
Abstract
Given the increased life expectancy of human immunodeficiency virus (HIV) infected individuals treated with combination antiretroviral therapy (cART) and the ongoing inflammation observed in the brains of these patients, it is likely that premature neurodegeneration as measured by phospho-tau (p-tau) or increased total tau (t-tau) protein may become an increasing problem. This review examines the seven human studies that have occurred over the past 14 years measuring p-tau and/or t-tau in cerebrospinal fluid (CSF) or via post-mortem brain immunohistochemistry. Although not all studies are in agreement as to the changes in p-and t-tau in HIV infected patients, HIV persists in the brain despite cART. Thus is it is suggested that those maintained on long-term cART may develop tau pathology beyond the extent seen in the studies reviewed herein and overtime may then reach the threshold for clinical manifestation.
Similar content being viewed by others


Introduction
HIV-associated neurocognitive disorders (HAND)
More than 33 million people worldwide are infected with the human immunodeficiency virus (HIV) [1]. HIV-1 infection of the central nervous system (CNS) can result in cognitive, motor, and behavioral deficits, collectively termed HAND [2, 3]. These deficits present along a spectrum from asymptomatic neurocognitive impairment (ANI) to HIV associated dementia (HAD). The latter is also termed HIV associated dementia complex (HADC), AIDS dementia complex (ADC), or HIV-encephalitis (HIVE; a diagnosis which can be made only upon brain autopsy exam) depending on the research report [3]. Soon after infection by the HIV, it rapidly moves into the brain via infected monocytes and lymphocytes [4] and, despite combination antiretroviral therapy (cART), persists in parenchymal microglia as well as the perivascular macrophages [5–7]. As HIV is unable to productively infect neurons, neuronal cell damage is largely promoted by neurotoxins secreted by these infected and/or activated macrophages, microglia, and astrocytes [5]. In spite of the fact that the clinical severity of HAND has been significantly reduced due to the widespread utilization of cART, the prevalence and associated morbidity still remains high (~50% [8, 9]). Why HAND still persists in the current era of cART, even in patients effectively controlled for systemic viremic load, is incompletely understood. Recent evidence suggests prolonged inflammation in both the brain and periphery may be responsible [10–13].
Chronic HIV infection promotes neuroinflammation and leads to neuronal death and damage [14]. In addition, it has been reported that subjects treated with combination cART have high levels of neuroinflammation, especially in the hippocampus [13, 15]. This type of neuroinflammation in the form of microglial activation is associated with a number of neurodegenerative diseases including Alzheimer’s disease (AD), Pick’s disease, and hereditary frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17) [16, 17].
Since the introduction of cART the neuropathology associated with HIV has changed. The prevalence of some opportunistic conditions including Cytomegalovirus and toxoplasmosis has decreased while that of others such as progressive multifocal leucoencephalopathy and lymphoma appears to have not changed [15, 18, 19]. The effect of cART on neurocognitive decline and dementia is, on the other hand, less clear, despite a reduced prevalence of the most severe form of HAND; HAD. While some studies report improved cognitive function in cART-treated individuals [20–24], others suggest cognitive impairment is still a significant clinical problem [9, 24–32].
Regardless of the effects of cART directly on HAND, HIV patients who are given this therapy are now living much longer. As humans age, HIV infected or not, there is an increase in the prevalence of signs of neurodegeneration [33] even in otherwise healthy individuals [34]. Since 2000 there has been an array of reports suggesting the possibility of advanced brain aging in the form of AD-like pathology in HIV patients [35–40]. Most of the recent literature indicates that there is an increased risked of advanced brain aging but perhaps not in the exact pattern of true AD. Rather the reports signal an increase in certain, but not all forms of the key proteins of AD, amyloid-beta (Aβ) and tau. This review focuses on tau protein and begins to identify what influence both chronic HIV infection and long-term cART will have on the process of advanced brain aging in the form of abnormal tau pathology and associated functional deficits.
Tau protein
Central to the formation and subsequent stabilization of microtubules as well as the movement of organelles along axons and dendrites, tau is a microtubule-associated protein which is largely expressed in central nervous system (CNS) neurons [41]. Inflammatory stimuli can facilitate tau phosphorylation [42–44] although it remains unclear where phosph-tau is the cause, result, or merely an correlation with inflammation. The hyperphosphorylation of tau has been associated with neurodegeneration [45–47]. Further, hyperphosphorylated tau (p-tau) can undergo cellular accumulation leading to the formation of insoluble neurofibrillary tangles (NFTs) and neuropil threads [48] and thus in some transgenic models of AD, deletion of tau is protective [49, 50].
