
Abstract
Throughout immeasurable time, microorganisms evolved and accumulated remarkable physiological and functional heterogeneity, and now constitute the major reserve for genetic diversity on earth. Using metagenomics, namely genetic material recovered directly from environmental samples, this biogenetic diversification can be accessed without the need to cultivate cells. Accordingly, microbial communities and their metagenomes, isolated from biotopes with high turnover rates of recalcitrant biomass, such as lignocellulosic plant cell walls, have become a major resource for bioprospecting; furthermore, this material is a major asset in the search for new biocatalytics (enzymes) for various industrial processes, including the production of biofuels from plant feedstocks. However, despite the contributions from metagenomics technologies consequent upon the discovery of novel enzymes, this relatively new enterprise requires major improvements. In this review, we compare function-based metagenome screening and sequence-based metagenome data mining, discussing the advantages and limitations of both methods. We also describe the unusual enzymes discovered via metagenomics approaches, and discuss the future prospects for metagenome technologies.
Similar content being viewed by others
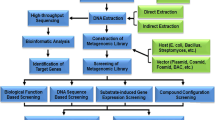

Background
In recent years, biofuels have attracted great interest as an alternative, renewable source of energy in the face of the ongoing depletion of fossil fuels, our energy dependence on them, and our growing environmental awareness of the critical consequences of burning such fuels. Plant biomass, the most abundant biopolymer on earth, has long been recognized as a potential sustainable source of mixed sugars for biofuel production. However, breakthrough technologies are still needed to overcome the several barriers to developing cost-effective processes for converting biomass to fuels and chemicals [1]. As yet, we have an incomplete understanding of the plant cell wall and its deconstruction and conversion; considerable research will be needed to better appreciate the fundamental and applied aspects of enzymatic hydrolysis and microbial hydrolysis and/or fermentation of plant cell walls.
Estimates suggest that approximately 4–6 × 1030 prokaryotes inhabit the earth [2]. Being the oldest life form, prokaryotic microorganisms have evolved and accumulated remarkable physiological and functional diversity, thereby constituting the world's major reserve of genetic diversity. Traditional methods to tap this information are by cultivating the microorganisms, subsequently screening individual ones for the requisite phenotypes. However, about 95% to 99.9% of microorganisms have not been cultured by standard laboratory techniques [3]. One way to overcome this limitation is by improving cultivation-based methodologies [4, 5].
As a cultivation independent approach, Pace and colleagues [6] proposed a way to isolate directly the collective genomes from all microorganisms in a given habitat, and, in 1991, Schmidt et al. [7] undertook the first metagenome-based community characterization on amplified 16S rRNA genes. The subsequent improvement of sequencing technologies made feasible the metagenome shot-gun sequencing of environmental samples; however, most environmental communities are far too complex to be fully sequenced in this manner. Initial attempts were made to reconstruct the metagenomes of viral communities in the ocean and human feces [8–10], and subsequently of samples from the Sargasso Sea [11] and a biofilm from an acid mine drainage (AMD) [12]. However, since most marine communities are far richer in species diversity than the AMD biofilm, on the order of 100 to 200 species per milliliter of water [13, 14], this further complicated their sequencing and assembly. Soil communities are even more complex, with an estimated species richness of about 4,000 species per gram of soil [13–15]. On the other hand, with recent developments in high-throughput sequencing technologies, such as the 454 pyrosequencing (GS FLX Titanium Series, 454 Life Science, Roche) partly mitigating this problem, metagenomics is becoming an increasingly sophisticated approach to the study of complex DNA samples directly isolated from defined habitats [16]. According to the Genomes OnLine Database (GOLD) [17] until January 2009, 137 metagenomics projects were in various stages of sequencing, 72% of which were derived from environmental samples, 23% from endobiotic samples, along with 5% synthetic metagenomes. Forty-six of these projects were completed; data are available on the website Integrated Microbial Genomes with Microbiome Samples [18]
Here, we review some recent metagenomic approaches to mining complex microbial communities, comprising both cultivable and non-cultivable microorganisms, for novel biocatalytic enzymes, such as glycosyl hydrolases (GHase) for industrial uses and biofuel production. We also discuss the advantages and limitations of the strategies and tools developed for targeted screening, as well as the future prospects of metagenomics in bioprospecting for new enzymes.
Strategies for target-gene enrichment
In principle, directly isolating metagenomic DNA from the environment implies unbiased genomic representation; however, biases are introduced during its isolation, for example, resulting from differences in cell lyses. In searching for relatively under-represented genes, enrichment can increase the probability of their cloning, and hasten the process of discovering new genes. By exposing microbial communities to a selective pressure expected to entail the enrichment of microorganisms displaying the desired phenotypes (including substrate utilization, physical-, chemical-, and nutritional-selective conditions), the numbers of those community members with the desired phenotypes and corresponding target genes are successfully boosted. For example, using DNA isolated from enrichment cultures grown on cellulose as their major carbon source increased from three- to four-fold the isolation of GHase with cellulase activity from metagenome libraries, compared with the isolates from libraries made directly from total environmental DNA [19]. Also, we can remove eukaryotic community members by size-selective filtration, leaving behind enriched prokaryotic and archaeal populations [11]. Other enrichment techniques include stable isotope probing, affording a means to isolate microorganisms actively metabolizing the substrate and undergoing replication [20, 21], suppressive subtractive hybridization [22–24], differential expression analysis [25], phage display, and affinity capture (reviewed by Cowan et al. [26]).
