
Abstract
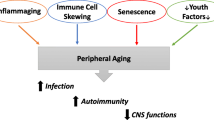
Recently, the term "inflammaging" was coined by Franceshci and colleagues to characterize a widely accepted paradigm that ageing is accompanied by a low-grade chronic up-regulation of certain pro-inflammatory responses. Inflammaging differs significantly from the traditional five cardinal features of acute inflammation in that it is characterized by a relative decline in adaptive immunity and T-helper 2 responses and is associated with increased innate immunity by cells of the mononuclear phagocyte lineage. While the over-active innate immunity characteristic of inflammaging may remain subclinical in many elderly individuals, a portion of individuals (postulated to have a "high responder inflammatory genotype") may shift from a state of "normal" or "subclinical" inflammaging to one or more of a number of age-associated diseases. We and others have found that IFN-γ and other pro-inflammatory cytokines interact with processing and production of Aβ peptide, the pathological hallmark feature of Alzheimer's disease (AD), suggesting that inflammaging may be a "prodrome" to AD. Although conditions of enhanced innate immune response with overproduction of pro-inflammatory proteins are associated with both healthy aging and AD, it is suggested that those who age "well" demonstrate anti-inflammaging mechanisms and biomarkers that likely counteract the adverse immune response of inflammaging. Thus, opposing the features of inflammaging may prevent or treat the symptoms of AD. In this review, we fully characterize the aging immune system. In addition, we explain how three novel treatments, (1) human umbilical cord blood cells (HUCBC), (2) flavanoids, and (3) Aβ vaccination oppose the forces of inflammaging and AD-like pathology in various mouse models.
Similar content being viewed by others


Introduction
Some two thousand years ago, Celsus first described 4 cardinal signs of inflammation: (1) redness, (2) swelling, (3) heat, and (4) pain. Shortly thereafter a fifth sign, loss of function, was added by Galen. Recently, the term "inflammaging" was coined by Franceshci and colleagues to characterize a widely accepted paradigm that ageing is accompanied by a low-grade chronic up-regulation of certain inflammatory responses [1–8]. Inflammaging differs significantly from the traditional five cardinal features of acute inflammation in that it is a (a) low-grade, (b) controlled, (c) asymptomatic, (d) chronic, and (e) systemic state of inflammation [9]. This systemic inflammatory response is evidenced by increased serum levels of pro-inflammatory cytokines (IL-6, IL-15, IL-8) [10–15] and other inflammatory biomarkers, such as coagulation factors [16–18]. Additionally, subclinical infection with common viruses such as cytomegalovirus (CMV) is a hallmark feature of inflammaging. There also may be a genetic component, as suggested by studies on alleles coding for mediators of inflammation including cytokines and coagulation factors [19–21]. Finally, reactive oxygen species (ROS) cause amplification of cytokine release, fueling a self-perpetuating positive feedback loop. The end result of this cycle is a chronic and systemic pro-inflammatory state where both tissue damaging and healing mechanisms operate simultaneously. Over decades, the opposing forces are likely critical perpetrators of ageing and age related disease, leading to an accumulation of subtle tissue damage [3, 5–8].
Inflammaging is characterized by a relative decline in adaptive immunity and T-helper 2 responses and is associated with increased cell mediated responses
According to Franceschi's original description of inflammaging, innate immunity progresses to a chronically active state secondary to exhaustion of the more evolutionary recent adaptive (specific) immune system [22]. This exhaustion is in large part due to age-associated reduction of T-cells for various reasons including thymic involution [23–25], as well as fewer bone marrow early progenitor B cells [26]. In early life, naïve T cells are activated by contact with antigens. They then differentiate into effector or memory cells. Because the quantity of T-cells in healthy individuals is stable over the adult lifespan, peripheral T cell turnover of pre-existing populations in the thymus is required for replacement of T cells in relatively young individuals [27–30].
Together with the diminution of adaptive immunity that occurs in inflammaging, there is also an increase in the number of antigen-experienced cells and a decrease in the number of naïve T cells in the circulation, which results in accumulation of incompetent memory lymphocytes [31]. These cells likely clonally expanded and became effector memory T-cells that were competent at one time, but then lost their antigen-specific function due to their age. This phenomenon is believed to be owed to life-long antigenic stress from immunosurveillance against persistent viruses, especially CMV [32]. This accumulation of a limited repertoire of "megaclones" of effector memory CD8+ T cells results from long-term, chronic exposure to antigens over time frames much longer than during evolution of the human immune system [28, 33–39]. The net result of this is (1) to reduce adaptive immunity to previously encountered pathogens, and (2) to weaken the host adaptive immune response to novel pathogens due to a reduction in the diversity of the antigen-recognition repertoire with age. In fact, based on analysis of human T-cell receptor (TCR) Vβ chain usage, the antigen-recognition repertoire decreases from approximately 108 in young adults to 106 in the elderly [40]. Moreover, CD8+ T-cells in the elderly display significantly decreased ability to secrete interferon-gamma (IFN-γ) when stimulated by cognate antigen in comparison to younger age groups [36, 38]. Also, naïve CD4+ T-cells from old humans and mice show decreased responsiveness to TCR stimulation and altered profiles of cytokine production versus naïve CD4+ T-cells from young hosts. Likewise, the helper function of naïve CD4+ T-cells for antibody production by B cells is also decreased [41]. The decline in responsiveness of naïve CD4+ T cells is due to the chronologic age of the cells themselves, and not of the individual host [41], suggesting that long-term maintenance of naïve CD4+ T-cells through homeostatic cytokines may not have a positive impact on their function. Indeed, naïve CD4+ T-cells that have undergone homeostatic cell divisions proliferate less and secrete less IL-2 in response to antigen than do naïve CD4+ T-cells that have not undergone homeostatic division [41].
Unlike naïve lymphocytes, memory CD4+ T lymphocytes are long-lived and relatively competent with age. These antigen-experienced T-cells, when isolated from healthy elderly humans and healthy old mice, respond normally to antigen-induced proliferation in vitro [42] and those generated at a young age respond well to antigens over time. Conversely, naïve CD4+ T-cells derived in old age respond poorly [43]. Together, these studies point to an age-associated defect in memory CD4+ T cells which may originate from defects of aged naïve CD4+ T-cells that have reduced clonal diversity and proliferation potential. Interestingly, changes of the ratio of memory to effector CD4+ T-cell subsets with age have also been implicated in deficient adaptive immune responses to viral infections and vaccines [44].
An additional important feature of inflammaging which contributes to defective T-cell function and adaptive immunity is accumulation of CD28-CD8+ T-cells and the loss of naïve CD8+ T-cells. These T-cells are absent in newborns while composing some 85% of circulating CD8+ T-cells in the elderly. The accumulation of CD28-CD8+ T-cells was also shown in patients with viral infections such as CMV [45]. CD28-CD8+ T-cells may signify terminal differentiation from the CD28+CD8+ subset after repeated antigenic stimulation. Functionally, CD28-CD8+ T-cells have a reduced proliferative response to TCR stimulation but exhibit normal or even enhanced cytotoxic capacity and are resistant to apoptosis [46]. Furthermore, several studies have demonstrated the virtual disappearance of naïve CD8+ T-cells in the elderly associated with a significant increase in the proportions of differentiated effector memory and effector CD8+ T-cells in comparison to younger individuals. Interestingly, most of these cells in the elderly do not have short telomeres. Taken together, these data suggest that, over the course of life, these T-cell populations have undergone a process of end-stage differentiation and that persistent infection with common pathogens, such as CMV, induces chronic stimulation of specific T cells that leads to terminal differentiation to senescent cells with an altered functional capability [30, 47–50]. Further, it seems that clonal expansion of CD28-CD8+T-cells seems directly responsible for increased infection rates and the common failed response to vaccines in the elderly [51].
Decreased adaptive immunity characteristic of inflammaging also involves changes in the B-cell repertoire not unlike those observed in the T-cell pool [52–54, 54]. The quality of the humoral immune response is decreased with age owing to low serum immunoglobulin concentrations and low numbers of antigen-specific, immunoglobulin-secreting plasma cells. Further, antibody specificity, isotype and affinity changes are typical features of old age [54]. For example, immunoglobulins produced in aged mice have lower affinity and are less protective versus those of young animals [55]. This may be secondary to aging's adverse effects on the germinal centre reaction in secondary lymphoid tissues [56], and leads to a diminished quantity of germinal centres in response to tetanus toxoid stimulation [57]. The quantity of B cells also decreases in the elderly (58). At the cellular level, alteration in immunoglobulin generation (through class switching) in B cells is observed in aged individuals [59], which may contribute to the decline of the adaptive immune response in the elderly. Also, age is positively associated with B cell malignancies in older adults with oligoclonally expanded B cells [30].
The weakened adaptive immune responses characteristic of inflammaging may also be caused by deficient MHC class II antigen presentation by dendritic cells (DCs) and macrophages, or in the case of the brain, microglia [60, 61]. Although these cells are part of the innate immune system, they are critical for initiation of adaptive immunity. That is, activation of these phagocytic cells via pattern recognition receptors (PRRs), such as toll-like receptors (TLRs), mediates their maturation and migration to secondary lymphoid organs where they present antigens to T-cells [62, 63]. Studies have indicated that this diminished MHC class II expression over the life span is typical in both humans and mice [64, 65]. Not only do innate immune cells down-regulate MHC II, but decreased numbers of Langerhans cells (LCs; the DCs of the skin) have been observed consistently in both elderly humans and old mice. LC migration to lymph nodes is also decreased in old mice; possibly secondary to reduced availability of IL-1β, a cytokine which provides autocrine stimulus for migration and induces TNF-α production by adjacent keratinocytes, which also promotes LC mobilization [66]. Additionally, DCs from elderly people may under-express costimulatory receptors and IL-12, thus impairing their ability to induce effector T-cell differentiation. In addition, the production of IL-10 is elevated in elderly individuals and may inhibit DC maturation and macrophage function [60]. Further, it was also demonstrated that IFN-γ-mediated induction of the MHC II gene (H2-Ab1) is impaired in macrophages from aged mice due to the decreased binding of transcription factors to the promoter [64]. This lack of proper activity of the innate arm of immunity has also been associated with upregulated pro-inflammatory responsiveness. That is, activated macrophages from aged humans and mice produce higher amounts of the proinflammatory paracrine molecule prostaglandin E2 than younger individuals, which inhibits surface expression of MHC class II [65]. In addition, we also found proinflammatory activation of mouse microglia inhibits their ability to phagocytose Alzheimer's amyloid-beta (Aβ) peptide [67].