In very young healthy individuals hyperphosphorylated tau is not commonly detected [51]. It has been reported that by the age of 55, half of individuals show some evidence of tau in the entorhinal cortex (EC) [51]. In these early stages, hyperphosphorylated tau is usually only found in the EC in non-demented individuals. By the age 75, almost all brains contain some hyperphosphorylated tau which has spread to involve the hippocampus (CA1) and neocortex. It should be noted however that tau accumulation is not an inevitable consequence of ageing since studies have identified extremely old, non-demented individuals with only mild tau pathology at autopsy [52]. Although a full understanding of any pathophysiological importance of increased tau (t-tau) is not yet established, it is seen in AD and to a lesser extent in vascular dementia and other neurodegenerative disorders [53]. Therefore, given the increased life expectancy of HIV-infected individuals treated with cART and, age being a major risk factor for increased phospho-tau, it likely that premature brain ageing as measured by p-tau or increased t-tau protein may become an increasing problem.
Epidemiology of Tau protein in HIV infection
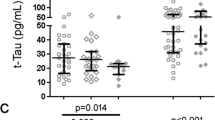
Over the past 14 years there have been several epidemiological studies examining cerebrospinal fluid (CSF) and post- mortem brain samples for changes of p-tau or t-tau in HIV infected patients (Table 1, Figure 1). Some studies have taken cART into account [39, 54–57] while others have not [35, 37]. A study by Green and colleagues prospectively measured CSF t-tau in 76 consecutive HIV infected patients who had acute neurological episodes. Twenty four patients had HADC, 10 had lymphoma, 20 had cerebral infections; 22 patients had miscellaneous conditions, including nine with self-limiting headache/fever [35]. The subjects had a median age of 37 years. Past history of cART was not taken into account. CSF tau levels were acquired by enzyme linked immunosorbent assay (ELISA) (INNOTEST, Fujirebio Europe/Innogenetics, Ghent, Belgium) [58] with an upper limit of normal in CSF set at 315 pg/ml and a lower limit of detection set at 75 pg/ml (Table 1). Results indicated CSF tau was not elevated in the majority (62/76) (82%) of patients, regardless of their CD4 lymphocyte count or their clinical diagnoses. Elevated CSF t-tau was significantly associated with poor outcome as six of eight patients who died within 4 weeks of lumbar puncture had elevated t-tau [35].

Synopsis of clinical studies examining the relationship between tau and HIV infection and/or HAND.
Subsequently there was an analysis of the prevalence of CSF t-tau and p-tau concentrations from 101 HIV-1 seropositive individuals, some of whom had ADC [37]. The CSF from 20 patients with moderate to severe AD was used as a positive control [37]. CSF from 20 patients not known to have HIV or AD was also analyzed. CSF t-tau was quantified using an hTau antigen assay, and p-tau concentrations were determined using a Phospho-Tau181 (p-tau181) assay that recognizes the p-Tau at Threonine-181. Both were measured using the Innotest ELISA with lower limits of 75 and 16 pg/mL. They found HAND subjects had significantly increased t-tau and p-tau at concentrations similar to patients with AD, suggesting that ADC may be associated with an AD-like process [37]. Overall, CSF t-tau levels were significantly increased in ADC Stage 2 while CSF p-tau levels were significantly increased in each of the ADC stages compared with the negative controls and the AD patients. The authors noted that these data indicate patients with ADC have significantly increased t-tau and p-tau concentrations which are of the same magnitude as found in AD patients [37]. In eight ADC patients (mean age 43 years), CSF t-tau was increased [37]. This is in contrast to the previous study by Green and colleagues [35] where 24 ADC patients (median age 37 years) were analyzed, finding only 5 with increased levels. One explanation for these conflicting results relates to the age of the patients [35] and this confounder also occurs in other studies in this review. In general studies indicating CSF t-tau concentrations that were increased were composed of older individuals (mean age of 43 years or older). Conversely those with negative results had a younger mean age, in this case [37] approximately 37 years. The study by Green and colleagues [35], did show a non-significant trend to higher values in those patients 40 years of age and older. Furthermore, Green and colleagues study [35] likely included more subjects with more severe ADC because which is in agreement with the results of Brew and colleagues who showed a relationship to Stage 2 ADC [37] (Table 1).