Strategies for prospecting novel enzymes from metagenomes
Having isolated metagenomic DNA, two complementary approaches can be used for prospecting novel enzymes from it; function-based screening of expression libraries and sequence-based gene searches. In the former, metagenomic expression libraries are constructed and screened for target enzyme activities. For the latter, target genes are cloned after being amplified from metagenomic DNA by using polymerase chain reaction with conserved sequences as primers; alternatively, they may be directly discovered from metagenome sequence databases using bioinformatics tools, subsequently amplified, and cloned in the appropriate expression systems. Below, we detail these two approaches.
Metagenome expression libraries (function-based screening)
Metagenome expression libraries are constructed by inserting fragmented metagenomic DNA into expression vectors based on plasmids, cosmids, fosmids, or phages, after which gene expression is examined in a suitable host system. The advantage of directly screening for enzymatic activities from metagenome libraries is that researchers access previously unknown genes and their encoded enzymes. Furthermore, the sequences and enzyme activities are functionally guaranteed. However, some potential drawbacks compromise this approach. Thus, before a clone correctly expresses an active enzyme, several requirements must be met. First, when functional enzymatic activity depends on more than one genetic subunit, the clone must contain the complete gene sequence, or even a gene cluster. This problem can be resolved by selecting suitable vector systems. For small target genes, DNA fragment libraries with inserts between 2 and 10 kilobase (kb) are constructed in plasmids or Lambda expression vectors, and then screened for enzyme expression. Larger gene clusters, preferentially necessitate expression libraries with inserts between 20 and 40 kb in cosmids and fosmids, and up to 100 to 200 kb in bacterial artificial chromosome vectors. Although common E. coli host strains have relaxed requirements for promoter recognition and translation initiation, many genes from environmental samples may not be expressed efficiently in heterologous hosts due to differences in codon usage, transcription and/or translation initiation signals, protein-folding elements, post-translational modifications, such as glycosylation, or toxicity of the active enzyme. This obstacle is overcome partly by selecting suitable vector systems containing apposite transcription and translation-initiation sequences, and using suitable expression hosts, such as the E. coli Rosetta strains (Novagen, Madison, Wisconsin, USA) that contain the tRNA genes for rare amino acid codons [27], or co-expression of the chaperone proteins, such as GroES, GroEL, and heat-shock proteins [28, 29]. Alternatively, host systems such as insect cells, the yeast Pichia pastoris, and bacterial hosts such as Pseudomonas putida, Streptomyces lividans, or Bacillus subtilis were suitably improved for heterologous gene expression [30]. Furthermore, several modified function-based methods exist specifically for exploring metagenome libraries. Thus, Uchiyama and colleagues [31] developed substrate-induced gene-expression screening to rapidly identify clones that can be induced by a target substrate and display catabolic gene expression, while metabolite-regulated expression detects clones generating quorum-sensing gene-inducing compounds [32].
Function-based metagenome library screening has uncovered a wide range of biocatalysts. Here, we highlight several published results that screened for polysaccharide and plant cell wall biomass-degrading enzymes, most belonging to GHase families. In most cases, colorimetric-based analyses on agar plates employing dye-linked substrates or reaction products staining were used for preliminary screening. Candidate clones were then confirmed by enzyme activity assays.
Amylases attract much industrial interest and are the focus of many metagenome studies. Richardson et al. [33], Voget et al. [34], Yun et al. [35], and Lämmle et al. [36] detailed novel amylolytic enzyme activities from metagenome libraries; some of these enzymes were purified and characterized [33, 35]. Cellulose is nature's most abundant biopolymer, and long has been recognized as a potential source of sugars for biofuel production. Voget and colleagues [37] obtained several cellulolytic clones by functionally screening a soil metagenome library from which they purified and characterized a cellulase. Rees et al. [38] screened a lake water metagenome library and retrieved four cellulolytic clones. From a metagenome library representing the microbial community present in the rabbit's cecum, several clones with cellulose activities were revealed [39]. Functional screening of metagenome libraries from extreme (high salinity and alkalinity) environmental samples (soil from Soda Lake, California, and lake sediments from Africa and Egypt) also disclosed cellulolytic clones [19]. Cellulolytic enzymes isolated from environments with extreme temperatures and pH values are receiving a lot of interest as these enzymes are expected to be better adapted to the conditions of industrial processes, such as the decomposition of recalcitrant plant cell wall biomass into fermentable sugars.