Coincident with this phenomenon, either due to a "release" of adaptive control on innate immunity and/or a compensatory mechanism for lack of effectiveness of adaptive immunity, is over-activation of certain pro-inflammatory responses [4, 7, 22] by mononuclear phagocytes (cells of the innate immune system) [68]. These cells are the first to arrive at sites of infection or tissue damage. Their physiologic role is to initiate an inflammatory response, phagocytose the pathogen or foreign body, recruit natural killer (NK) cells, and facilitate the terminal differentiation and migration of DCs that will modulate the T cell-mediated adaptive immune response. [32]. These cells, including neutrophils and macrophages, activate innate immunity via their PRRs [69] which induce a pro-inflammatory response via downstream nuclear factor-kappa beta (NF-kβ) and mitogen associated protein kinase (MAPK) signaling [70, 71]. Indeed, studies in both elderly humans and old mice substantiate the notion that inflammaging constitutes a gradual loss of adaptive and increased activity of mononuclear cell innate immunity demonstrated by pro-inflammatory cell responses to mitogens [30, 32]. For example, peripheral blood monocytes from elderly individuals generated higher amounts of tumor necrosis factor alpha (TNF-α) and interleukin 1 beta (IL-1β) in comparison to cells from young subjects [72]. Furthermore, the data regarding cytokine and chemokine secretion in several studies using human monocytes from aged or young individuals activated in vitro with mitogens or the bacterial endotoxin lipopolysacharride (LPS), for the most part, demonstrate an increase in the levels of pro-inflammatory factors such as IL-6 and IL-8 [73–79]. As noted above, activated macrophages from aged mice and humans produce more prostaglandin E2 than younger individuals, and this proinflammatory coagulation factor suppresses IL-12 secretion, decreases APC surface expression of MHC II, and enhances IL-10 secretion which inhibits T-cell function. This increased prostaglandin E2 is due to enhanced expression of inducible cyclooxygenase [80]. We suggest that, underlying this increased proinflammatory signaling by mononuclear cells of the innate immune system is the previously mentioned increase in the number of antigen-experienced T cells and the decrease in the number of naïve cells in the elderly. The antigen-experienced T cells (observed as large oligoclonal expansions which dominate the repertoire) have lost the important costimulatory molecule CD28 and have a polarized pro-inflammatory T-helper 1 (Th1) profile [81]. Cytokines derived from Th1 cells are associated with cell mediated pro-inflammatory responses, while Th2 cytokines act on B cells to enhance adaptive immune responses (including antibody production) by promoting activation and differentiation. Importantly, an imbalanced Th1/Th2 ratio can change the cytokine microenvironment in lymphatic tissues and trigger the chronic pro-inflammatory innate immune processes characteristic of inflammaging [22]. It is important to note that there are conflicting reports on the basic notion that an imbalance in Th1/Th2 immunity is best characterized in the elderly by overactive Th1 and underactive Th2 cytokine production. For instance, the production of the Th1 cytokine IFN- has been reported to be high [82, 83] or low [84–87] during aging. Such discrepancies may result from variations among species, strains, organs, culture systems, or inter-individual differences during the course of the aging process. To circumvent such confounds, studies focusing on provoking an antigen-specific immune response, for example by vaccination, have proved powerful. For example, influenza vaccination induces protective antibodies in only 40–70% of the elderly [88, 89]. Saurwein-Teissl and colleagues (2002) analyzed a very homogenous "model" cohort of elderly persons, none of whom produced specific antibodies one month after influenza vaccination. Using this homogenous cohort, the lack of antibody production following immunization was positively associated with the increased occurrence of the expanded CD8+CD28- T cell subset. These clones, which produce large amounts of IFN-γ, are strongly suggested to be the basis for a change in the polarization of the immune system toward a Th1 profile in elderly persons and the development of age-related immune deficiency [90]. Moreover, we would suggest that the response to a vaccine may be used as an assessment of inflammaging, and thus risk for age-related disease.
As further detailed below, proinflammatory cytokines associated with reduced adaptive and upregulated innate mononuclear cells (an integral feature of inflammaging) are now believed to exacerbate the pathology and disease course of age-related disorders including Alzheimer's disease (AD) [91–93]. For example, IFN-γ in combination with TNF-α triggers the production of Aβ peptide in human neuronal and extraneuronal cells [3] and increases the production of ROS by microglia. TNF-α can also further exacerbate Aβ toxicity [4].
Does inflammaging perpetrate and/or exacerbate AD?
While the over-active innate immunity characteristic of inflammaging may remain sub-clinical in many elderly individuals for the reasons stated above, a portion of individuals (postulated to have a "high responder inflammatory genotype") may shift from a state of "normal" or "subclinical" inflammaging to one or more of any age-associated diseases. Simultaneously, aging is associated with an increase in production of oxygen-derived radicals, i.e., reactive oxygen species (ROS), with a concurrent decrease in the ability to defend against ROS. Indeed, many works have established oxidative stress and damage not only in the lesions of AD but also in neurons at risk of death [94–100].
It is interesting to note that Alois Alzheimer himself first noted what he termed "gliose" (English translation: gliosis, or inflammation of glia) when characterizing brain pathology in 1907 in the first AD case. Indeed microglia are the common source of pro-inflammatory and oxidative stress in AD as shown by consistent gross observations of upregulated inflammatory responses in brain (including products of activated microglia and astrocytes) regions that exhibit high levels of AD pathology such as the frontal and limbic cortices [101–103]. Conversely, these markers of inflammation or oxidative stress are absent or minimal in brain regions which are typically less susceptible to AD pathology, such as cerebellum. Upon analysis, inflammatory mediators and signs of oxidative stress are most robustly observed in regions of Aβ deposits and neurofibrillary tangles (NFT), classical AD hallmarks often associated with neurodegeneration [104]. For example, significant increases of an oxidized nucleoside derived from RNA, 8-hydroxyguanosine (8OHG), and an oxidized amino acid, nitrotyrosine, have been demonstrated in vulnerable neurons of AD patients [105]. Importantly, it was shown that oxidative damage is quantitatively greatest early in the disease and reduces with disease progression. Interestingly however, increases in Aβ deposition were associated with decreased oxidative damage [105]. Moreover, neurons with NFT show a 40%–56% decrease in relative 8OHG levels compared with neurons free of NFT. We would suggest that inflammaging involves increased oxidative damage prior to a diagnosis of AD (or at least early in AD [105, 208] due to over-activation of certain pro-inflammatory responses [4, 7, 22] by mononuclear phagocytes (cells of the innate immune system) [68]. As AD worsens, amyloid deposition increases simultaneously with compensatory changes that reduce damage from ROS, which could include disregulation of T-cell responses [106]. This is further supported by findings in Tg2576 AD transgenic mice in which oxidative stress precedes Aβ deposition [107]. Also supporting this notion, patients with MCI or very mild AD show increased levels of lipid peroxidation and nucleic acid oxidation in postmortem brain, CSF, plasma, urine and peripheral leukocytes in conjunction with decreased levels of plasma antioxidants and total plasma antioxidant capacity [108–110].
Perhaps the most important piece of evidence to substantiate the pathogenic importance of inflammaging in AD comes from patients without history of dementia but who nonetheless exhibit sufficient Aβ/NFT at autopsy to otherwise qualify an AD diagnosis (so-called "high pathology controls") [111]. Interestingly, these patients demonstrate only modest elevations of proinflammatory markers compared with typical non-demented patients but dramatically less than AD patients [112]. Furthermore, there is a plethora of evidence indicating direct inflammatory toxicity in AD brain. For example, assembly of complement components ultimately leading to membrane attack complex formation and lysis of neuronal cells has been shown ultrastructurally [113]. In seeking to elaborate and extend upon the original theories postulated by Franceshci, it appears that inflammaging may serve as a prodrome or an exacerbating factor for development of AD [9, 22]. Understanding AD in this context may shed new light on immunological treatment targets for AD, which may also apply to other neurodegenerative diseases including HIV associated dementia (HAD), Parkinson's Disease, and Multiple Sclerosis (MS).
Conditions of enhanced innate immune response with overproduction of pro-inflammatory proteins are associated with both healthy aging and AD [114–116]. The difference between these groups seems to be that those who age "well" demonstrate anti-inflammaging mechanisms and biomarkers that likely counteract the adverse immunity characteristic of inflammaging. For example, it seems that centenarians demonstrate anti-inflammatory responses which may delay or even stop emergence of many age-related diseases [9, 117, 118]. Some examples in centenarians are as follows: reduction of pro-inflammatory, pro-atherosclerotic properties of platelets [119], increased resistance to inhibitory actions of homocysteine on nitric oxide generation [119], low plasma membrane lipid peroxide levels, high membrane fluidity with normal content of sialic acid [120–123] and increased plasma cortisol, one of the most potent anti-inflammatory molecules [124, 106]. Further, an example of a genetic anti-inflammaging mechanism in centenarians is increased frequency in males of the IL-10-1082GG genotype, which is associated with high production of this important anti-inflammatory cytokine [125]. Conversely, the same genotype is much less frequent in patients affected by AD [126]. Thus, it is believed that individuals who carry such "high responder" genotypes may suffer from diseases in which inflammaging could be a key eitiological disease component. This notion is supported by many studies indicating an increase of inflammatory parameters, such as complement factors, proinflammatory cytokines, and activation of microglia co-localized with amyloid plaques in the AD brain [127].
Others have also reported what would seem to be biomarkers of inflammaging in AD. One of these markers is neopterin, a blood compound produced by monocyte-derived macrophages upon stimulation with IFN-γ. Although it is virtually impossible to determine whether activation of the innate or acquired arms of the immune system is primary based on measurements of neopterin, in contrast to the well known marker of systemic immune activation, c-reactive protein (CRP), neopterin seems to be more associated with cell-mediated responses and is commonly used as a biomarker for this purpose [128, 129]. Three studies to date have shown elevated levels of neopterin in AD patients compared to age-matched controls [130–132]. The authors suggest this elevated neopterin may be a vestigial result of serum immunity to CMV. That is, in the AD patients, 70% were positive for CMV antibody and CMV antibody concentrations correlated with concentrations of neopterin and CRP. As previously mentioned, CMV infection seems to be a critical factor in the diminution of adaptive immunity in the elderly as it confers expansion of a limited repertoire of CD8+ T cells, which afterward lack full functionality [45, 90]. A similar response to influenza vaccination in the elderly was described [90]. Thus, it has been proposed that enhanced cell-mediated innate immunity reflected by neopterin production in AD patients might be secondary to senescence of CMV-expanded T-cell clones [114]. It is of course difficult to determine from these clinical studies whether such factors as neopterin, CRP, and humoral responses are etiologically involved in AD, but it certainly appears that there is a positive correlation between these factors and AD.
Several studies have examined T-cell populations and activation markers in AD in peripheral blood (for a review see 188). Although total percentage of T-cells was not found to be altered [133, 134], alterations of their function, differentiation and subset distribution have been demonstrated. Abnormal functioning of suppressor as well as Th cells has been described in AD patients [135, 136]. Significantly decreased activation response of this adaptive immune cell in AD compared to age-matched controls has also been observed. The proliferative response of T-cells in AD seems reduced; however, this was not universally observed. While two studies found no alterations in the proliferative response of AD patient T-cells [137, 138], four studies report a decrease in proliferation of AD patient T-cells [139–141]. Most recently, one study [109] found a significant decrease of CD3+ T lymphocytes and CD19+ B lymphocytes in AD patients. A slight increase of the CD4+ and a decrease of the CD8+ subpopulation was also observed, but this was not accompanied by a significant change in the CD4+/CD8+ ratio, and CD16+CD56+ cells were not altered. Schindowski and colleagues found increased apoptosis in CD4+ T cells as well as NK cells in AD patients, while in aged individuals, all subsets were affected. This may be explained by the levels of Bcl-2 in T-cells, which were significantly increased in patients with mild AD. Apoptosis and Bcl-2 levels were also elevated in the APP751SL × PS1M146L transgenic mouse model of AD [142]. Together, these finding suggest a diminution of adaptive immunity reflected by dysregulated and altered T- cell distributions in AD [127]. However, as mentioned above, it is difficult to conclude from these clinical studies whether such alterations in the leukocyte repertoire of AD patients are etiologically involved in disease progression or simply epiphenomena. For this reason, future studies in mouse models of AD deficient in key innate/adaptive immune/inflammatory molecules will be important.
In vitro and in vivo studies by us and other groups suggest that IFN-γ and other pro-inflammatory cytokines interact with processing and production of the Aβ peptide [143, 144]. For example, IFN-γ is a strong stimulator of Aβ1–40 and Aβ1–42 production in vitro in neurons and in other brain cells [143, 144]. Even if IFN-γ produced in the periphery does not directly affect brain Aβ levels, increased peripheral production of Aβ at sites where IFN-γ levels are elevated might influence Aβ levels in the brain [145, 146]. Thus, we and others have proposed adverse effects of altered immunity (upregulated innate mononuclear cell immunity/Th1 cytokine secretion and downregulated adaptive immunity) in AD patients, and this has led us and others to focus on three diverse treatment modalities in AD transgenic mice models which seem to "re-balance" pro- versus anti-inflammatory responses in the AD brain. These therapies are: (1) non-steroidal anti-inflammatory drugs (NSAIDs), (2) Aβ immunization, (3) human umbilical cord blood cells (HUCBC), and (4) plant-derived compounds known as flavonoids.
