508-517. 10.1002/mrc.2201
Kupče E, Freeman R: Compensated adiabatic inversion pulses: Broadband INEPT and HSQC. J Magn Reson 2007, 187: 258-265. 10.1016/j.jmr.2007.05.009
Hu KF, Westler WM, Markley JL: Simultaneous quantification and identification of individual chemicals in metabolite mixtures by two-dimensional extrapolated time-zero 1H-13C HSQC (HSQC 0 ). J Am Chem Soc 2011, 133: 1662-1665. 10.1021/ja1095304
Koskela H, Heikkilä O, Kilpeläinen I, Heikkinen S: Quantitative two-dimensional HSQC experiment for high magnetic field NMR spectrometers. J Magn Reson 2010, 202: 24-33. 10.1016/j.jmr.2009.09.021
Xia J, Mandal R, Sinelnikov IV, Broadhurst D, Wishart DS: MetaboAnalyst 2.0—a comprehensive server for metabolomic data analysis. Nucleic Acids Res 2012, 40: W127-W133. 10.1093/nar/gks374
Acknowledgements
This work was supported by the US Department of Energy’s Great Lakes Bioenergy Research Center (DOE Office of Science BER DE-FC02-07ER64494) and the Ghent University’s Multidisciplinary Research Partnership ‘Biotechnology for a Sustainable Economy’. RVA is indebted to the Agency for Innovation by Science and Technology (IWT) for a pre-doctoral fellowship. We are grateful to Gustav Sundqvist and Prof. Vincent Bulone for preparing the cell wall materials from all samples that were used in this study; these were used for preparing the NMR samples.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RAC developed the FMLR approach, its application to cell wall profiling, implemented the Newton software, and wrote the manuscript, RVA produced the Arabidopsis mutants, HK developed and ran the whole-CW NMR methodology, AA processed and prepared the NMR data in the required format, PM prepared and assisted with running the samples, JLM supervised RAC, VS aided in statistical analysis, WB conceived the Arabidopsis project and supervised RVA, and JR supervised PM, AA, and HK and aided in the analysis of the NMR spectra, manuscript writing, etc. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Chylla, R.A., Van Acker, R., Kim, H. et al. Plant cell wall profiling by fast maximum likelihood reconstruction (FMLR) and region-of-interest (ROI) segmentation of solution-state 2D 1H–13C NMR spectra. Biotechnol Biofuels 6, 45 (2013). https://doi.org/10.1186/1754-6834-6-45
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1754-6834-6-45