Chitin, a compound of the fungal cell wall, is the second most abundant natural biopolymer that is broken down by chitinases. Cottrell et al. [40] acquired clones with chitinase activities from metagenome libraries derived from marine samples (filtrated from coastal sea water and estuarine water near the Delaware Bay). Hemicellulose consists primarily of xylan and constitutes the second most abundant polymer in plant biomass. Xylanase activities were detected and expressed from metagenome libraries representing the microbial communities of an insect gut [41], and the waste water from a dairy farm [42].
The ester linkage between the 4-O-methyl-D-glucuronic acid of glucuronoxylan and lignin alcohols is one type of covalent linkage connecting lignin and hemicellulose in plant cell walls. Esterases, which belong to the group of carboxylester hydrolases, hydrolyze such linkages. Esterase activities were detected from metagenome libraries of soil [43, 44], lake water [38], drinking water [43], and the micro flora from bovine rumen [45]. From these libraries, clones with endo-β-1,4-glucanase activity and a clone with cyclodextrinase activity were identified [45]. Agarases are enzymes that liquefy agar by cleaving either the polymer's α-L-(1,3) linkage or its β-D-(1,4) linkage. Voget et al. [34] discovered six agarase genes in a soil metagenome library. The same library yielded two clones with pectate lyase activity, and one clone with 1,4-α-glucan branching enzyme activity [34]. Table 1 summarizes the enzymes discovered via function-based screening, their metagenome origin, and the library types and sizes.
Metagenome sequencing (homology-based identification)
Sequence-based screening methods rely on known conserved sequences, and cannot uncover non-homologous enzymes. Therefore, the drawback of this 'closed approach' is its failure to detect fundamentally different 'new' genes. However, unlike function-based methods, it can disclose target genes, regardless of gene expression and protein folding in the host, and irrespective of the completeness of the target gene's sequence. The success of this approach rests on meeting several conditions:
(1) The more complex the community, the larger must be the sequencing effort. Here, the development of new sequencing technology, such as the next-generation 454-pyrosequencing, has changed the outcome. For instance, one of the first metagenome projects was the exploration of microbial communities in the drainage from acid mines [12], wherein only three bacterial and three archaeal lineages were detected. Nowadays, metagenome projects using new sequencing technologies not only generate greater total base pair reads but also have more even coverage of species within the community [17].
(2) While the metagenomic approach captures representative DNA samples from diverse organisms, many sequence reads remain unassembled due to the variety of sizes of the environmental genomes, and their abundance. Therefore, a shift in focus emerged, from complete metagenome sequencing to bulk sequencing of as many possible genes and/or functions. In this latter approach, where there is less need to assemble sequences into contigs, the limiting factor becomes the lengths of the fragments that can be obtained for high-throughput screening and cloning. Ideally, the fragments must be long enough to contain the full open reading frame for the functions of interest. Accordingly, optimized 454 sequencing (approximately 450 nucleotide (nt) sequence length) looks more promising than extremely high-volume short-run (25 nt) sequencing [46, 47], but still has its limitations for downstream cloning and expression of genes like GHase that vary in length from less than 1 kb to more than 20 kb. Gene-finding tools, such as MetaGene, were demonstrated to predict 90% of shotgun sequences [48].
(3) New bioinformatics tools are needed for data mining, based not only on primary sequence homology but also able to predict protein structures, putative catalytic sites, and activities. With the betterment of protein classification tools, models might be designed to correlate enzyme mechanisms and protein folding. Based on this folding and the creation of putative active sites, gene function can be predicted [49–54]. We anticipate that soon sequence-based metagenome databases searches combined with bioinformatics tools will have a greater influence on mining novel biocatalyst genes than function-based methods.
Several publications describe searching metagenome sequence databases in prospecting for genes and their enzymes that will be useful in biofuel production. For example, in sequencing a metagenome library of hindgut microbiota from the largest family of wood-feeding termites (Termitidae), Warnecke and colleagues [55] generated 71 million base pairs of sequence data. By detecting complete domains using global alignment, they identified more than 700 domains homologous to glycoside-hydrolase catalytic corresponding to 45 different carbohydrate-active enzymes (CAZy) families [56], including a rich diversity of putative cellulases and hemicellulases. Schlüter and colleagues [57, 58] sequenced, using 454-pyrosequencing technology, a metagenome library of the microbial community from the biogas fermenter of an agricultural biogas plant. From among the 141 million base pair sequences generated, bacteria that played dominant roles in methanogenesis and gene-encoding cellulolytic functions were identified from among the Clostridia spp. [57, 58]. In the near future, we anticipate more publications on mining novel biocatalysts using sequence-based metagenome searches.