Finally,, we suggest that that inflammaging as a prodrome to AD fits well with the "2-hit hypothesis" of AD put forth by Zhu and colleagues stating that both oxidative stress and mitogenic dysregulation are necessary and sufficient to cause AD [147]. The first "hit" required for AD development is suggested to be the above discussed oxidative stress during MCI and early in the course of AD. This goes hand in hand with upregulation of innate immunity as part of inflammaging. The second "hit" is also suggested to occur during this same prodromal time period during inflammaging and disregulation of neuronal mitotic proteins [148, 149]. Indeed, cell cycle markers occur prior to the appearance of gross cytopathological changes in AD [150]. In further support of this idea, mitotic events have been shown to occur in pre-AD patients with MCI which represents a time point where inflammaging effects are near their maximum, ie during the prodromal stage of AD [151].
Re-balancing innate and adaptive immunity as a treatment paradigm for AD NSAIDs and Aβ immunization: from mice to humans
Several observational studies [152–154] have shown that use of various anti-inflammatory agents is associated with reduced risk of developing AD. After preliminary studies produced encouraging results with nonsteroidal anti-inflammatory drugs (NSAIDs) for AD treatment, [155, 156] a series of larger studies were launched that tested glucocorticoid therapy, [157] NSAIDs, [158–160] and hydroxychloroquine [161] on patients with AD and MCI and also cognitively normal individuals at risk for AD. The results of these trials to date have been negative. None of the NSAIDs tested in early trials had amyloid-reducing activity. Newer NSAIDS including LY450139 [162, 163] and R-flurbiprofen [164] do have this activity. LY450139, given in doses ranging from 5–50 mg/day over 14 days to normal human volunteers, decreased plasma Aβ concentrations up to 40% in a dose-dependent manner. Unfortunately however, CSF Aβ concentrations were unchanged. Furthermore, at the 50 mg/d dose, adverse events that were possibly drug-related were noted. R-flurbiprofen is currently in phase III testing (N = 2400). In a one-year phase II study (N = 207) of subjects with mild-to-moderate AD receiving 400 mg b.i.d., 800 mg b.i.d., or placebo, statistical significance was not reached in any memory measures. However, a subset of mild patients receiving the 800 mg b.i.d dose (who developed high blood levels of the drug) demonstrated significant benefits in activities of daily living and overall cognitive function [164]. A recent trial of the new NSAID, trifusal, which also has γ-secretase inhibition activity, was also negative [165]. The primary outcome measure was the change in score on a cognitive assessment (an expanded version of the ADAScog). Of course, these above-named negative trials do not offer evidence that a different NSAID, another dose, a different duration, or another study population would not yield positive results. We and others have pre-clinically examined three diverse therapies; all which lower cerebral amyloid deposits: (1) Aβ immunization, (2) human umbilical cord blood cell (HUBC) transplantation, and (3) flavonoid supplementation, which we suggest may show promise as combination or single treatment(s) in AD patients.
Immunization against Aβ peptide was first explored by Schenk and colleagues at Elan Pharmaceuticals. The vaccine was prepared from Aβ1–42 peptide (the presumed pathogenic form of Aβ found in AD plaques) and complete Freund's adjuvant. This original Aβ vaccine conferred both therapeutic and prophylactic value in terms of reducing CNS amyloid deposits in a transgenic mouse model that overexpressed mutant human APP. In addition to augmenting adaptive immunity, the Elan vaccine generated high-titre Aβ-specific antibodies [166]. The following year, another member of the team at Elan Pharmaceuticals reported a different version of the vaccine using passive transfer of Aβ-specific antibodies to transgenic AD mice [167]. This version of the vaccine was also efficacious, although less-so than the original "active" Aβ vaccine developed by Schenk and co-workers. Elan/Wyeth then developed the vaccine for clinical trials in 2001 (AN-1792), but approximately 6% of patients developed aseptic meningoencephalitis, which was most likely attributed to brain infiltration of activated Aβ-specific T-cells. Two years later, consent for long-term clinical follow-up, post-mortem neuropathological examination, or both, was sought from 80 patients (or their caretakers) who had entered the original Elan AN-1792 phase I trial. The follow-up study was completed in 2006. Twenty participants-15 in the AN1792 group, 5 in the placebo group-died before follow-up started. A further 22 patients-19 in the AN1792 group, three in the placebo group-died during follow-up. Nine of the deceased subjects, all in the AN1792 group, had given consent for post-mortem analysis; one of these who did not die with AD was excluded [168]. In the remaining eight patients who received immunization and who were examined neuropathologically, mean Aβ load was lower than in an unimmunised control group matched for age at death (2.1% [SE 0.7] in treated participants vs 5.1% [0.9] in controls; mean difference 3.0%, 95% CI 0.6–5.4; p = 0.02). Although there was variation in load and degree of plaque removal among immunized subjects, the degree of plaque removal varied significantly with mean antibody response attained during the treatment study period (Kruskal-Wallis p = 0.02). Seven of the eight immunised patients who underwent post-mortem assessment, including those with essentially complete plaque removal, still had severe end stage dementia before death. Further, in the whole cohort, there was no evidence of improved survival (hazard ratio 0.93, 95% CI 0.43–3.11; p = 0.86) or of an extension of the time until severe dementia (1.18, 0.45–3.11; p = 0.73) in the AN1792 group versus the placebo group. Thus, it seems that immunization lowered amyloid load but not progressive neurodegeneration [168].
There have been a number of other attempts at Aβ vaccination [169–172]. However, it should be noted that none of these other Aβ vaccines have yet to be tested in humans, and none of the Aβ vaccines tested in mice (with the exception of one unconfirmed report) have produced the meningoencephalitis that occurred in a subset of vaccinated AD patients [173]. Despite this limitation of the mouse models, we have developed a transdermal method to effectively deliver an Aβ vaccine. This version of the vaccine decreases cerebral amyloidosis and does not promote infiltration of T-cells into the brain; but, again, the latter data should be interpreted in the context of lack of brain T-cell infiltrates in most AD mouse models receiving the Aβ vaccine. Specifically, we designed and tested a novel transcutaneous (t.c) Aβ1–42 vaccination strategy along with cholera toxin (CT) as the adjuvant. The vaccine was tested on wild-type mice as well as on PSAPP and Tg2657 transgenic mouse models of AD, which over-express mutant human APP and develop brain β-amyloid plaques. Additionally, the Tg2657 model develops AD-like cerebral amyloid angiopathy (CAA); a pathology which occurs in the majority of AD patients. We found that the vaccine impacted both innate and adaptive immunity in both wild-type and AD transgenic mice and mitigated AD pathology in the transgenic mice [174].
The t.c. Aβ vaccine stimulated an innate immune response in the non-transgenic mice as it caused LCs to migrate to dermal layers containing the Aβ peptide. This response was less frequently observed with administrations of CT only or with PBS, suggesting that this effect may mediate the initial immune response to the vaccine. In addition, there is evidence from our experiments that the immunization stimulated an adaptive immune response as there was an increase in Aβ antibody concentrations (particularly of the IgG1 subtype) as well as Aβ-specific immune responses in splenocytes from vaccinated mice. Since IgG1 antibodies were produced at higher levels than IgG2a or IgG2b, and since there was an 8-fold increase in the secretion of the cardinal Th2 cytokine IL-4 from Aβ "recall" challenged splenocytes, it seems that the t.c. Aβ vaccine promotes an anti-inflammatory Th2 adaptive immune response, which appears similar to the immune responses observed by our group when employing an i.p. Aβ vaccine protocol similar to the original Schenk and colleagues report [175, 176]. The AD mouse models produced a similar adaptive immune response after t.c. Aβ vaccination, with increased concentrations of Aβ antibody and Aβ "recall" challenge-induced production of cytokines by splenocytes.
The above-mentioned alterations in adaptive immunity were associated with reduction of cerebral β-amyloid deposits by 4G8 immunohistochemistry and Congo red histochemistry, and levels of insoluble Aβ1–40,42 were reduced by 50% and 54%, respectively. In addition, Aβ deposits were reduced by 42–58% across hippocampal and cortical areas, brain regions classically regarded as most sensitive to AD-type pathological changes. It deserves mentioning that we did not observe the negative side-effects of cerebral T-cell infiltration (as detected by CD3 antibody) or cerebral microhemorrhage; but, as mentioned above for the former, most Aβ vaccines do not produce this effect in mice. The lack of microhemorrhage in the brains of mice receiving the t.c. Aβ vaccine suggests that the Aβ antibody response was not pathogenic, as the vaccine did not promote break-down of the blood-brain barrier. We also observed an inverse correlation between plasma and brain-soluble Aβ, signifying that circulating Aβ antibodies may be important for clearing Aβ from the brain to the blood; consistent with the "peripheral sink hypothesis" of DeMattos and colleagues [177].
Modulating immunity in AD with human umbilical cord blood cells
Just as vaccination strategies modulate immunity in AD by boosting adaptive immunity, human umbilical cord blood cells (HUCBC) have been shown to have immunomodulatory properties by opposing pro-inflammatory Th1 and stimulating anti-inflammatory Th2 responses in animal models of stroke and AD [178, 179]. HUCBCs have also shown therapeutic benefit in other neuroinflammatory conditions including multiple sclerosis, amyotrophic lateral sclerosis, neurodegenerative macular degeneration, and Parkinson's disease [180–182]. Our recent study showed that intravenous HUCBC infusion into transgenic AD mice resulted in diminished cerebral Aβ/β-amyloid pathology and down-regulation of pro-inflammatory responses in the brain and in the periphery [183]. We attempted to localize HUCBCs to the brain, but were unable to find CNS penetration of these cells, leading us to hypothesize that HUCBCs exerted their effect on reducing cerebral amyloidosis by impacting host immune responses. Although the exact mechanism is still unknown, evidence strongly suggests this therapeutic effect is accomplished by a diminution of Th1 and augmentation of Th2 responses. Importantly, the CD40-CD40L pathway is a key receptor/ligand dyad in initiating and promoting the adaptive immune Th1 response and innate immune responses in mononuclear cells. CD40 receptor, a member of the TNF and nerve growth factor receptor super-family, is expressed on many professional and non-professional APCs, including dendritic cells, B cells, monocytes/macrophages and microglial cells [184–188]. Furthermore, soluble CD40L was shown to be elevated in MS and AD patients [189, 190]. Based on the conspicuous role of the CD40-CD40L interaction in mediating brain pro-inflammatory responses and exacerbating AD-like pathology, we investigated the impact of HUCBC administration to AD mice on CD40 pathway-mediated immune responses [186]. Our results show decreased expression of microglial CD40 and reduction in both CNS and peripheral sCD40L concomitant with HUCBC-induced diminished AD-like pathology, raising the possibility that disruption of CD40-CD40L interaction may be responsible for mitigation of AD-like pathology in this scenario [183]. These results are in accord with those of Vendrame and colleagues, who observed that HUCBC administration conferred rescue of behavioral impairment and brain pathology in mouse models of stroke via upregulation of Th2, and downregulation of Th1 cytokine responses [178].
Flavonoids: a natural strategy to modulate immune responses in AD
A third means by which the aberrant pro-inflammatory responses of inflammaging may be countered thereby reducing AD severity is a group of natural plant-derived compounds known as polyphenols; specifically those known as "flavonoids" from the green tea plant. We and others have focused on the treatment potential of a group of flavonoids, which have been demonstrated to be both anti-inflammatory and anti-amyloidogenic in the context of AD [191–195]. Flavonoids are a large family of compounds synthesized by plants that have a common chemical structure [196]. Extensive analysis of the polyphenolic components of green tea have been conducted. Green tea flavonoids like epigallocatechin gallate (EGCG) appear to promote downregulation of innate immune cell functions. Putative mechanisms of flavonoid action on the innate immune system include direct free radical scavenging [197, 198] as well as reduction of inflammatory cytokine production including TNF-α, IL-1β, and prostaglandin E2 in response to LPS-induced activation [199]. In accord with these findings, activated microglia co-cultured with neuroblastoma cells were less neurotoxic in the presence of the flavonoid fisetin, suggesting that some flavonoids may act to inhibit proinflammatory innate immune responses [200, 201]. Some flavonoids, including silibinin and EGCG, may modulate adaptive T-cell mediated immune function by downregulating innate immune stimulating cytokines that promote Th1 immunity (e.g., TNF-α) and by promoting Th2 cytokines. These effects are thought to be mediated in part via downregulation of NFκB signaling [202–206]. Recently, we have focused on three flavonoids as having therapeutic potential for AD: EGCG, luteolin, and diosmin.