Following, there was an investigation into investigated the prevalence of tau-associated neurodegeneration in nine cART-treated individuals who had a history of excellent therapeutic compliance for at least 18 months and compared 20 pre-cART HIV-infected subjects and 14 control cases [54]. The pre-cART groups included: pre-symptomatic HIV-positive and AIDS cases. The nine cART-treated cases had a history of excellent therapeutic compliance for at least 18 months. Two groups of HIV-negative control cases with no CNS pathology were selected to provide a baseline. The first group was age matched (mean age 40) with the HIV cases while the second group represented older individuals (mean age 70) who had no history of cognitive decline or other neurodegenerative disorder. This older group allowed for analyzing the extent of any premature ageing effect in a much younger HIV cohort. Paraffin blocks of hippocampus and pons were selected for each case. Immunohistochemistry was performed using AT8 antibody, which recognizes p-tau at serine 202. Tyramide signal amplification (TSA) was used to enhance staining in experiments because the standard avidin-biotin complex (ABC) techniques were not sensitive enough for several of the antibodies. Diaminobenzidine (DAB) was then used to visualize all antibodies. Results of this study showed higher levels of p-tau in the EC, hippocampus, and neocortex in HIV infected subjects compared to aged matched controls. Interestingly, the greatest levels of p-tau were noted in cART-treated subjects. Increased levels of t-tau were also seen in the hippocampal region of pre-cART HIV-infected groups compared to HIV-negative age-matched controls [54]. The increases in tau were independent of a history of drug abuse, although the number of cases studied in each group was low when subdivided into drug abusers and non-drug abusers. This is interesting given past finding of increased levels of p-tau in young HIV-negative drug abusers (mean age 27) [59]. The group of pre-symptomatic subjects studied were all HIV-positive drug abusers and therefore in this group it was not possible to determine whether p-tau levels were increased compared to controls as a result of drug abuse, as reported by Ramage et al., [59] or whether factors related to HIV infection were responsible. No evidence of significant head injury in patients’ life history or on routine neuropathological examination was found and it was thus conclude that head injury was unlikely to be a contributing factor in the observed tau pathology [54]. The authors note that if two control groups represent different ends of the spectrum of normal deposition of hyperphosphorylated tau in non-demented individuals, then these results suggest that HIV-infected individuals have shifted significantly away from normal age-matched controls towards the levels seen normally in ‘old age’ [54]. This would suggest these individuals are subject to advanced brain aging, which is of particular concern in the cART-treated group and may promote future HAND [33]. Thus those maintained on long term cART may develop tau pathology beyond the extent seen in this study and that this may then reach the threshold for clinical manifestation [54].
Clifford and colleagues [39] examined CSF from a total of 188 subjects clinically categorized with normal cognition from the general population (mean age 50 years), HIV + subjects with normal cognition (mean age 43 years), HIV + subjects with impaired cognition (mean age 48), or presumed HIV - subjects with mild AD (mean age 74). CSF samples were analyzed for t-tau, and p-tau by ELISA (INNOTEST, Fujirebio Europe/Innogenetics, Ghent, Belgium), however, upper and lower limits of detection were not supplied in the report. In contrast to Brew and colleagues [37], HIV-positive subjects with HAND did not have CSF t-tau and p-tau characteristic of AD however, this report in congruence with Brew and colleagues analyses in which also indicates t-tau was significantly elevated in CSF of this HIV positive population versus non-HIV infected subjects [37]. Overall there was a trend for HAND patients to have higher p-tau and t-tau than HIV positive non-HAND patients. It is notable that this report of non-significantly elevated p-tau in the AD patients is also not in agreement with the AD literature.
Using both an epidemiological approach to identify tau pathology in HIV infection, and then modeling this phenomenon in vivo can lead to useful prevalence and mechanistic data regarding the influence of abnormal tau on HAND. Patrick and colleagues [55], using double labeled using the Tyramide Signal Amplification-Direct (Red) system (NEN Life Sciences, Boston, MA). Using this system, FITC tagged p-tau immunohistochemistry antibodies: AT8 and PHF-1 were analyzed, in the brains of 43 patients with HIVE (median age 43.13 years) and in gp120 Tg mice, showing that increased expression of cyclin-dependent kinase 5 (CDK5) can lead to neurotoxicity via promotion of abnormal tau phosphorylation. Researchers utilized FITC tagged manufactured p-tau immunohistochemistry antibodies: AT8 (Ser 202) and PHF-1, which detects p-tau at Ser396 and Ser404. Subjects were excluded if they had a history of CNS opportunistic infections or non-HIV-related developmental, neurological, psychiatric, or metabolic conditions that might affect CNS functioning. For inclusion, a total of 16 age-matched cases were identified with and without encephalitis or other complications. cART status was not taken into account. Overall the resulting data are consistent with other studies indicating aberrant activation of the CDK5 [60, 61] and glycogen sythanse kinase (GSK)3β [60, 62–65] signaling promoting neurodegenerative changes in HAND patients. The p25/CDK5 complex associates with NFT in AD patients [66], and CDK5 has been shown to abnormally phosphorylate tau [67]. These are recognized by the AT8 and PHF-1 antibodies [68]. Both of these phosphorylation epitope-specific tau antibodies were elevated in patients with HIVE and in gp120 Tg mice. The patterns of AT8 and PHF-1 immunostaining in HIVE patients differed from what has been reported in AD patients. That is, in advanced AD cases the p-tau immunoreactivity is associated with dystrophic neurites, neuropil threads, and NFT [69–73]. On the other hand in the HIVE cases and in gp120 Tg mouse model there was diffuse nonfibrillar p-tau immunostaining detected in neurons and throughout the neuropil. Increased CDK5 and p35/p25 immunoreactivities were also detected throughout the neuropil of HIVE brains and gp120 Tg mice, indicating the expression of these proteins in cellular compartments that extend into the dendritic arbor and synapses. Data on whether the patients were on cART was not provided. The diffuse pattern of p-tau immunostaining was similar to what has been described in patients with preclinical AD or in the pretangle stage [70, 74], suggesting that in HIVE some of the initial triggering events are present [55] which is in agreement with the previous study by Anthony et al. [54] and suggests HIV patients, whether on cART or not, may develop tau pathology beyond the extent seen at the age relatively young ages of the patients of these studies and that tau pathology may reach the threshold for clinical manifestation as the aging process continues [54].