A survey of available metagenome databases
According to GOLD [17], of the 137 metagenomic projects in the various stages of sequencing, 46 were finished (including 43 projects from 22 different environmental samples and 3 simulated communities), and the resulting data are available through the IMG/M website [18, 56]. By searching through the list of 'genes with Pfam' (the protein family database) from every metagenome on the IMG/M website, our group retrieved 4,874 glycosyl/GHase homologues from these 46 completed metagenome databases. Then, to gain better insight into the diversity and representation of putative glycosyl hydrolases in these metagenomes, we downloaded the databases of translated sequences from all 43 environmental metagenome projects, and blast-searched them against the CAZy sequences for homologues of GHases (van der Lelie et al., unpublished data). As shown in Table 2, using an e value < 10-40 as a cut-off threshold, we recognized 7,338 putative GHase homologues. The table also gives the metagenome size of each environmental sample, the number of homologues, and the number of putative GHases found per million base pairs. Generally, metagenome samples taken from environments that are characterized by a steady input and turnover of complex plant cell wall biomass have an increased abundance of putative GHases: the metagenomes from microbial communities derived from termite, human, and mouse guts displayed more putative GHase homologues (approximately 1.5% total gene count) than those from other samples, such as human oral microflora, uranium-contaminated groundwater or Singapore air sample (approximately 0.3% total gene count). Many of these metagenomic projects originally were targeted on different subjects, such as sulfate reduction, metal tolerance or marine archaeal anaerobic methane oxidation (denoted in descriptions of metagenome sources in Table 2). Table 3 lists the five most abundant GHase families for each environment (except the marine archaeal anaerobic methane-oxidation community that had only three GHase matches on 2.1 million base pairs). In most metagenomes, GHase family 13 represents the most abundant family. Its known activities include the following: α-amylase; pullulanase; cyclomaltodextrin glucanotransferase; cyclomaltodextrinase; trehalose-6-phosphate hydrolase; oligo-α-glucosidase; maltogenic amylase; neopullulanase; α-glucosidase; maltotetraose-forming α-amylase; isoamylase; glucodextranase; maltohexaose-forming α-amylase; maltotriose-forming α-amylase; branching enzyme; trehalose synthase; 4-α-glucanotransferase; maltopentaose-forming α-amylase; amylosucrase; sucrose phosphorylase; malto-oligosyltrehalose trehalohydrolase; isomaltulose synthase; and, amino acid transporter. The next most abundant is GHase family 23 (lysozyme type G; peptidoglycan lyase; also known in the literature as peptidoglycan lytic transglycosylase). Additionally, we found that members of the GHase family 2 (β-galactosidase; β-mannosidase; β-glucuronidase; mannosylglycoprotein endo-β-mannosidase; exo-β-glucosaminidase), and GHase family 3 (β-glucosidase; xylan 1,4-β-xylosidase; β-N-acetylhexosaminidase; glucan 1,3-β-glucosidase; glucan 1,4-β-glucosidase; exo-1,3-1,4-glucanase; α-L-arabinofuranosidase) are abundant in most environments. In fact, GHase family 13 (also known as the α-amylase family) is the largest sequence-based family of GHases, and encompasses several different enzyme activities and substrate specificities acting on α-glycosidic bonds. This might be a reason why GHase family 13 seemingly is the dominant family in most metagenomes. Clearly, homology, enzyme activity, and substrate specificity are not always well linked for GHases of the same family, thereby highlighting one weak point of homology-based screening for new GHase activities. Better classification and functional prediction of GHases should improve future bioprospecting of new ones for biofuel production.
Future prospects
(i) Development of high through-put screening methods
Although the new ultra-fast sequencing technologies quickly generate a remarkable number of target gene candidates, functional assays are still needed to confirm them. Assays for protein function represent one of the most reliable and irreplaceable tools for mining target genes, and, therefore, developing high through-put functional screening methods is a priority for reducing the time exhausted in primary screening. Furthermore, such future screening methods might valuably be combined with other technologies, such as micro-arrays, biosensors, or proteomics tools to accelerate the discovery of new biocatalyst genes.
(ii) Advances in bioinformatics tools
The metagenomics approach provided valuable insight into a full range of microbial diversity in the environment, regardless of their cultivability. However, the complexity of microbial species, together with the limitations of the technology to cover fully whole genome sequences of every species present still pose a great challenge for metagenome research. A few bioinformatics programs are established for assembling and binning metagenome sequences, for gene prediction and annotation, estimating community composition, and data management (see Kunin et al. [60] for review). In addition, the European Union-funded 'MetaFunctions' project [61] also covers the development of 'metagenomes Mapserver', a data-mining system that correlates genetic patterns in genomes and metagenomes with contextual environmental data. Nevertheless, more innovative and sophisticated bioinformatics tools must be devised to assure continued valuable progress in the field of metagenomics.
Conclusion
With the depletion of fossil fuels and growing environmental awareness, bioenergy production from renewable, non-food resources more and more enters into public focus. The natural gene diversity and complexity found in metagenomes is remarkable, affording us an ideal resource for mining of novel biocatalytics that efficiently break down recalcitrant plant biomass into fermentable sugars for generating biofuels and other chemical commodities. With the development of new biotechnologies and bioinformatics tools, our discovery of, and access to novel enzymes via metagenomic approaches potentially may significantly contribute to their future economical production from renewable resources.
Abbreviations
- AMD:
-
acid mine drainage
- CAZy:
-
carbohydrate-active enzymes
- GHase:
-
glycosyl hydrolase
- GOLD:
-
Genomes OnLine Database
- IMG/M:
-
Integrated Microbial Genomes with Microbiome Samples
- kb:
-
kilobase
- nt:
-
nucleotide.