EGCG inhibits TNF-α-induced production of monocyte chemotactic protein-1 from vascular endothelial cells [207]. Furthermore, EGCG also displays the ability to suppress neuron death mediated by activated microglia [208]. Interestingly, green tea polyphenols and unripened apple polyphenols appear to downregulate CD11b on bovine peripheral monocytes, suggesting that leukocyte adhesion and migration mediated in part by CD11b/CD18 could be inhibited after treatment with these compounds. In the same study, unripened apple polyphenols upregulated CD11b on γδ T cells, indicating that these compounds also act to modulate the adaptive immune response [209]. In addition, EGCG suppresses endocytotic activity, and inhibits stimulatory activity toward allogeneic T cells [166].
Most recently, we found that treatment of AD model neurons (murine N2a cells transfected with the human "Swedish" mutant form of APP) and primary neuronal cells derived from AD model mice (the Tg2576 mouse model of AD [211] with luteolin yielded significant reduction in Aβ generation [212]. This flavonoid achieves this effect via selective inactivation of glycogen synthase kinase 3-alpha (GSK-3α). Additionally, we observed that administration of a structurally similar flavonoid, diosmin, to Tg2576 mice similarly reduced Aβ generation, and this effect seems to also occur through GSK-3 inhibition. Luteolin has been previously been shown to be a potent free radical scavenger [213, 214], anti-inflammatory agent [215, 216], and immunomodulator [217, 218], and it is possible that these latter two properties are owed to inhibition of GSK-3. Luteolin is also associated with inhibition of NF-κB nuclear translocation, and p38 MAPK signaling [219–221].
Although flavonoid-rich diets and flavonoid administration prevent cognitive impairment associated with inflammaging in animal studies [222–225], retrospective cohort studies are inconsistent in showing an inverse association between dietary flavonoid (e.g., green tea) intake and dementia or neurodegenerative disease risk in humans [226–229]. For example, an epidemiological study of Dutch adults found total dietary flavonoid intake was not associated with the risk of developing AD [226, 227], except in current smokers whose risk of AD decreased by half for every 12 mg increase in daily flavonoid intake. On the other hand, elderly French men and women with the lowest flavonoid intakes had a risk of developing dementia over the next 5 years that was 50% higher than those with the highest intakes [228]. Thus, future human studies (ideally randomized clinical trials) will be required which involve supplementation with relatively high doses of specific purified flavanoids to shed light on the apparent inverse risk relationship with AD (and whether this occurs by reducing inflammaging) and to determine if such compounds are therapeutically beneficial.
Conclusion
In this review we have attempted to give a perspective on the immunologic aspects of aging as they may relate to AD, particularly the increased innate immunity by cells of the mononuclear phagocyte lineage. While the over-active innate immunity characteristic of inflammaging may remain sub-clinical in many elderly individuals, in others it may represent a significant risk for development of one or more aging-associated diseases, including AD. Bringing all this together, it is clear that the association between inflammation, aging, and AD, as suggested by the wealth of epidemiological, clinical and laboratory data, is based on a series of complex molecular and cellular changes that we are only just beginning to uncover and understand. The initial signs are promising but further work is needed in this area to research dietary, cell-based, and pharmacological strategies which oppose the features of inflammaging and thus allow for prevention or treatment of AD.
Abbreviations
- AD:
-
Alzheimer's disease
- IL:
-
Interleukin
- TNF:
-
Tumor necrosis factor
- EGCG:
-
Epigallocatchin gallate
- HUCBC:
-
Human umbilical cord blood cells.
References
Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M, Cevenini E, Castellani GC, Salvioli S: Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007, 128: 92-105. 10.1016/j.mad.2006.11.016.
Salvioli S, Capri M, Valensin S, Tieri P, Monti D, Ottaviani E, Franceschi C: Inflamm-aging, cytokines and aging: State of the art, new hypotheses on the role of mitochondria and new perspectives from systems biology. Curr Pharm Des. 2006, 12: 3161-3171. 10.2174/138161206777947470.
De Martinis M, Franceschi C, Monti D, Ginaldi L: Inflamm-ageing and lifelong antigenic load as major determinants of ageing rate and longevity. FEBS Lett. 2005, 579: 2035-2039. 10.1016/j.febslet.2005.02.055.
Fagiolo U, Cossarizza A, Scala E, Fanales-Belasio E, Ortolani C, Cozzi E, Monti D, Franceschi C, Paganelli R: Increased cytokine production in mononuclear cells of healthy elderly people. Eur J Immunol. 1993, 23: 2375-2378. 10.1002/eji.1830230950.
Franceschi C, Monti D, Sansoni P, Cossarizza A: The immunology of exceptional individuals: The lesson of centenarians. Immunol Today. 1995, 16: 12-16. 10.1016/0167-5699(95)80064-6.
Lio D, Scola L, Crivello A, Colonna-Romano G, Candore G, Bonafé M, Cavallone L, Marchegiani F, Olivieri F, Franceschi C, Caruso C: Inflammation, genetics, and longevity: Further studies on the protective effects in men of IL-10-1082 promoter SNP and its interaction with TNF-alpha -308 promoter SNP. J Med Genet. 2003, 40: 296-299. 10.1136/jmg.40.4.296.
Candore G, Colonna-Romano G, Balistreri CR, Di Carlo D, Grimaldi MP, Listì F, Nuzzo D, Vasto S, Lio D, Caruso C: Biology of longevity: Role of the innate immune system. Rejuvenation Res. 2006, 9: 143-148. 10.1089/rej.2006.9.143.
Vasto S, Candore G, Balistreri CR, Caruso M, Colonna-Romano G, Grimaldi MP, Listi F, Nuzzo D, Lio D, Caruso C: Inflammatory networks in ageing, age-related diseases and longevity. Mech Ageing Dev. 2007, 128: 83-91. 10.1016/j.mad.2006.11.015.
Giunta S: Is inflammaging an auto[innate]immunity subclinical syndrome?. Immun Ageing. 2006, 3: 12-10.1186/1742-4933-3-12.
Bruunsgaard H, Skinhoj P, Qvist J, Pedersen BK: Elderly humans show prolonged in vivo inflammatory activity during pneumococcal infections. J Infect Dis. 1999, 180: 551-554. 10.1086/314873.
Bruunsgaard H, Pedersen BK: The senile immune system. Ugeskr Laeger. 1999, 16: 4740-4743.
Bruunsgaard H, Andersen-Ranberg K, Jeune B, Pedersen AN, Skinhøj P, Pedersen BK: A high plasma concentration of TNF-alpha is associated with dementia in centenarians. J Gerontol A Biol Sci Med Sci. 1999, 54: M357-64.
Zanni F, Vescovini R, Biasini C, Fagnoni F, Zanlari L, Telera A, Di Pede P, Passeri G, Pedrazzoni M, Passeri M, Franceschi C, Sansoni P: Marked increase with age of type 1 cytokines within memory and effector/cytotoxic CD8+ T cells in humans: A contribution to understand the relationship between inflammation and immunosenescence. Exp Gerontol. 2003, 38: 981-987. 10.1016/S0531-5565(03)00160-8.
Giacconi R, Cipriano C, Albanese F, Boccoli G, Saba V, Olivieri F, Franceschi C, Mocchegiani E: The -174G/C polymorphism of IL-6 is useful to screen old subjects at risk for atherosclerosis or to reach successful ageing. Exp Gerontol. 2004, 39: 621-628. 10.1016/j.exger.2003.12.013.
Wikby A, Nilsson BO, Forsey R, Thompson J, Strindhall J, Löfgren S, Ernerudh J, Pawelec G, Ferguson F, Johansson B: The immune risk phenotype is associated with IL-6 in the terminal decline stage: Findings from the swedish NONA immune longitudinal study of very late life functioning. Mech Ageing Dev. 2006, 127: 695-704. 10.1016/j.mad.2006.04.003.
Mannucci PM, Mari D, Merati G, Peyvandi F, Tagliabue L, Sacchi E, Taioli E, Sansoni P, Bertolini S, Franceschi C: Gene polymorphisms predicting high plasma levels of coagulation and fibrinolysis proteins. A study in centenarians. Arterioscler Thromb Vasc Biol. 1997, 17: 755-759.
Mari D, Mannucci PM, Coppola R, Bottasso B, Bauer KA, Rosenbergm R: Hypercoagulability in centenarians: The paradox of successful aging. Blood. 1995, 85: 3144-3149.
Coppola R, Mari D, Lattuada A, Franceschi C: Von willebrand factor in italian centenarians. Haematologica. 2003, 88: 39-43.
Bonfigli AR, Sirolla C, Cenerelli S, Marra M, Boemi M, Franceschi C, Testa I, Mari D, Sacchi E, Testa R: Plasminogen activator inhibitor-1 plasma level increases with age in subjects with the 4G allele at position -675 in the promoter region. Thromb Haemost. 2004, 92: 1164-1165.
Bonafe M, Olivieri F, Cavallone L, Giovagnetti S, Mayegiani F, Cardelli M, Pieri C, Marra M, Antonicelli R, Lisa R, Rizzo MR, Paolisso G, Monti D, Franceschi C: A gender–dependent genetic predisposition to produce high levels of IL-6 is detrimental for longevity. Eur J Immunol. 2001, 31: 2357-2361. 10.1002/1521-4141(200108)31:8<2357::AID-IMMU2357>3.0.CO;2-X.
Cipriano C, Caruso C, Lio D, Giacconi R, Malavolta M, Muti E, Gasparini M, Franceschi C, Mocchegiani E: The -308G/A polymorphism of TNF-alpha influences immunological parameters in old subjects affected by infectious diseases. Int J Immunogenet. 2005, 32: 13-18. 10.1111/j.1744-313X.2005.00490.x.
Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, De Benedictis G: Inflamm-aging. an evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000, 908: 244-254.
Aspinall R, Andrew D: Immunosenescence: Potential causes and strategies for reversal. Biochem Soc Trans. 2000, 28: 250-254.
Aspinall R: Longevity and the immune response. Biogerontology. 2000, 1: 273-278. 10.1023/A:1010046532657.
Linton PJ, Li SP, Zhang Y, Bautista B, Huynh Q, Trinh T, Linton PJ: Intrinsic versus environmental influences on T-cell responses in aging. Immunol Rev. 2005, 205: 207-219. 10.1111/j.0105-2896.2005.00266.x.
Allman D, Miller JP: B cell development and receptor diversity during aging. Curr Opin Immunol. 2005, 17: 463-467.
Effros RB, Dagarag M, Spaulding C, Man J: The role of CD8+ T-cell replicative senescence in human aging. Immunol Rev. 2005, 205: 147-157. 10.1111/j.0105-2896.2005.00259.x.
Pawelec G: When T cells get old. Sci Aging Knowledge Environ. 2005, 2005 (50): pe39-10.1126/sageke.2005.50.pe39.
Fann M, Chiu WK, Wood WH, Levine BL, Becker KG, Weng NP: Gene expression characteristics of CD28null memory phenotype CD8+ T cells and its implication in T-cell aging. Immunol Rev. 2005, 205: 190-206. 10.1111/j.0105-2896.2005.00262.x.
Weng NP: Aging of the immune system: How much can the adaptive immune system adapt?. Immunity. 2006, 24: 495-499. 10.1016/j.immuni.2006.05.001.
Cossarizza A, Ortolani C, Paganelli R, Barbieri D, Monti D, Sansoni P, Fagiolo U, Castellani G, Bersani F, Londei M, Franceschi C: CD45 isoforms expression on CD4+ and CD8+ T cells throughout life, from newborns to centenarians: Implications for T cell memory. Mech Ageing Dev. 1996, 86: 173-195. 10.1016/0047-6374(95)01691-0.