A recent cross-sectional investigation examined 94 patients (mean age 45 ± 10 years) via CSF analysis for t-tau and p-tau [56]. This study only enrolled patients who never received cART because of unimpaired immune state or who were taking their first cART regimen. In total, 68% of the cohort was cART-treated. Seventy-two percent (72%) of the patients with cART were virologically suppressed. Upper and lower limits of detection were not supplied for the t-tau or p-tau ELISA assays (INNOTEST, Fujirebio Europe/Innogenetics, Ghent, Belgium). Patients with signs of opportunistic CNS infection by MRI or CSF analysis were excluded from further analysis as well as those with psychiatric disorders or severe impairment of consciousness. A significant correlation between HAND and t-tau was uncovered, but not for p-tau [56]. Interestingly HAND severity as measured by the Memorial Sloan-Kettering scale, HIV dementia scale and Mosaic test correlated significantly with the total tau level in CSF but not p-tau levels [56]. Although measures of global brain atrophy via MRI in this study did not significantly correlate with the increase in t-tau, the results suggest t-tau might be a non-specific marker of ongoing subcortical CNS damage particularly in the region of the periventricular white matter and the basal ganglia in HIV infection.
A most recent study by Smith et al. [57] examined p-tau in ten HIV positive and ten HIV negative subjects. A portion of both groups were IDU. All HIV positive patients had been treated with cART for up to 7.9 years and most had received Zidovudine before the start of cART. Cases with opportunistic infections or tumors in the brain were excluded from this study. All six HIV positive IDU, but only one HIV positive non-DU, were HCV positive while the HIV negative IDU and non-IDU were all HCV-negative. Using the AT8 (Ser202) antibody as the primary method for tau quantification, the results revealed declining positivity for tau (neuropil threads and NFT) across all four groups, with IDU having more tau than the non-DU and HIV positive subjects more than HIV negative, but these differences did not achieve statistical significance. A likely reason for the non-significance may be that the study was underpowered [57].
Conclusion
Together, this review suggests that the measurement of t-tau and p-tau in CSF maybe a useful tool for monitor HAND risk at relatively young ages, or for predicting prognosis in later ages (Table 1). Though differing methods of tau quantification were utilized across a portion of the reviewed reports, the research is beneficial in emphasizing the relationship between the presence of t-tau and p-tau in the brain or CSF and the patient’s HAND status. Because of the aging of the HIV-positive population, this biomarker may well be of increasing utility. Although the incidence of HAD has decreased with the widespread use of cART, patients continue to experience varying degrees of HAND [75]. Since even mild impairment affects quality of life [76], the neurological consequences of HIV infection have gained increased attention, as has the need to distinguish those at risk for impairing neurodegeneration through dynamic changes in tau. Future studies with larger numbers of patients across a wider age range may fine-tune this suggested form of evaluation for HAND risk and as a possible treatment target.
Abbreviations
- Aβ:
-
Amyloid-beta
- AD:
-
Alzheimer’s disease
- ADC:
-
AIDS dementia complex
- AIDS:
-
Acquired immunodeficiency virus
- ADC:
-
AIDS dementia complex
- ANI:
-
Asymptomatic neurocognitive impairment
- CNS:
-
Central nervous system
- cART:
-
Combination antiretroviral therapy
- CSF:
-
Cerebrospinal fluid
- CDK5:
-
Cyclin-dependent kinase 5
- EC:
-
Entorhinal cortex
- GSK:
-
Glycogen synthase kinase
- HAD:
-
HIV associated dementia
- HADC:
-
HIV associated dementia complex
- HAND:
-
HIV associated neurocognitive disorders
- HIV:
-
Human immunodeficiency virus
- HIVE:
-
HIV-encephalitis
- IDU:
-
Intravenous drug user
- NFT:
-
Neurofibrillary tangle
- p-tau:
-
Phospho tau
- t-tau:
-
Total tau.