References
Himmel ME: Biomass Recalcitrance – Deconstructing the Plant Cell Wall for Bioenergy. Oxford: Blackwell Publishing; 2008.
Whitman WB, Coleman DC, Wiebe WJ: Prokaryotes: The unseen majority. Proc Natl Acad Sci USA 1998, 95: 6578-6583. 10.1073/pnas.95.12.6578
Amann RJ, Binder BL, Chisholm SW, Devereux R, Stahl DA: Combination of 16S rRNA targeted oligonucleotide probes with flow-cemetry for analysing mixed microbial populations. Appl Environ Microbiol 1990, 56: 1910-1925.
Zengler K, Toledo G, Rappe M, Elkins J, Mathur EJ, Short JM, Keller M: Cultivating the uncultured. Proc Natl Acad Sci USA 2002, 99: 15681-15686. 10.1073/pnas.252630999
Looser V, Hammes F, Keller M, Berney M, Kovar K, Egli T: Flow-cytometric detection of changes in the physiological state of E. coli expressing a heterologous membrane protein during carbon-limited fedbatch cultivation. Biotechnol Bioeng 2005, 92: 69-78. 10.1002/bit.20575
Pace NR, Stahl DA, Lane DJ, Olsen GJ: The analysis of natural microbial populations by ribosomal RNA sequences. Adv Microb Ecol 1986, 9: 1-55.
Schmidt TM, DeLong EF, Pace NR: Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing. J Bacteriol 1991, 173: 4371-4378.
Breitbart M, Hewson I, Felts B, Mahaffy JM, Nulton J, Salamon P, Rohwer F: Metagenomic analyses of an uncultured viral community from human feces. J Bacteriol 2003, 185: 6220-6223. 10.1128/JB.185.20.6220-6223.2003
Breitbart M, Salamon P, Andresen B, Mahaffy JM, Segall AM, Mead D, Azam F, Rohwer F: Genomic analysis of uncultured marine viral communities. Proc Natl Acad Sci USA 2002, 99: 14250-14255. 10.1073/pnas.202488399
Breitbart M, Felts B, Kelley S, Mahaffy JM, Nulton J, Salamon P, Rohwer F: Diversity and population structure of a near-shore marine-sediment viral community. Proc Biol Sci 2004, 271: 565-574. 10.1098/rspb.2003.2628
Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, Wu D, Paulsen I, Nelson KE, Nelson W, Fouts DE, Levy S, Knap AH, Lomas MW, Nealson K, White O, Peterson J, Hoffman J, Parsons R, Baden-Tillson H, Pfannkoch C, Rogers YH, Smith HO: Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304: 66-74. 10.1126/science.1093857
Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF: Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 2004, 428: 37-43. 10.1038/nature02340
Curtis TP, Sloan WT: Prokaryotic diversity and its limits: microbial community structure in nature and implications for microbial ecology. Curr Opin Microbiol 2004, 7: 221-226. 10.1016/j.mib.2004.04.010
Curtis TP, Sloan WT, Scannell JW: Estimating prokaryotic diversity and its limits. Proc Natl Acad Sci USA 2002, 99: 10494-10499. 10.1073/pnas.142680199
Torsvik V, Goksøyr J, Daae FL: High diversity in DNA of soil bacteria. Appl Environ Microbiol 1990, 56: 782-787.
Pooja Sharma HK, Mukesh Kumar, Mansi Verma, Kirti Kumari, Shweta Malhotra, Jitendra Khurana RL: From bacterial genomics to metagenomics: concept, tools and recent advances. Indian J Microbiol 2008, 48: 173-194. 10.1007/s12088-008-0031-4
Genomes OnLine Database[http://www.genomesonline.org/]
Integrated Microbial Genomes with Microbiome Samples[http://img.jgi.doe.gov/cgi-bin/m/main.cgi]
Grant S, Sorokin DY, Grant WD, Jones BE, Heaphy S: A phylogenetic analysis of Wadi el Natrun soda lake cellulase enrichment cultures and identification of cellulase genes from these cultures. Extremophiles 2004, 8: 421-429. 10.1007/s00792-004-0402-7
Radajewski S, Ineson P, Parekh NR, Murrell JC: Stable-isotope probing as a tool in microbial ecology. Nature 2000, 403: 646-649. 10.1038/35001054
Kalyuzhnaya MG, Lapidus A, Ivanova N, Copeland AC, McHardy AC, Szeto E, Salamov A, Grigoriev IV, Suciu D, Levine SR, Markowitz VM, Rigoutsos I, Tringe SG, Bruce DC, Richardson PM, Lidstrom ME, Chistoserdova L: High-resolution metagenomics targets specific functional types in complex microbial communities. Nat Biotechnol 2008, 26: 1029-1034. 10.1038/nbt.1488
Akopyants NS, Fradkov A, Diatchenko L, Hill JE, Siebert PD, Lukyanov SA, Sverdlov ED, Berg DE: PCR-based subtractive hybridization and differences in gene content among strains of Helicobacter pylori . Proc Natl Acad Sci USA 1998, 95: 13108-13113. 10.1073/pnas.95.22.13108