Solana R, Pawelec G, Tarazona R: Aging and innate immunity. Immunity. 2006, 24: 491-494. 10.1016/j.immuni.2006.05.003.
Tarazona R, DelaRosa O, Alonso C, Ostos B, Espejo J, Pena J, Solana R: Increased expression of NK cell markers on T lymphocytes in aging and chronic activation of the immune system reflects the accumulation of effector/senescent T cells. Mech Ageing Dev. 2000, 121: 77-88. 10.1016/S0047-6374(00)00199-8.
Khan N, Shariff N, Cobbold M, Bruton R, Ainsworth JA, Sinclair AJ, Nayak L, Moss PA: Cytomegalovirus seropositivity drives the CD8 T cell repertoire toward greater clonality in healthy elderly individuals. J Immunol. 2002, 169: 1984-1992.
Koch S, Solana R, Dela Rosa O, Pawelec G: Human cytomegalovirus infection and T cell immunosenescence: A mini review. Mech Ageing Dev. 2006, 127: 538-543. 10.1016/j.mad.2006.01.011.
Ouyang Q, Wagner WM, Voehringer D, Wikby A, Klatt T, Walter S, Muller CA, Pircher H, Pawelec G: Age-associated accumulation of CMV-specific CD8+ T cells expressing the inhibitory killer cell lectin-like receptor G1 (KLRG1). Exp Gerontol. 2003, 38: 911-920. 10.1016/S0531-5565(03)00134-7.
Vsecovini R, Telera A, Fagnoni FF, Biasini C, Medici MC, Valcavi P, di Pede P, Lucchini G, Zanlari L, Passeri G, Zanni F, Chezzi C, Franchesci C, Sansoni P: Different contribution of EBV and CMV infections in very long-term carriers to age-related alterations of CD8+ T cells. Exp Gerontol. 2004, 39: 1233-1243. 10.1016/j.exger.2004.04.004.
Ouyang Q, Wanger WM, Wikby A, Walter S, Aubert G, Dodi AI, Travers P, Pawelec G: Large numbers of dysfunctional CD8+ T lymphocytes bearing receptors for a single dominant CMV epitope in the very old. J Clin Immunol. 2003, 23: 247-257. 10.1023/A:1024580531705.
Franceschi C, Valensin S, Fagnoni F, Barbi C, Bonafe M: Biomarkers of immunosenescence within an evolutionary perspective: The challenge of heterogeneity and the role of antigenic load. Exp Gerontol. 1999, 34: 911-921. 10.1016/S0531-5565(99)00068-6.
Goronzy JJ, Weyand CM: T cell development and receptor diversity during aging. Curr Opin Immunol. 2005, 17: 468-475.
Swain S, Clise-Dwyer K, Haynes L: Homeostasis and the age-associated defect of CD4 T cells. Semin Immunol. 2005, 17: 370-377. 10.1016/j.smim.2005.05.007.
Kovaiou RD, Weiskirchner I, Keller M, Pfister G, Cioca DP, Grubeck-Loebensten B: Age-related differences in phenotype and function of CD4+ T cells are due to a phenotypic shift from naive to memory effector CD4+ T cells. Int Immunol. 2005, 17: 1359-1366. 10.1093/intimm/dxh314.
Haynes L, Eaton SM, Burns EM, Randall TD, Swain SL: Newly generated CD4 T cells in aged animals do not exhibit age-related defects in response to antigen. J Exp Med. 2005, 201: 845-851. 10.1084/jem.20041933.
Kang I, Hong MS, Nolasco H, Park SH, Dan JM, Choi JY, Craft Kang I: Age-associated change in the frequency of memory CD4+ T cells impairs long term CD4+ T cell responses to influenza vaccine. J Immunol. 2004, 173: 673-681.
Almanzar G, Schwaiger S, Jenewein B, Keller M, Herndler-Brandstetter D, Wurzner R, Schonitzer D, Grubeck-Lobenstein B: Long-term cytomegalovirus infection leads to significant changes in the composition of the CD8+ T-cell repertoire, which may be the basis for an imbalance in the cytokine production profile in elderly persons. J Virol. 2005, 79: 3675-3683. 10.1128/JVI.79.6.3675-3683.2005.
Azuma M, Phillips JH, Lanier LL: CD28- T lymphocytes. antigenic and functional properties. J Immunol. 1993, 150: 1147-1159.
Effros RB, Dagarag M, Spaulding C, Man J: The role of CD8+ T-cell replicative senescence in human aging. Immunol Rev. 2005, 205: 147-157. 10.1111/j.0105-2896.2005.00259.x.
Tarazona R, DelaRosa O, Alonso C, Ostos B, Espejo J, Pena J, Solana R: Increased expression of NK cell markers on T lymphocytes in aging and chronic activation of the immune system reflects the accumulation of effector/senescent T cells. Mech Ageing Dev. 2000, 121: 77-88. 10.1016/S0047-6374(00)00199-8.
Pawelec G, Akbar A, Caruso C, Solana R, Grubeck-Loebenstein B, Wikby A, Pawelec G: Human immunosenescence: Is it infectious?. Immunol Rev. 2005, 205: 257-268. 10.1111/j.0105-2896.2005.00271.x.
Fann M, Chiu WK, Wood WH, Levine BL, Becker KG, Weng NP: Gene expression characteristics of CD28null memory phenotype CD8+ T cells and its implication in T-cell aging. Immunol Rev. 2005, 205: 190-206. 10.1111/j.0105-2896.2005.00262.x.
Almanzar G, Schwaiger S, Jenewein B, Keller M, Herndler-Brandstetter D, Wurzner R, Schonitzer D, Grubeck-Loebenstein B: Long-term cytomegalovirus infection leads to significant changes in the composition of the CD8+ T-cell repertoire, which may be the basis for an imbalance in the cytokine production profile in elderly persons. J Virol. 2005, 79: 3675-3683. 10.1128/JVI.79.6.3675-3683.2005.
Ghia P, Melchers F, Rolink AG: Age-dependent changes in B lymphocyte development in man and mouse. Exp Gerontol. 2000, 35: 159-165. 10.1016/S0531-5565(99)00095-9.
Szabo P, Shen S, Weksler ME: Age-associated defects in B lymphocyte development. Exp Gerontol. 1999, 34: 431-434. 10.1016/S0531-5565(99)00023-6.
Weksler ME: Changes in the B-cell repertoire with age. Vaccine. 2000, 18: 1624-1628. 10.1016/S0264-410X(99)00497-1.
Yang X, Stedra J, Cerny J: Relative contribution of T and B cells to hypermutation and selection of the antibody repertoire in germinal centers of aged mice. J Exp Med. 1996, 183: 959-970. 10.1084/jem.183.3.959.
Zheng B, Han S, Takahashi Y, Kelsoe G: Immunosenescence and germinal center reaction. Immunol Rev. 1997, 160: 63-77. 10.1111/j.1600-065X.1997.tb01028.x.
Kraft R, Bachmann M, Bachmann K, Buerki H, Hess MW, Cottier H, Stoner RD: Satisfactory primary tetanus antitoxin responses but markedly reduced germinal centre formation in first draining lymph nodes of ageing mice. Clin Exp Immunol. 1987, 67: 447-453.
Franceschi C, Cossarizza A: Introduction: The reshaping of the immune system with age. Int Rev Immunol. 1995, 12: 1-4. 10.3109/08830189509056697.
Frasca D, Riley RL, Blomberg BB: Humoral immune response and B-cell functions including immunoglobulin class switch are downregulated in aged mice and humans. Semin Immunol. 2005, 17: 378-384. 10.1016/j.smim.2005.05.005.
Uyemura K, Castle SC, Makinodan T: The frail elderly: Role of dendritic cells in the susceptibility of infection. Mech Ageing Dev. 2002, 123: 955-962. 10.1016/S0047-6374(02)00033-7.
Perlmutter LS, Scott SA, Barron E, Chui HC: MHC class II-positive microglia in human brain: Association with alzheimer lesions. J Neurosci Res. 1992, 33: 549-558. 10.1002/jnr.490330407.
O'Brien K, Fitzgerald DC, Naiken K, Alugupalli KR, Rostami AM, Gran B: Role of the innate immune system in autoimmune inflammatory demyelination. Curr Med Chem. 2008, 15: 1105-1115. 10.2174/092986708784221458.
Brehin AC, Mouries J, Frenkiel MP, Dadaglio G, Despres P, Lafon M, Couderc T: Dynamics of immune cell recruitment during west nile encephalitis and identification of a new CD19+B220-BST-2+ leukocyte population. J Immunol. 2008, 180: 6760-6767.
Herrero C, Marques L, Lloberas J, Celada A: IFN-gamma-dependent transcription of MHC class II IA is impaired in macrophages from aged mice. J Clin Invest. 2001, 107: 485-493. 10.1172/JCI11696.
Plowden J, Renshaw-Hoelscher M, Engleman C, Katz J, Sambhara S: Innate immunity in aging: Impact on macrophage function. Aging Cell. 2004, 3: 161-167. 10.1111/j.1474-9728.2004.00102.x.
Cumberbatch M, Dearman RJ, Kimber I: Influence of ageing on langerhans cell migration in mice: Identification of a putative deficiency of epidermal interleukin-1beta. Immunology. 2002, 105: 466-477. 10.1046/j.1365-2567.2002.01381.x.
Tan J, Town T, Crawford F, Mori T, DelleDonne A, Crescentini R, Obregon D, Flavell RA, Mullan MJ: Role of CD40 ligand in amyloidosis in transgenic alzheimer's mice. Nat Neurosci. 2002, 5: 1288-1293. 10.1038/nn968.
Medzhitov R, Janeway C: Innate immunity. N Engl J Med. 2000, 343: 338-344. 10.1056/NEJM200008033430506.
Krieger M: The other side of scavenger receptors: Pattern recognition for host defense. Curr Opin Lipidol. 1997, 8: 275-280.
Muzio M, Natoli G, Saccani S, Levrero M, Mantovani A: The human toll signaling pathway: Divergence of nuclear factor kappaB and JNK/SAPK activation upstream of tumor necrosis factor receptor-associated factor 6 (TRAF6). J Exp Med. 1998, 187: 2097-2101. 10.1084/jem.187.12.2097.
Guha M, Mackman N: LPS induction of gene expression in human monocytes. Cell Signal. 2001, 13: 85-94. 10.1016/S0898-6568(00)00149-2.
Fagiolo U, Cossarizza A, Scala E, Fanales-Belasio E, Ortolani C, Cozzi E, Monti D, Franceschi C, Paganelli R: Increased cytokine production in mononuclear cells of healthy elderly people. Eur J Immunol. 1993, 23: 2375-2378. 10.1002/eji.1830230950.
Ershler WB, Sun WH, Binkley N, Gravenstein S, Volk MJ, Kamoske G, Klopp RG, Roecker EB, Daynes RA, Weindruch R: Interleukin-6 and aging: Blood levels and mononuclear cell production increase with advancing age and in vitro production is modifiable by dietary restriction. Lymphokine Cytokine Res. 1993, 12: 225-230.
Clark JA, Peterson TC: Cytokine production and aging: Overproduction of IL-8 in elderly males in response to lipopolysaccharide. Mech Ageing Dev. 1994, 77: 127-139. 10.1016/0047-6374(94)90020-5.
O'Mahony L, Holland J, Jackson J, Feighery C, Hennessy TP, Mealy K: Quantitative intracellular cytokine measurement: Age-related changes in proinflammatory cytokine production. Clin Exp Immunol. 1998, 113: 213-219. 10.1046/j.1365-2249.1998.00641.x.
Roubenoff R, Harris TB, Abad LW, Wilson PW, Dallal GE, Dinarello CA: Monocyte cytokine production in an elderly population: Effect of age and inflammation. J Gerontol A Biol Sci Med Sci. 1998, 53: M20-6.