References
AIDS Epidemic Update. 2009, Geneva: Joint United Nations Programme on HIV/AIDS and World Health Organization, http://data.unaids.org/pub/report/2009/jc1700_epi_update_2009_en.pdf, accessed 12 July 2014
McArthur JC, Steiner J, Sacktor N, Nath A: Human immunodeficiency virus-associated neurocognitive disorders: mind the gap. Ann Neurol. 2010, 67: 699-714.
Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, Clifford DB, Cinque P, Epstein LG, Goodkin K, Gisslen M, Grant I, Heaton RK, Joseph J, Marder K, Marra CM, McArthur JC, Nunn M, Price RW, Pulliam L, Robertson KR, Sacktor N, Valcour V, Wojna VE: Updated research nosology for HIV-associated neurocognitive disorders. Neurology. 2007, 69: 1789-1799. 10.1212/01.WNL.0000287431.88658.8b.
Dunfee R, Thomas ER, Gorry PR, Wang J, Ancuta P, Gabuzda D: Mechanisms of HIV-1 neurotropism. Curr HIV Res. 2006, 4: 267-278. 10.2174/157016206777709500.
Salemi J, Obregon DF, Cobb A, Reed S, Sadic E, Jin J, Fernandez F, Tan J, Giunta B: Flipping the switches: CD40 and CD45 modulation of microglial activation states in HIV associated dementia (HAD). Mol Neurodegener. 2011, 6: 3-10.1186/1750-1326-6-3.
Petito CK, Cho ES, Lemann W, Navia BA, Price RW: Neuropathology of acquired immunodeficiency syndrome (AIDS): an autopsy review. J Neuropathol Exp Neurol. 1986, 45: 635-646. 10.1097/00005072-198611000-00003.
Ho DD, Rota TR, Schooley RT, Kaplan JC, Allan JD, Groopman JE, Resnick L, Felsenstein D, Andrews CA, Hirsch MS: Isolation of HTLV-III from cerebrospinal fluid and neural tissues of patients with neurologic syndromes related to the acquired immunodeficiency syndrome. N Engl J Med. 1985, 313: 1493-1497. 10.1056/NEJM198512123132401.
Robertson KR, Smurzynski M, Parsons TD, Wu K, Bosch RJ, Wu J, McArthur JC, Collier AC, Evans SR, Ellis RJ: The prevalence and incidence of neurocognitive impairment in the HAART era. Aids. 2007, 21: 1915-1921. 10.1097/QAD.0b013e32828e4e27.
Sacktor N, McDermott MP, Marder K, Schifitto G, Selnes OA, McArthur JC, Stern Y, Albert S, Palumbo D, Kieburtz K, De Marcaida JA, Cohen B, Epstein L: HIV-associated cognitive impairment before and after the advent of combination therapy. J Neurovirol. 2002, 8: 136-142. 10.1080/13550280290049615.
Dreyer EB, Kaiser PK, Offermann JT, Lipton SA: HIV-1 coat protein neurotoxicity prevented by calcium channel antagonists. Science. 1990, 248: 364-367. 10.1126/science.2326646.
Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek DC: Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006, 12: 1365-1371.
Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A, Zaman T, Stone D, Mefford M, Morgello S, Singer EJ, Wolinsky SM, Gabuzda D: Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS One. 2008, 3: e2516-10.1371/journal.pone.0002516.
Eden A, Price RW, Spudich S, Fuchs D, Hagberg L, Gisslen M: Immune activation of the central nervous system is still present after >4 years of effective highly active antiretroviral therapy. J Infect Dis. 2007, 196: 1779-1783. 10.1086/523648.
Gartner S, Liu Y: Insights into the role of immune activation in HIV neuropathogenesis. J Neurovirol. 2002, 8: 69-75. 10.1080/13550280290049525.
Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE: Influence of HAART on HIV-related CNS disease and neuroinflammation. J Neuropathol Exp Neurol. 2005, 64: 529-536.
McGeer PL, McGeer EG: Local neuroinflammation and the progression of Alzheimer's disease. J Neurovirol. 2002, 8: 529-538. 10.1080/13550280290100969.
Perry VH: The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain Behav Immun. 2004, 18: 407-413. 10.1016/j.bbi.2004.01.004.
Gray F, Chretien F, Vallat-Decouvelaere AV, Scaravilli F: The changing pattern of HIV neuropathology in the HAART era. J Neuropathol Exp Neurol. 2003, 62: 429-440.
Gray F, Keohane C: The neuropathology of HIV infection in the era of Highly Active AntiRetroviral Therapy (HAART). Brain Pathol. 2003, 13: 79-83.
Cohen RA, Boland R, Paul R, Tashima KT, Schoenbaum EE, Celentano DD, Schuman P, Smith DK, Carpenter CC: Neurocognitive performance enhanced by highly active antiretroviral therapy in HIV-infected women. Aids. 2001, 15: 341-345. 10.1097/00002030-200102160-00007.