Qi M, Nelson KE, Daugherty SC, Nelson WC, Hance IR, Morrison M, Forsberg CW: Novel molecular features of the fibrolytic intestinal bacterium Fibrobacter intestinalis not shared with Fibrobacter succinogenes as determined by suppressive subtractive hybridization. J Bacteriol 2005, 187: 3739-3751. 10.1128/JB.187.11.3739-3751.2005
Galbraith EA, Antonopoulos DA, White BA: Suppressive subtractive hybridization as a tool for identifying genetic diversity in an environmental metagenome: the rumen as a model. Environ Microbiol 2004, 6: 928-937. 10.1111/j.1462-2920.2004.00575.x
Green CD, Simons JF, Taillon BE, Lewin DA: Open systems: panoramic views of gene expression. J. Immunol Methods 2001, 250: 67-79. 10.1016/S0022-1759(01)00306-4
Cowan D, Meyer Q, Stafford W, Muyanga S, Cameron R, Wittwer P: Metagenomic gene discovery: past, present and future. Trends Biotechnol 2005, 23: 321-329. 10.1016/j.tibtech.2005.04.001
Goldman E, Rosenberg AH, Zubay G, Studier FW: Consecutive low-usage leucine codons block translation only when near the 5' end of a message in Escherichia coli . J Mol Biol 1995, 245: 467-473. 10.1006/jmbi.1994.0038
Nishihara K, Kanemori M, Kitagawa M, Yanagi H, Yura T: Chaperone coexpression plasmids: differential and synergistic roles of DnaK-DnaJ-GrpE and GroEL-GroES in assisting folding of an allergen of Japanese cedar pollen, Cryj2, in Escherichia coli . Appl Environ Microbiol 1998, 64: 1694-1699.
Wall JG, Plückthun A: Effects of overexpressing folding modulators on the in vivo folding of heterologous proteins in Escherichia coli . Curr Opin Biotechnol 1995, 6: 507-516. 10.1016/0958-1669(95)80084-0
Martinez A, Kolvek SJ, Yip CLT, Hopke J, Brown KA, MacNeil IA, Osburne MS: Genetically modified bacterial strains and novel bacterial artificial chromosome shuttle vectors for constructing environmental libraries and detecting heterologous natural products in multiple expression hosts. Appl Environ Microbiol 2004, 70: 2452-2463. 10.1128/AEM.70.4.2452-2463.2004
Uchiyama T, Abe T, Ikemura T, Watanabe K: Substrate-induced gene-expression screening of environmental metagenome libraries for isolation of catabolic genes. Nat Biotechnol 2005, 23: 88-93. 10.1038/nbt1048
Williamson LL, Borlee BR, Schloss PD, Guan C, Allen HK, Handelsman J: Intracellular screen to identify metagenomic clones that induce or inhibit a quorum-sensing biosensor. Appl Environ Microbiol 2005, 71: 6335-6344. 10.1128/AEM.71.10.6335-6344.2005
Richardson TH, Tan X, Frey G, Callen W, Cabell M, Lam D, Macomber J, Short JM, Robertson DE, Miller C: A novel, high performance enzyme for starch liquefaction. Discovery and optimization of a low pH, thermostable alpha-amylase. J Biol Chem 2002, 277: 26501-26507. 10.1074/jbc.M203183200
Voget S, Leggewie C, Uesbeck A, Raasch C, Jaeger KE, Streit WR: Prospecting for novel biocatalysts in a soil metagenome. Appl Environ Microbiol 2003, 69: 6235-6242. 10.1128/AEM.69.10.6235-6242.2003
Yun J, Kang S, Park S, Yoon H, Kim MJ, Heu S, Ryu S: Characterization of a novel amylolytic enzyme encoded by a gene from a soil-derived metagenomic library. Appl Environ Microbiol 2004, 70: 7229-7235. 10.1128/AEM.70.12.7229-7235.2004
Lämmle K, Zipper H, Breuer M, Hauer B, Buta C, Brunner H, Rupp S: Identification of novel enzymes with different hydrolytic activities by metagenome expression cloning. J Biotechnol 2007, 127: 575-592. 10.1016/j.jbiotec.2006.07.036
Voget S, Steele HL, Streit WR: Characterization of a metagenome-derived halotolerant cellulase. J Biotechnol 2006, 126: 26-36. 10.1016/j.jbiotec.2006.02.011
Rees HC, Grant S, Jones B, Grant WD, Heaphy S: Detecting cellulase and esterase enzyme activities encoded by novel genes present in environmental DNA libraries. Extremophiles 2003, 7: 415-421. 10.1007/s00792-003-0339-2
Feng YD, Cheng-Jie , Hao Pang, Xin-Chun Mo, Chun-Feng Wu, Yuan Yu, Ya-Lin Hu, Jie Wei, Ji-Liang Tang, Jia-Xun Feng: Cloning and identification of novel cellulase genes from uncultured microorganisms in rabbit cecum and characterization of the expressed cellulases. Appl Microbiol Biotechnol 2007, 75: 319-328. 10.1007/s00253-006-0820-9
Cottrell MT, Moore JA, Kirchman DL: Chitinases from uncultured marine microorganisms. Appl Environ Microbiol 1999, 65: 2553-2557.