Sadeghi HM, Schnelle JF, Thoma JK, Nishanian P, Fahey JL: Phenotypic and functional characteristics of circulating monocytes of elderly persons. Exp Gerontol. 1999, 34: 959-970. 10.1016/S0531-5565(99)00065-0.
Hasegawa Y, Sawada M, Ozaki N, Inagaki T, Suzumura A: Increased soluble tumor necrosis factor receptor levels in the serum of elderly people. Gerontology. 2000, 46: 185-188. 10.1159/000022157.
Mariani E, Meneghetti A, Neri S, Ravaglia G, Forti P, Cattini L, Facchini A: Chemokine production by natural killer cells from nonagenarians. Eur J Immunol. 2002, 32: 1524-1529. 10.1002/1521-4141(200206)32:6<1524::AID-IMMU1524>3.0.CO;2-E.
Hayek MG, Mura C, Wu D, Beharka AA, Han SN, Paulson KE, Hwang D, Meydani SN: Enhanced expression of inducible cyclooxygenase with age in murine macrophages. J Immunol. 1997, 159: 2445-2451.
Saurwein-Teissl M, Lung TL, Marx F, Gschosser C, Asch E, Blasko I, Parson W, Bock G, Schonitzer D, Trannoy E, Grubeck-Loebenstein B: Lack of antibody production following immunization in old age: Association with CD8(+)CD28(-) T cell clonal expansions and an imbalance in the production of Th1 and Th2 cytokines. J Immunol. 2002, 168: 5893-5899.
Bandres E, Merino J, Vazquez B, Inoges S, Moreno C, Subira ML, Sanchez-Ibarrola A: The increase of IFN-gamma production through aging correlates with the expanded CD8(+high)CD28(-)CD57(+) subpopulation. Clin Immunol. 2000, 96: 230-235. 10.1006/clim.2000.4894.
Sakata-Kaneko S, Wakatsuki Y, Matsunaga Y, Usui T, Kita T: Altered Th1/Th2 commitment in human CD4+ T cells with ageing. Clin Exp Immunol. 2000, 120: 267-273. 10.1046/j.1365-2249.2000.01224.x.
Karanfilov CI, Liu B, Fox CC, Lakshmanan RR, Whisler RL: Age-related defects in Th1 and Th2 cytokine production by human T cells can be dissociated from altered frequencies of CD45RA+ and CD45RO+ T cell subsets. Mech Ageing Dev. 1999, 109: 97-112. 10.1016/S0047-6374(99)00030-5.
Mbawuike IN, Acuna CL, Walz KC, Atmar RL, Greenberg SB, Couch RB: Cytokines and impaired CD8+ CTL activity among elderly persons and the enhancing effect of IL-12. Mech Ageing Dev. 1997, 94: 25-39. 10.1016/S0047-6374(96)01855-6.
Rink L, Cakman I, Kirchner H: Altered cytokine production in the elderly. Mech Ageing Dev. 1998, 102: 199-209. 10.1016/S0047-6374(97)00153-X.
Bernstein ED, Gardner EM, Abrutyn E, Gross P, Murasko DM: Cytokine production after influenza vaccination in a healthy elderly population. Vaccine. 1998, 16: 1722-1731. 10.1016/S0264-410X(98)00140-6.
Gross PA, Hermogenes AW, Sacks HS, Lau J, Levandowski RA: The efficacy of influenza vaccine in elderly persons. A meta-analysis and review of the literature. Ann Intern Med. 1995, 123: 518-527.
Nichol KL, Margolis KL, Wuorenma J, von Sternberg T: The efficacy and cost effectiveness of vaccination against influenza among elderly persons living in the community. N Engl J Med. 1994, 331: 778-784. 10.1056/NEJM199409223311206.
Saurwein-Teissl M, Lung TL, Marx F, Gschosser C, Asch E, Blasko I, Parson W, Bock G, Schonitzer D, Trannoy E, Grubeck-Loebenstein B: Lack of antibody production following immunization in old age: Association with CD8(+)CD28(-) T cell clonal expansions and an imbalance in the production of Th1 and Th2 cytokines. J Immunol. 2002, 168: 5893-5899.
McGeer PL, McGeer EG: Anti-inflammatory drugs in the fight against alzheimer's disease. Ann N Y Acad Sci. 1996, 777: 213-220. 10.1111/j.1749-6632.1996.tb34421.x.
McGeer PL, McGeer EG: The inflammatory response system of brain: Implications for therapy of alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev. 1995, 21: 195-218. 10.1016/0165-0173(95)00011-9.
Shoenfeld Y, Sherer Y, Harats D: Artherosclerosis as an infectious, inflammatory and autoimmune disease. Trends Immunol. 2001, 22: 293-295. 10.1016/S1471-4906(01)01922-6.
Nguyen TT, Cho SO, Ban JY, Kim JY, Ju HS, Koh SB, Song KS, Seong YH: Neuroprotective Effect of Sanguisorbae Radix against Oxidative Stress-Induced Brain Damage: in Vitro and in Vivo. Biol Pharm Bull. 2008, 31: 2028-35. 10.1248/bpb.31.2028.
Smith M, Richey P, Taneda S, Kutty R, Sayre L, Monnier V, Perry G: Advanced Maillard reaction end products, free radicals, and protein oxidation in Alzheimer's disease. Ann N Y Acad Sci. 1994, 738: 447-454.
Smith M, Sayre L, Monnier V, Perry G: Radical AGEing in Alzheimer's disease. Trends Neurosci. 1995, 18: 172-176. 10.1016/0166-2236(95)93897-7.
Smith M, Rudnicka-Nawro M, Richey P, Praprotnik D, Mulvihill P, Miller C, Sayre L, Perry G: Carbonyl-related posttranslational modification of neurofilament protein in the neurofibrillary pathology of Alzheimer's disease. J Neurochem. 1995, 64: 2660-2666.
Smith M, Perry G, Richey P, Sayre L, Anderson V, Beal M, Kowall N: Oxidative damage in Alzheimer's. Nature. 1996, 382: 120-121. 10.1038/382120b0.
Smith M, Harris P, Sayre L, Perry G: Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci. 1997, 94: 9866-9868. 10.1073/pnas.94.18.9866.
Smith M, Richey Harris P, Sayre L, Beckman J, Perry G: Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997, 17 (8): 2653-2657.
Cras M, Kawai S, Siedlak P, Mulvihill P, Gambetti D, Lowery P, Gonzalez-DeWhitt B, Perry G: Neuronal and microglial involvement in beta-amyloid protein deposition in Alzheimer's disease. Am J Pathol. 1990, 137: 241-246.
Colton C, Gilbert D: Production of superoxide anions by a CNS macrophage, the microglia. FEBS Lett. 1987, 223: 284-288. 10.1016/0014-5793(87)80305-8.
Good P, Werner P, Hsu A, Olanow C, Perl D: Evidence of neuronal oxidative damage in Alzheimer's disease. Am J Pathol. 1996, 149: 21-28.
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T: Inflammation and alzheimer's disease. Neurobiol Aging. 2000, 21: 383-421. 10.1016/S0197-4580(00)00124-X.
Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA: Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001, 60: 759-67.
Richartz-Salzburger E, Batra A, Stransky E, Laske C, Kohler N, Bartels M, Buchkremer G, Schott K: Altered lymphocyte distribution in alzheimer's disease. J Psychiatr Res. 2007, 41: 174-178. 10.1016/j.jpsychires.2006.01.010.
Smith M, Hirai K, Hsiao K, Pappolla M, Harris P, Siedlak S, Tabaton M, Perry G: Amyloid-beta deposition in Alzheimer transgenic mice is associated with oxidative stress. J Neurochem. 1998, 70: 2212-2215.
Butterfield A, Poon H, St Clair D, Keller J, Pierce W, Klein J, Markesbery W: Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer's disease. Neurobiol Dis. 2006, 22: 223-232. 10.1016/j.nbd.2005.11.002.
Keller J, Schmitt F, Scheff S, Ding Q, Chen Q, Butterfield D, Markesbery W: Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005, 64: 1152-1156.
Butterfield D, Reed T, Perluigi M, De Marco C, Coccia R, Cini C, Sultana R: Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci Lett. 2006, 397: 170-173. 10.1016/j.neulet.2005.12.017.
Smith M, Kutty R, Richey P, Yan S, Stern D, Chader G, Wiggert B, Petersen R, Perry G: Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer's disease. Am J Pathol. 1994, 145: 42-47.
Lue LF, Brachova L, Civin WH, Rogers J: Inflammation, A beta deposition, and neurofibrillary tangle formation as correlates of alzheimer's disease neurodegeneration. J Neuropathol Exp Neurol. 1996, 55: 1083-1088.
Webster S, Lue LF, Brachova L, Tenner AJ, McGeer PL, Terai K, Walker DG, Bradt B, Cooper NR, Rogers J: Molecular and cellular characterization of the membrane attack complex, C5b-9, in alzheimer's disease. Neurobiol Aging. 1997, 18: 415-421. 10.1016/S0197-4580(97)00042-0.
Blasko I, Knaus G, Weiss E, Kemmler G, Winkler C, Falkensammer G, Griesmacher A, Wurzner R, Marksteiner J, Fuchs D: Cognitive deterioration in alzheimer's disease is accompanied by increase of plasma neopterin. J Psychiatr Res. 2007, 41: 694-701. 10.1016/j.jpsychires.2006.02.001.
Mrak RE, Griffin WS: Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging. 2005, 26: 349-354. 10.1016/j.neurobiolaging.2004.05.010.
Blasko I, Stampfer-Kountchev M, Robatscher P, Veerhuis R, Eikelenboom P, Grubeck-Loebenstein B: How chronic inflammation can affect the brain and support the development of alzheimer's disease in old age: The role of microglia and astrocytes. Aging Cell. 2004, 3: 169-176. 10.1111/j.1474-9728.2004.00101.x.
Franceschi C, Bonafe M: Centenarians as a model for healthy aging. Biochem Soc Trans. 2003, 31: 457-461. 10.1042/BST0310457.
Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M, Cevenini E, Castellani GC, Salvioli S: Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007, 128: 92-105. 10.1016/j.mad.2006.11.016.
Mutus B, Rabini RA, Franceschi C, Paolisso G, Rizzo MR, Ragno E, Rappelli A, Braconi M, Mazzanti L: Cellular resistance to homocysteine: A key for longevity. Atherosclerosis. 2000, 152: 527-528. 10.1016/S0021-9150(00)00546-3.
Rabini RA, Vignini A, Martarelli D, Nanetti L, Salvolini E, Rizzo MR, Ragno E, Paolisso G, Franceschi C, Mazzanti L: Evidence for reduction of pro-atherosclerotic properties in platelets from healthy centenarians. Exp Gerontol. 2003, 38: 367-371. 10.1016/S0531-5565(02)00268-1.
Nanetti L, Vignini A, Moroni C, Bartolini M, Luzzi S, Provinciali L, Mazzanti L: Peroxynitrite production and NOS expression in astrocytes U373MG incubated with lipoproteins from alzheimer patients. Brain Res. 2005, 1054: 38-44. 10.1016/j.brainres.2005.06.025.
Nanetti L, Moroni C, Vignini A, Vannini P, Franceschi C, Mazzanti L: Age-related changes on platelet membrane: A study on elderly and centenarian monozygotic twins. Exp Gerontol. 2005, 40: 519-525. 10.1016/j.exger.2005.07.002.
Kario K, Matsuo T: Relation between sialic acid concentrations and the haemostatic system in the elderly. BMJ. 1993, 306: 1650-1651.
Troiano L, Pini G, Petruzzi E, Ognibene A, Franceschi C, Monti D, Masotti G, Cilotti A, Forti G: Evaluation of adrenal function in aging. J Endocrinol Invest. 1999, 22: 74-75.
Lio D, Scola L, Crivello A, Colonna-Romano G, Candore G, Bonafe M, Cavallone L, Franceschi C, Caruso C: Gender-specific association between -1082 IL-10 promoter polymorphism and longevity. Genes Immun. 2002, 3: 30-33. 10.1038/sj.gene.6363827.