Kandanearatchi A, Williams B, Everall IP: Assessing the efficacy of highly active antiretroviral therapy in the brain. Brain Pathol. 2003, 13: 104-110.
Robertson KR, Robertson WT, Ford S, Watson D, Fiscus S, Harp AG, Hall CD: Highly active antiretroviral therapy improves neurocognitive functioning. J Acquir Immune Defic Syndr. 2004, 36: 562-566. 10.1097/00126334-200405010-00003.
Tozzi V, Balestra P, Galgani S, Narciso P, Ferri F, Sebastiani G, D'Amato C, Affricano C, Pigorini F, Pau FM, De Felici A, Benedetto A: Positive and sustained effects of highly active antiretroviral therapy on HIV-1-associated neurocognitive impairment. Aids. 1999, 13: 1889-1897. 10.1097/00002030-199910010-00011.
Cysique LA, Maruff P, Brew BJ: Variable benefit in neuropsychological function in HIV-infected HAART-treated patients. Neurology. 2006, 66: 1447-1450. 10.1212/01.wnl.0000210477.63851.d3.
Dore GJ, Correll PK, Li Y, Kaldor JM, Cooper DA, Brew BJ: Changes to AIDS dementia complex in the era of highly active antiretroviral therapy. Aids. 1999, 13: 1249-1253. 10.1097/00002030-199907090-00015.
Ciccarelli N, Fabbiani M, Di Giambenedetto S, Fanti I, Baldonero E, Bracciale L, Tamburrini E, Cauda R, De Luca A, Silveri MC: Efavirenz associated with cognitive disorders in otherwise asymptomatic HIV-infected patients. Neurology. 2011, 76: 1403-1409. 10.1212/WNL.0b013e31821670fb.
Liner KJ, Ro MJ, Robertson KR: HIV, antiretroviral therapies, and the brain. Curr HIV/AIDS Rep. 2010, 7: 85-91. 10.1007/s11904-010-0042-8.
Velasco M, Pareja JA, Losa JE, Valverde JF, Espinosa A, Gujarro C: Dream changes following initiation of efavirenz treatment. Med Clin (Barc). 2011, 136: 103-105. 10.1016/j.medcli.2010.06.011.
Del Palacio M, Alvarez S, Munoz-Fernandez MA: HIV-1 infection and neurocognitive impairment in the current era. Rev Med Virol. 2012, 22: 33-45. 10.1002/rmv.711.
Clark US, Cohen RA: Brain dysfunction in the era of combination antiretroviral therapy: implications for the treatment of the aging population of HIV-infected individuals. Curr Opin Investig Drugs. 2010, 11: 884-900.
Schouten EJ, Jahn A, Ben-Smith A, Makombe SD, Harries AD, Aboagye-Nyame F, Chimbwandira F: Antiretroviral drug supply challenges in the era of scaling up ART in Malawi. J Int AIDS Soc. 2011, 14 (Suppl 1): S4-10.1186/1758-2652-14-S1-S4.
Blas-Garcia A, Esplugues JV, Apostolova N: Twenty years of HIV-1 non-nucleoside reverse transcriptase inhibitors: time to reevaluate their toxicity. Curr Med Chem. 2011, 18: 2186-2195. 10.2174/092986711795656180.
Giunta B, Fernandez F, Nikolic WV, Obregon D, Rrapo E, Town T, Tan J: Inflammaging as a prodrome to Alzheimer's disease. J Neuroinflammation. 2008, 5: 51-10.1186/1742-2094-5-51.
Band GP, Ridderinkhof KR, Segalowitz S: Explaining neurocognitive aging: is one factor enough?. Brain Cogn. 2002, 49: 259-267. 10.1006/brcg.2001.1499.
Green AJ, Giovannoni G, Hall-Craggs MA, Thompson EJ, Miller RF: Cerebrospinal fluid tau concentrations in HIV infected patients with suspected neurological disease. Sex Transm Infect. 2000, 76: 443-446. 10.1136/sti.76.6.443.
Green DA, Masliah E, Vinters HV, Beizai P, Moore DJ, Achim CL: Brain deposition of beta-amyloid is a common pathologic feature in HIV positive patients. Aids. 2005, 19: 407-411. 10.1097/01.aids.0000161770.06158.5c.
Brew BJ, Pemberton L, Blennow K, Wallin A, Hagberg L: CSF amyloid beta42 and tau levels correlate with AIDS dementia complex. Neurology. 2005, 65: 1490-1492. 10.1212/01.wnl.0000183293.95787.b7.
Alisky JM: The coming problem of HIV-associated Alzheimer's disease. Med Hypotheses. 2007, 69: 1140-1143. 10.1016/j.mehy.2007.02.030.