Brennan Y, Callen WN, Christoffersen L, Dupree P, Goubet F, Healey S, Hernádez M, Keller M, Li K, Palackal N, Sittenfeld A, Tamayo G, Wells S, Hazlewood GP, Mathur EJ, Short JM, Robertson DE, Steer BA: Unusual microbial xylanases from insect guts. Appl Environ Microbiol 2004, 70: 3609-3617. 10.1128/AEM.70.6.3609-3617.2004
Lee CC, Kibblewhite-Accinelli RE, Wagschal K, Robertson GH, Wong DW: Cloning and characterization of a cold-active xylanase enzyme from an environmental DNA library. Extremophiles 2006, 10: 295-300. 10.1007/s00792-005-0499-3
Elend C, Schmeisser C, Leggewie C, Babiak P, Carballeira JD, Steele HL, Reymond JL, Jaeger KE, Streit WR: Isolation and biochemical characterization of two novel metagenome-derived esterases. Appl Environ Microbiol 2006, 72: 3637-3645. 10.1128/AEM.72.5.3637-3645.2006
Kim Y-J, Choi G-S, Kim S-B, Yoon G-S, Kim Y-S, Ryu Y-W: Screening and characterization of a novel esterase from a metagenomic library. Protein Expr Purif 2006, 45: 315-323. 10.1016/j.pep.2005.06.008
Ferrer M, Golyshina OV, Chernikova TN, Khachane AN, Reyes-Duarte D, Martins Dos Santos VA, Strompl C, Elborough K, Jarvis G, Neef A, Yakimov MM, Timmis KN, Golyshin PN: Novel hydrolase diversity retrieved from a metagenome library of bovine rumen microflora. Environ Microbiol. 2005,7(12):1966-2010. 10.1111/j.1462-2920.2005.00920.x
Dalevi D, Ivanova NN, Mavromatis K, Hooper SD, Szeto E, Hugenholtz P, Kyrpides NC, Markowitz VM: Annotation of metagenome short reads using proxygenes. Bioinformatics 2008, 24: i7-i13. 10.1093/bioinformatics/btn276
Edwards RA, Rodriguez-Brito B, Wegley L, Haynes M, Breitbart M, Peterson DM, Saar MO, Alexander S, Alexander EC, Rohwer F: Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics 2006, 7: 57-69. 10.1186/1471-2164-7-57
Noguchi H, Park J, Takagi T: MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res 2006, 34: 5623-5630. 10.1093/nar/gkl723
Claudel-Renard C, Chevalet C, Faraut T, Kahn D: Enzyme-specific profiles for genome annotation: PRIAM. Nucleic Acids Res 2003, 31: 6633-6639. 10.1093/nar/gkg847
Rost B, Yachdav G, Liu J: The PredictProtein Server. Nucleic Acids Res 2004, 32: W321-W326. 10.1093/nar/gkh377
Henrissat B: A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 1991, 280: 309-316.
Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B: The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 2009, 37: D233-D238. 10.1093/nar/gkn663
Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer EL, Studholme DJ, Yeats C, Eddy SR: The Pfam protein families database. Nucleic Acids Res 2004, 32: D138-D141. 10.1093/nar/gkh121
Selengut JD, Haft DH, Davidsen T, Ganapathy A, Gwinn-Giglio M, Nelson WC, Richter AR, White O: TIGRFAMs and genome properties: tools for the assignment of molecular function and biological process in prokaryotic genomes. Nucleic Acids Res 2007, 35: D260-D264. 10.1093/nar/gkl1043
Warnecke F, Luginbuhl P, Ivanova N, Ghassemian M, Richardson TH, Stege JT, Cayouette M, McHardy AC, Djordjevic G, Aboushadi N, Sorek R, Tringe SG, Podar M, Martin HG, Kunin V, Dalevi D, Madejska J, Kirton E, Platt D, Szeto E, Salamov A, Barry K, Mikhailova N, Kyrpides NC, Matson EG, Ottesen EA, Zhang XN, Hernandez M, Murillo C, Acosta LG, et al.: Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 2007, 450: 560-565. 10.1038/nature06269
Carbohydrate-active Enzymes (CAZy) Database[http://www.cazy.org]
Schlüter A, Bekel T, Diaz NN, Dondrup M, Eichenlaub R, Gartemann KH, Krahn I, Krause L, Krömeke H, Kruse O, Mussgnug JH, Neuweger H, Niehaus K, Pühler A, Runte KJ, Szczepanowski R, Tauch A, Tilker A, Viehöver P, Goesmann A: The metagenome of a biogas-producing microbial community of a production-scale biogas plant fermenter analysed by the 454-pyrosequencing technology. J Biotechnol 2008, 136: 77-90. 10.1016/j.jbiotec.2008.05.008
Krause L, Diaz NN, Edwards RA, Gartemann KH, Krömeke H, Neuweger H, Pühler A, Runte KJ, Schlüter A, Stoye J, Szczepanowski R, Tauch A, Goesmann A: Taxonomic composition and gene content of a methane-producing microbial community isolated from a biogas reactor. J Biotechnol 2008, 136: 91-101. 10.1016/j.jbiotec.2008.06.003