Lio D, Licastro F, Scola L, Chiappelli M, Grimaldi LM, Crivello A, Colonna-Romano G, Candore G, Franceschi C, Caruso C: Interleukin-10 promoter polymorphism in sporadic alzheimer's disease. Genes Immun. 2003, 4: 234-238. 10.1038/sj.gene.6363964.
Richartz-Salzburger E, Batra A, Stransky E, Laske C, Kohler N, Bartels M, Buchkremer G, Schott K: Altered lymphocyte distribution in alzheimer's disease. J Psychiatr Res. 2007, 41: 174-178. 10.1016/j.jpsychires.2006.01.010.
Murr C, Hainz U, Asch E, Berger P, Jenewein B, Saurwein-Teissl M, Grubeck-Loebenstein B, Fuchs D: Association of increased neopterin production with decreased humoral immunity in the elderly. Exp Gerontol. 2003, 38: 583-587. 10.1016/S0531-5565(03)00062-7.
Wirleitner B, Schroecksnadel K, Winkler C, Fuchs D: Neopterin in HIV-1 infection. Mol Immunol. 2005, 42: 183-194. 10.1016/j.molimm.2004.06.017.
Blasko I, Knaus G, Weiss E, Kemmler G, Winkler C, Falkensammer G, Griesmacher A, Wurzner R, Marksteiner J, Fuchs D: Cognitive deterioration in alzheimer's disease is accompanied by increase of plasma neopterin. J Psychiatr Res. 2007, 41: 694-701. 10.1016/j.jpsychires.2006.02.001.
Leblhuber F, Walli J, Demel U, Tilz GP, Widner B, Fuchs D: Increased serum neopterin concentrations in patients with alzheimer's disease. Clin Chem Lab Med. 1999, 37: 429-431. 10.1515/CCLM.1999.070.
Hull M, Pasinetti GM, Aisen PS: Elevated plasma neopterin levels in alzheimer disease. Alzheimer Dis Assoc Disord. 2000, 14: 228-230. 10.1097/00002093-200010000-00007.
Ikeda T, Yamamoto K, Takahashi K, Yamada M: Immune system-associated antigens on the surface of peripheral blood lymphocytes in patients with alzheimer's disease. Acta Psychiatr Scand. 1991, 83: 444-448. 10.1111/j.1600-0447.1991.tb05573.x.
Singh VK: Studies of neuroimmune markers in alzheimer's disease. Mol Neurobiol. 1994, 9: 73-81. 10.1007/BF02816106.
Skias D, Bania M, Reder AT, Luchins D, Antel JP: Senile dementia of alzheimer's type (SDAT): Reduced T8+-cell-mediated suppressor activity. Neurology. 1985, 35: 1635-1638.
Singh VK, Fudenberg HH, Brown FR: Immunologic dysfunction: Simultaneous study of alzheimer's and older down's patients. Mech Ageing Dev. 1986, 37: 257-264. 10.1016/0047-6374(86)90043-6.
Leffell MS, Lumsden L, Steiger WA: An analysis of T lymphocyte subpopulations in patients with alzheimer's disease. J Am Geriatr Soc. 1985, 33: 4-8.
Shalit F, Sredni B, Brodie C, Kott E, Huberman M: T lymphocyte subpopulations and activation markers correlate with severity of alzheimer's disease. Clin Immunol Immunopathol. 1995, 75: 246-250. 10.1006/clin.1995.1078.
Trieb K, Ransmayr G, Sgonc R, Lassmann H, Grubeck-Loebenstein B: APP peptides stimulate lymphocyte proliferation in normals, but not in patients with alzheimer's disease. Neurobiol Aging. 1996, 17: 541-547. 10.1016/0197-4580(96)00068-1.
Singh VK: tudies of neuroimmune markers in alzheimer's disease. Mol Neurobiol. 1994, 9: 73-81. 10.1007/BF02816106.
Xu J, Hong Y, Shen W, Wang L, Di Q, Zhang Y, Sun Y, Xu W, Hu X: P1–369: The impaired suppression function of peripheral blood CD4+ CD25hi regulatory T cells in patients with Alzheimer's disease. Alz and dementia. 2008, 4: T327-10.1016/j.jalz.2008.05.951.
Schindowski K, Peters J, Gorriz C, Schramm U, Weinandi T, Leutner S, Maurer K, Frolich L, Muller WE, Eckert A: Apoptosis of CD4+ T and natural killer cells in alzheimer's disease. Pharmacopsychiatry. 2006, 39: 220-228. 10.1055/s-2006-954591.
Sastre M, Dewachter I, Landreth GE, Willson TM, Klockgether T, van Leuven F, Heneka MT: Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J Neurosci. 2003, 23: 9796-9804.
Blasko I, Marx F, Steiner E, Hartmann T, Grubeck-Loebenstein B: TNFalpha plus IFNgamma induce the production of alzheimer beta-amyloid peptides and decrease the secretion of APPs. FASEB J. 1999, 13: 63-68.
Mackic JB, Bading J, Ghiso J, Walker L, Wisniewski T, Frangione B, Zlokovic BV: Circulating amyloid-beta peptide crosses the blood-brain barrier in aged monkeys and contributes to alzheimer's disease lesions. Vascul Pharmacol. 2002, 38: 303-313. 10.1016/S1537-1891(02)00198-2.
Zlokovic BV, Deane R, Sallstrom J, Chow N, Miano JM: Neurovascular pathways and alzheimer amyloid beta-peptide. Brain Pathol. 2005, 15: 78-83.
Zhu X, Lee HG, Perry G, Smith MA: Alzheimer disease, the two-hit hypothesis: an update. Biochim Biophys Acta. 2007, 1772: 494-502.
Nagy Z, Esiri M, Smith A: Expression of cell division markers in the hippocampus in Alzheimer's disease and other neurodegenerative conditions. Acta Neuropathol (Berl). 1997, 93: 294-300. 10.1007/s004010050617.
McShea A, Harris P, Webster K, Wahl A, Smith M: Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Am J Pathol. 1997, 150: 1933-1939.
Vincent J, Zheng D, Dickson D, Kress Y, Davies P: Mitotic phosphoepitopes precede paired helical filaments in Alzheimer's disease. Neurobiol Aging. 1998, 19: 287-296. 10.1016/S0197-4580(98)00071-2.
Yang Y, Mufson E, Herrup K: Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer's disease. J Neurosci. 2003, 23: 2557-2563.
McGeer PL, Schulzer M, McGeer EG: Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies. Neurology. 1996, 47: 425-432.
Breitner JC, Gau BA, Welsh KA: Inverse association of anti-inflammatory treatments and Alzheimer's disease: initial results of a co-twin control study. Neurology. 1994, 44: 227-232.
Breitner JCS, Welsh KA, Helms MJ: Delayed onset of Alzheimer's disease with nonsteroidal anti-inflammatory and histamine H2 blocking drugs. Neurobiol Aging. 1995, 16: 523-530. 10.1016/0197-4580(95)00049-K.
Rogers J, Kirby LC, Hempelman SR: Clinical trial of indomethacin in Alzheimer's disease. Neurology. 1993, 43: 1609-1611.
Scharf S, Mander A, Ugoni A: A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer's disease. Neurology. 1999, 53: 197-201.
Aisen PS, Davis KL, Berg JD: A randomized controlled trial of prednisone in Alzheimer's disease. Alzheimer's Disease Cooperative Study. Neurology. 2000, 54: 588-593.
Aisen PS, Schafer KA, Grundman M: Effects of rofecoxib or naproxen versus placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003, 289: 2819-2826. 10.1001/jama.289.21.2819.
Thal LJ, Ferris SH, Kirby L: A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology. 2005, 30: 1204-1215. 10.1038/sj.npp.1300690.
Group AR, Lyketsos CG, Breitner JC: Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology. 2007, 68: 1800-1808. 10.1212/01.wnl.0000260269.93245.d2.
van Gool WA, Weinstein HC, Scheltens PK: Effect of hydroxychloroquine on progression of dementia in early Alzheimer's disease: an 18-month randomised, double-blind, placebo- controlled study. Lancet. 2001, 358: 455-460. 10.1016/S0140-6736(01)05623-9.
Siemers E, Skinner M, Dean R, Gonzales C, Satterwhite J, Farlow M, Ness D, May P: Safety, tolerability, and changes in amyloid beta concentrations after administration of a gamma-secretase inhibitor in volunteers. Clin Neuropharmacol. 2005, 28: 126-132. 10.1097/01.wnf.0000167360.27670.29.
Siemers E, Quinn J, Kaye J, Farlow M, Porsteinsson A, Tariot P, Zoulnouni P, Galvin J, Holtzman D, Knopman D, Satterwhite J, Gonzales C, Dean R, May P: Effects of a gamma-secretase inhibitor in a randomized study of patients with alzheimers disease. Neurology. 2006, 66: 602-604. 10.1212/01.WNL.0000198762.41312.E1.
Geerts H: Drug evaluation: (R)-flurbiprofen–an enantiomer of flurbiprofen for the treatment of alzheimer's disease. IDrugs. 2007, 10: 121-133.
Gómez-Isla T, Blesa R, Boada M, Clarimon J, Del Ser T, Domenech G, Ferro J, Gomez-Anson B, Manubens J, Martinez-Lage J, Munoz D, Pena-Casanova J, Torres F: A randomized, double-blind, placebo controlled-trial of triflusal in mild cognitive impairment: the TRIMCI study. Alzheimer Dis Assoc Disord. 2008, 22: 21-29.
Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P: Immunization with amyloid-beta attenuates alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999, 400: 173-177. 10.1038/22124.
Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T: Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nature medicine. 2000, 6: 916-919. 10.1038/78682.
Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA: Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008, 372: 216-23. 10.1016/S0140-6736(08)61075-2.
Koller MF, Mohajeri MH, Huber M, Wollmer MA, Roth Z'graggen BV, Sandmeier E, Moritz E, Tracy J, Nitsch RM, Christen P: Active immunization of mice with an abeta-Hsp70 vaccine. Neurodegener Dis. 2004, 1: 20-28. 10.1159/000076666.
Frenkel D, Dewachter I, van Leuven F, Solomon B: Reduction of beta-amyloid plaques in brain of transgenic mouse model of alzheimer's disease by EFRH-phage immunization. Vaccine. 2003, 21: 1060-1065. 10.1016/S0264-410X(02)00609-6.
Hara H, Monsonego A, Yuasa K, Adachi K, Xiao X, Takeda S, Takahashi K, Weiner HL, Tabira T: Development of a safe oral abeta vaccine using recombinant adeno-associated virus vector for alzheimer's disease. J Alzheimers Dis. 2004, 6: 483-488.
Zhang J, Wu X, Qin C, Qi J, Ma S, Zhang H, Kong Q, Chen D, Ba D, He W: A novel recombinant adeno-associated virus vaccine reduces behavioral impairment and beta-amyloid plaques in a mouse model of alzheimer's disease. Neurobiol Dis. 2003, 14: 365-379. 10.1016/j.nbd.2003.07.005.
Furlan R, Brambilla E, Sanvito F, Roccatagliata L, Olivieri S, Bergami A, Pluchino S, Uccelli A, Comi G, Martino G: Vaccination with amyloid-beta peptide induces autoimmune encephalomyelitis in C57/BL6 mice. Brain. 2003, 126: 285-291. 10.1093/brain/awg031.
Nikolic WV, Bai Y, Obregon D, Hou H, Mori T, Zeng J, Ehrhart J, Shytle RD, Giunta B, Morgan D, Town T, Tan J: Transcutaneous beta-amyloid immunization reduces cerebral beta-amyloid deposits without T cell infiltration and microhemorrhage. Proc Natl Acad Sci USA. 2007, 104: 2507-2512. 10.1073/pnas.0609377104.
Town T, Vendrame M, Patel A, Poetter D, DelleDonne A, Mori T, Smeed R, Crawford F, Klein T, Tan J, Mullan M: Reduced Th1 and enhanced Th2 immunity after immunization with Alzheimer's beta-amyloid(1–42). Reduced Th1 and enhanced Th2 immunity. J Neuroimmunol. 2002, 132: 49-59. 10.1016/S0165-5728(02)00307-7.