Clifford DB, Fagan AM, Holtzman DM, Morris JC, Teshome M, Shah AR, Kauwe JS: CSF biomarkers of Alzheimer disease in HIV-associated neurologic disease. Neurology. 2009, 73: 1982-1987. 10.1212/WNL.0b013e3181c5b445.
Giunta B, Hou H, Zhu Y, Rrapo E, Tian J, Takashi M, Commins D, Singer E, He J, Fernandez F, Tan J: HIV-1 Tat contributes to Alzheimer's disease-like pathology in PSAPP mice. Int J Clin Exp Pathol. 2009, 2: 433-443.
Sato-Harada R, Okabe S, Umeyama T, Kanai Y, Hirokawa N: Microtubule-associated proteins regulate microtubule function as the track for intracellular membrane organelle transports. Cell Struct Funct. 1996, 21: 283-295. 10.1247/csf.21.283.
Lee DC, Rizer J, Selenica ML, Reid P, Kraft C, Johnson A, Blair L, Gordon MN, Dickey CA, Morgan D: LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J Neuroinflammation. 2010, 7: 56-10.1186/1742-2094-7-56.
Sy M, Kitazawa M, Medeiros R, Whitman L, Cheng D, Lane TE, Laferla FM: Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am J Pathol. 2011, 178: 2811-2822. 10.1016/j.ajpath.2011.02.012.
Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM: Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007, 53: 337-351. 10.1016/j.neuron.2007.01.010.
Mookherjee P, Johnson GV: Tau phosphorylation during apoptosis of human SH-SY5Y neuroblastoma cells. Brain Res. 2001, 921: 31-43. 10.1016/S0006-8993(01)03074-8.
Mattson MP: Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000, 1: 120-129. 10.1038/35040009.
Augustinack JC, Schneider A, Mandelkow EM, Hyman BT: Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol. 2002, 103: 26-35. 10.1007/s004010100423.
Bancher C, Brunner C, Lassmann H, Budka H, Jellinger K, Wiche G, Seitelberger F, Grundke-Iqbal I, Iqbal K, Wisniewski HM: Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer's disease. Brain Res. 1989, 477: 90-99. 10.1016/0006-8993(89)91396-6.
Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L: Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007, 316: 750-754. 10.1126/science.1141736.
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J: Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010, 142: 387-397. 10.1016/j.cell.2010.06.036.
Braak H, Braak E: Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997, 18: 351-357. 10.1016/S0197-4580(97)00056-0.
Delacourte A, Sergeant N, Wattez A, Maurage CA, Lebert F, Pasquier F, David JP: Tau aggregation in the hippocampal formation: an ageing or a pathological process?. Exp Gerontol. 2002, 37: 1291-1296. 10.1016/S0531-5565(02)00141-9.
Blennow K, Wallin A, Agren H, Spenger C, Siegfried J, Vanmechelen E: Tau protein in cerebrospinal fluid: a biochemical marker for axonal degeneration in Alzheimer disease?. Mol Chem Neuropathol. 1995, 26: 231-245. 10.1007/BF02815140.
Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE: Accelerated Tau deposition in the brains of individuals infected with human immunodeficiency virus-1 before and after the advent of highly active anti-retroviral therapy. Acta Neuropathol. 2006, 111: 529-538. 10.1007/s00401-006-0037-0.
Patrick C, Crews L, Desplats P, Dumaop W, Rockenstein E, Achim CL, Everall IP, Masliah E: Increased CDK5 expression in HIV encephalitis contributes to neurodegeneration via tau phosphorylation and is reversed with Roscovitine. Am J Pathol. 2011, 178: 1646-1661. 10.1016/j.ajpath.2010.12.033.
Steinbrink F, Evers S, Buerke B, Young P, Arendt G, Koutsilieri E, Reichelt D, Lohmann H, Husstedt IW: Cognitive impairment in HIV infection is associated with MRI and CSF pattern of neurodegeneration. Eur J Neurol. 2013, 20: 420-428. 10.1111/ene.12006.
Smith DB, Simmonds P, Bell JE: Brain viral burden, neuroinflammation and neurodegeneration in HAART-treated HIV positive injecting drug users. J Neurovirol. 2014, 20: 28-38. 10.1007/s13365-013-0225-3.
Vandermeeren M, Mercken M, Vanmechelen E, Six J, van de Voorde A, Martin JJ, Cras P: Detection of tau proteins in normal and Alzheimer's disease cerebrospinal fluid with a sensitive sandwich enzyme-linked immunosorbent assay. J Neurochem. 1993, 61: 1828-1834. 10.1111/j.1471-4159.1993.tb09823.x.