Markowitz VM, Ivanova NN, Szeto E, Palaniappan K, Chu K, Dalevi D, Chen IMA, Grechkin Y, Dubchak I, Anderson I, Lykidis A, Mavromatis K, Hugenholtz P, Kyrpides NC: IMG/M: a data management and analysis system for metagenomes. Nucleic Acids Res 2008, 36: D534-D538. 10.1093/nar/gkm869
Kunin V, Copeland A, Lapidus A, Mavromatis K, Hugenholtz P: A Bioinformatician's Guide to Metagenomics. Microbiol Mol Biol Rev 2008, 72: 557-578. 10.1128/MMBR.00009-08
Meta-functions[http://www.grid.unep.ch/activities/assessment/metafunctions/index.php]
Hallam SJ, Putnam N, Preston CM, Detter JC, Rokhsar D, Richardson PM, DeLong EF: Reverse methanogenesis: testing the hypothesis with environmental genomics. Science. 2005,305(5689):1457-1462. 10.1126/science.1100025
Gill SR, Pop M, DeBoy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE: Metagenomic analysis of the human distal gut microbiome. Science 2006, 312: 1355-1359. 10.1126/science.1124234
Kunin V, Raes J, Harris JK, Spear JR, Walker JJ, Ivanova N, von Mering C, Bebout BM, Pace NR, Bork P, Hugenholtz P: Millimeter-scale genetic gradients and community-level molecular convergence in a hypersaline microbial mat. Mol Syst Biol 2008, 4: 198-203. 10.1038/msb.2008.35
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI: An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444: 1027-1031. 10.1038/nature05414
Woyke T, Teeling H, Ivanova NN, Huntemann M, Richter M, Gloeckner FO, Boffelli D, Anderson IJ, Barry KW, Shapiro HJ, Szeto E, Kyrpides NC, Mussmann M, Amann R, Bergin C, Ruehland C, Rubin EM, Dubilier N: Symbiosis insights through metagenomic analysis of a microbial consortium. Nature 2006, 443: 950-955. 10.1038/nature05192
Tringe SG, Zhang T, Liu X, Yu Y, Lee WH, Yap J, Yao F, Suan ST, Ing SK, Haynes M, Rohwer F, Wei CL, Tan P, Bristow J, Rubin EM, Ruan Y: The airborne metagenome in an indoor urban environment. PLoS ONE 2008, 3: e1862. 10.1371/journal.pone.0001862
Martin HG, Ivanova N, Kunin V, Warnecke F, Barry KW, McHardy AC, Yeates C, He SM, Salamov AA, Szeto E, Dalin E, Putnam NH, Shapiro HJ, Pangilinan JL, Rigoutsos I, Kyrpides NC, Blackall LL, McMahon KD, Hugenholtz P: Metagenomic analysis of two enhanced biological phosphorus removal (EBPR) sludge communities. Nat Biotechnol 2006, 24: 1263-1269. 10.1038/nbt1247
Tringe SG, von Mering C, Kobayashi A, Salamov AA, Chen K, Chang HW, Podar M, Short JM, Mathur EJ, Detter JC, Bork P, Hugenholtz P, Rubin EM: Comparative metagenomics of microbial communities. Science 2005, 308: 554-557. 10.1126/science.1107851
Marcy Y, Ouverney C, Bik EM, Lösekann T, Ivanova N, Martin HG, Szeto E, Platt D, Hugenholtz P, Relman DA, Quake SR: Dissecting biological 'dark matter' with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. Proc Natl Acad Sci USA 2007, 104: 11889-11894. 10.1073/pnas.0704662104
Smith CR, Baco AR: Ecology of whale falls at the deep-sea floor. In Oceanography and Marine Biology: An Annual Review. Volume 41. Edited by: Gibson RN. London: CRC Press; 2003:311-354.
Acknowledgements
The BioEnergy Science Center is a Bioenergy Research Center supported by the Office of Biological and Environmental Research in the Department of Energy Office of Science. We are grateful to Avril Woodhead for commenting and carefully reviewing this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
LLL participated in the metagenome data analysis and wrote the manuscript. SRM wrote the scripts and run the metagenome BLAST analysis. SM participated in the data analysis and commented on the manuscript. ST provided input and commented on the manuscript. DvdL participated in data analysis, set the outlines for the manuscript, and critically reviewed and commented on the manuscript.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Li, LL., McCorkle, S.R., Monchy, S. et al. Bioprospecting metagenomes: glycosyl hydrolases for converting biomass. Biotechnol Biofuels 2, 10 (2009). https://doi.org/10.1186/1754-6834-2-10
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1754-6834-2-10