Ramage SN, Anthony IC, Carnie FW, Busuttil A, Robertson R, Bell JE: Hyperphosphorylated tau and amyloid precursor protein deposition is increased in the brains of young drug abusers. Neuropathol Appl Neurobiol. 2005, 31: 439-448. 10.1111/j.1365-2990.2005.00670.x.
Crews L, Patrick C, Achim CL, Everall IP, Masliah E: Molecular pathology of neuro-AIDS (CNS-HIV). Int J Mol Sci. 2009, 10: 1045-1063. 10.3390/ijms10031045.
Wang Y, White MG, Akay C, Chodroff RA, Robinson J, Lindl KA, Dichter MA, Qian Y, Mao Z, Kolson DL, Jordan-Sciutto KL: Activation of cyclin-dependent kinase 5 by calpains contributes to human immunodeficiency virus-induced neurotoxicity. J Neurochem. 2007, 103: 439-455. 10.1111/j.1471-4159.2007.04746.x.
Dewhurst S, Maggirwar SB, Schifitto G, Gendelman HE, Gelbard HA: Glycogen synthase kinase 3 beta (GSK-3 beta) as a therapeutic target in neuroAIDS. J Neuroimmune Pharmacol. 2007, 2: 93-96. 10.1007/s11481-006-9051-1.
Hashimoto M, Sagara Y, Langford D, Everall IP, Mallory M, Everson A, Digicaylioglu M, Masliah E: Fibroblast growth factor 1 regulates signaling via the glycogen synthase kinase-3beta pathway: implications for neuroprotection. J Biol Chem. 2002, 277: 32985-32991. 10.1074/jbc.M202803200.
Maggirwar SB, Tong N, Ramirez S, Gelbard HA, Dewhurst S: HIV-1 Tat-mediated activation of glycogen synthase kinase-3beta contributes to Tat-mediated neurotoxicity. J Neurochem. 1999, 73: 578-586.
Schifitto G, Zhong J, Gill D, Peterson DR, Gaugh MD, Zhu T, Tivarus M, Cruttenden K, Maggirwar SB, Gendelman HE, Dewhurst S, Gelbard HA: Lithium therapy for human immunodeficiency virus type 1-associated neurocognitive impairment. J Neurovirol. 2009, 15: 176-186. 10.1080/13550280902758973.
Augustinack JC, Sanders JL, Tsai LH, Hyman BT: Colocalization and fluorescence resonance energy transfer between cdk5 and AT8 suggests a close association in pre-neurofibrillary tangles and neurofibrillary tangles. J Neuropathol Exp Neurol. 2002, 61: 557-564.
Gong CX, Liu F, Grundke-Iqbal I, Iqbal K: Post-translational modifications of tau protein in Alzheimer's disease. J Neural Transm. 2005, 112: 813-838. 10.1007/s00702-004-0221-0.
Plattner F, Angelo M, Giese KP: The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J Biol Chem. 2006, 281: 25457-25465. 10.1074/jbc.M603469200.
Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM: Microtubule-associated protein tau: a component of Alzheimer paired helical filaments. J Biol Chem. 1986, 261: 6084-6089.
Vincent I, Rosado M, Davies P: Mitotic mechanisms in Alzheimer's disease?. J Cell Biol. 1996, 132: 413-425. 10.1083/jcb.132.3.413.
Schmidt ML, Lee VM, Trojanowski JQ: Relative abundance of tau and neurofilament epitopes in hippocampal neurofibrillary tangles. Am J Pathol. 1990, 136: 1069-1075.
Mandelkow EM, Mandelkow E: Tau in Alzheimer's disease. Trends Cell Biol. 1998, 8: 425-427. 10.1016/S0962-8924(98)01368-3.
Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke-Iqbal I: Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005, 1739: 198-210. 10.1016/j.bbadis.2004.09.008.
Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, Yen SH, Aronson MK: Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging. 1992, 13: 179-189. 10.1016/0197-4580(92)90027-U.
Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, Simpson DM, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I, CHARTER Group; HNRC Group: HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol. 2011, 17: 3-16. 10.1007/s13365-010-0006-1.
Sadek JR, Vigil O, Grant I, Heaton RK: The impact of neuropsychological functioning and depressed mood on functional complaints in HIV-1 infection and methamphetamine dependence. J Clin Exp Neuropsychol. 2007, 29: 266-276. 10.1080/13803390600659384.
Funding and disclosure
BG is supported by NIMH/NIH grant (R01MH098737) (BG).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LAMB generated the table and figure in addition to assisting with the literature search and response to critiques. JS assisted with the response to critiques. AJS, PRS, and JT assisted with the literature search. BG conceived of the idea and wrote the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Brown, L.A., Scarola, J., Smith, A.J. et al. The role of tau protein in HIV-associated neurocognitive disorders. Mol Neurodegeneration 9, 40 (2014). https://doi.org/10.1186/1750-1326-9-40
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1750-1326-9-40