Town T, Tan J, Sansone N, Obregon D, Klein T, Mullan M: Characterization of murine immunoglobulin G antibodies against human amyloid-beta1–42. Neurosci Lett. 2002, 307: 101-104. 10.1016/S0304-3940(01)01951-6.
DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM: Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science. 2002, 295: 2264-2267. 10.1126/science.1067568.
Vendrame M: Anti-inflammatory effects of human cord blood cells in a rat model of stroke. Stem Cells Dev. 2005, 14: 595-604. 10.1089/scd.2005.14.595.
Vendrame M, Cassady J, Newcomb J, Butler T, Pennypacker KR, Zigova T, Sanberg CD, Sanberg PR, Willing AE: Infusion of human umbilical cord blood cells in a rat model of stroke dose-dependently rescues behavioral deficits and reduces infarct volume. Stroke. 2004, 35: 2390-2395. 10.1161/01.STR.0000141681.06735.9b.
El-Badri NS, Hakki A, Saporta S, Liang X, Madhusodanan S, Willing AE, Sanberg CD, Sanberg PR: Cord blood mesenchymal stem cells: Potential use in neurological disorders. Stem Cells Dev. 2006, 15: 497-506. 10.1089/scd.2006.15.497.
Garbuzova-Davis S, Gografe SJ, Sanberg CD, Willing AE, Saporta S, Cameron DF, Desjarlais T, Daily J, Kuzmin-Nichols N, Chamizo W, Klasko SK, Sanberg PR: Maternal transplantation of human umbilical cord blood cells provides prenatal therapy in sanfilippo type B mouse model. FASEB J. 2006, 20: 485-487.
Henning SM, Aronson W, Niu Y, Conde F, Lee NH, Seeram NP, Lee RP, Lu J, Harris DM, Moro A, Hong J, Pak-Shan L, Barnard RJ, Ziaee HG, Csathy G, Go VL, Wang H, Heber D: Tea polyphenols and theaflavins are present in prostate tissue of humans and mice after green and black tea consumption. J Nutr. 2006, 136: 1839-1843.
Nikolic WV, Hou H, Town T, Zhu Y, Giunta B, Sanberg CD, Zeng J, Luo D, Ehrhart J, Mori T, Sanberg PR, Tan J: Peripherally administered human umbilical cord blood cells reduce parenchymal and vascular beta-amyloid deposits in alzheimer mice. Stem Cells Dev. 2008
Tan J, Town T, Paris D, Placzek A, Parker T, Crawford F, Yu H, Humphrey J, Mullan M: Activation of microglial cells by the CD40 pathway: Relevance to multiple sclerosis. J Neuroimmunol. 1999, 97: 77-85. 10.1016/S0165-5728(99)00053-3.
Tan J, Town T, Saxe M, Paris D, Wu Y, Mullan M: Ligation of microglial CD40 results in p44/42 mitogen-activated protein kinase-dependent TNF-alpha production that is opposed by TGF-beta 1 and IL-10. J Immunol. 1999, 163: 6614-6621.
Tan J, Town T, Paris D, Mori T, Suo Z, Crawford F, Mattson MP, Flavell RA, Mullan M: Microglial activation resulting from CD40-CD40L interaction after beta-amyloid stimulation. Science. 1999, 286: 2352-2355. 10.1126/science.286.5448.2352.
Nguyen VT, Walker WS, Benveniste EN: Post-transcriptional inhibition of CD40 gene expression in microglia by transforming growth factor-beta. Eur J Immunol. 1998, 28: 2537-2548. 10.1002/(SICI)1521-4141(199808)28:08<2537::AID-IMMU2537>3.0.CO;2-1.
Carson MJ, Reilly CR, Sutcliffe JG, Lo D: Mature microglia resemble immature antigen-presenting cells. Glia. 1998, 22: 72-85. 10.1002/(SICI)1098-1136(199801)22:1<72::AID-GLIA7>3.0.CO;2-A.
Gerritse K, Laman JD, Noelle RJ, Aruffo A, Ledbetter JA, Boersma WJ, Claassen E: CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci USA. 1996, 93: 2499-2504. 10.1073/pnas.93.6.2499.
Desideri G, Cipollone F, Necozione S, Marini C, Lechiara MC, Taglieri G, Zuliani G, Fellin R, Mezzetti A, di Orio F, Ferri C: Enhanced soluble CD40 ligand and alzheimer's disease: Evidence of a possible pathogenetic role. Neurobiol Aging. 2008, 29: 348-356. 10.1016/j.neurobiolaging.2006.10.019.
Marambaud P, Zhao H, Davies P: Resveratrol promotes clearance of alzheimer's disease amyloid-beta peptides. J Biol Chem. 2005, 280: 37377-37382. 10.1074/jbc.M508246200.
Rezai-Zadeh K, Shytle D, Sun N, Mori T, Hou H, Jeanniton D, Ehrhart J, Townsend K, Zeng J, Morgan D, Hardy J, Town T, Tan J: Green tea epigallocatechin-3-gallate (EGCG) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in alzheimer transgenic mice. J Neurosci. 2005, 25: 8807-8814. 10.1523/JNEUROSCI.1521-05.2005.
Obregon DF, Rezai-Zadeh K, Bai Y, Sun N, Hou H, Ehrhart J, Zeng J, Mori T, Arendash GW, Shytle D, Town T, Tan J: ADAM10 activation is required for green tea (-)-epigallocatechin-3-gallate-induced alpha-secretase cleavage of amyloid precursor protein. J Biol Chem. 2006, 281: 16419-16427. 10.1074/jbc.M600617200.
Giunta B, Obregon D, Hou H, Zeng J, Sun N, Nikolic V, Ehrhart J, Shytle D, Fernandez F, Tan J: EGCG mitigates neurotoxicity mediated by HIV-1 proteins gp120 and tat in the presence of IFN-gamma: Role of JAK/STAT1 signaling and implications for HIV-associated dementia. Brain Res. 2006, 1123: 216-225. 10.1016/j.brainres.2006.09.057.
Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM: Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005, 280: 5892-5901. 10.1074/jbc.M404751200.
Beecher GR: Overview of dietary flavonoids: Nomenclature, occurrence and intake. J Nutr. 2003, 133: 3248S-3254S.
Hashimoto F, Ono M, Masuoka C, Ito Y, Sakata Y, Shimizu K, Nonaka G, Nishioka I, Nohara T: Evaluation of the anti-oxidative effect (in vitro) of tea polyphenols. Biosci Biotechnol Biochem. 2003, 67: 396-401. 10.1271/bbb.67.396.
Bors W, Saran M: Radical scavenging by flavonoid antioxidants. Free Radic Res Commun. 1987, 2: 289-294. 10.3109/10715768709065294.
Zheng LT, Ock J, Kwon BM, Suk K: Suppressive effects of flavonoid fisetin on lipopolysaccharide-induced microglial activation and neurotoxicity. Int Immunopharmacol. 2008, 8: 484-494. 10.1016/j.intimp.2007.12.012.
Zheng LT, Ock J, Kwon BM, Suk K: Suppressive effects of flavonoid fisetin on lipopolysaccharide-induced microglial activation and neurotoxicity. Int Immunopharmacol. 2008, 8: 484-494. 10.1016/j.intimp.2007.12.012.
Kim JS, Jobin C: The flavonoid luteolin prevents lipopolysaccharide-induced NF-kappaB signalling and gene expression by blocking IkappaB kinase activity in intestinal epithelial cells and bone-marrow derived dendritic cells. Immunology. 2005, 115: 375-387. 10.1111/j.1365-2567.2005.02156.x.
Kim JY, Kina T, Iwanaga Y, Noguchi H, Matsumura K, Hyon SH: Tea polyphenol inhibits allostimulation in mixed lymphocyte culture. Cell Transplant. 2007, 16: 75-83. 10.3727/000000007783472381.
Mantena SK, Roy AM, Katiyar SK: Epigallocatechin-3-gallate inhibits photocarcinogenesis through inhibition of angiogenic factors and activation of CD8+ T cells in tumors. Photochem Photobiol. 2005, 81: 1174-1179. 10.1562/2005-04-11-RA-487.
Kang TH, Lee JH, Song CK, Han HD, Shin BC, Pai SI, Hung CF, Trimble C, Lim JS, Kim TW, Wu TC: Epigallocatechin-3-gallate enhances CD8+ T cell-mediated antitumor immunity induced by DNA vaccination. Cancer Res. 2007, 67: 802-811. 10.1158/0008-5472.CAN-06-2638.
Min K, Yoon WK, Kim SK, Kim BH: Immunosuppressive effect of silibinin in experimental autoimmune encephalomyelitis. Arch Pharm Res. 2007, 30: 1265-1272. 10.1007/BF02977779.
Watson JL, Ansari S, Cameron H, Wang A, Akhtar M, McKay DM: Green tea polyphenol (-)-epigallocatechin gallate blocks epithelial barrier dysfunction provoked by IFN-gamma but not by IL-4. Am J Physiol Gastrointest Liver Physiol. 2004, 287: G954-961. 10.1152/ajpgi.00302.2003.
Ahn HY, Xu Y, Davidge ST: Epigallocatechin-3-O-gallate inhibits TNFalpha-induced monocyte chemotactic protein-1 production from vascular endothelial cells. Life Sci. 2008, 82: 964-968. 10.1016/j.lfs.2008.02.018.
Xu Z, Chen S, Li X, Luo G, Li L, Le W: Neuroprotective effects of (-)-epigallocatechin-3-gallate in a transgenic mouse model of amyotrophic lateral sclerosis. Neurochem Res. 2006, 31: 1263-1269. 10.1007/s11064-006-9166-z.
Graff JC, Jutila MA: Differential regulation of CD11b on gammadelta T cells and monocytes in response to unripe apple polyphenols. J Leukoc Biol. 2007, 82: 603-607. 10.1189/jlb.0207125.
Yoneyama S, Kawai K, Tsuno NH, Okaji Y, Asakage M, Tsuchiya T, Yamada J, Sunami E, Osada T, Kitayama J, Takahashi K, Nagawa H: Epigallocatechin gallate affects human dendritic cell differentiation and maturation. J Allergy Clin Immunol. 2008, 121: 209-214. 10.1016/j.jaci.2007.08.026.
Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G: Correlative memory deficits, abeta elevation, and amyloid plaques in transgenic mice. Science. 1996, 274: 99-102. 10.1126/science.274.5284.99.
Rezai-Zadeh K, Shytle RD, Bai Y, Tian J, Hou H, Mori T, Zeng J, Obregon D, Town T, Tan J: Flavonoid-mediated presenilin-1 phosphorylation reduces alzheimer's disease beta-amyloid production. J Cell Mol Med. 2008
Hyun SK, Jung HA, Chung HY, Choi JS: In vitro peroxynitrite scavenging activity of 6-hydroxykynurenic acid and other flavonoids from gingko biloba yellow leaves. Arch Pharm Res. 2006, 29: 1074-1079. 10.1007/BF02969294.
Horvathova K, Novotny L, Tothova D, Vachalkova A: Determination of free radical scavenging activity of quercetin, rutin, luteolin and apigenin in H2O2-treated human ML cells K562. Neoplasma. 2004, 51: 395-399.
Hougee S, Sanders A, Faber J, Graus YM, Berg van den WB, Garssen J, Smit HF, Hoijer MA: Decreased pro-inflammatory cytokine production by LPS-stimulated PBMC upon in vitro incubation with the flavonoids apigenin, luteolin or chrysin, due to selective elimination of monocytes/macrophages. Biochem Pharmacol. 2005, 69: 241-248. 10.1016/j.bcp.2004.10.002.
Verbeek R, Plomp AC, van Tol EA, van Noort JM: The flavones luteolin and apigenin inhibit in vitro antigen-specific proliferation and interferon-gamma production by murine and human autoimmune T cells. Biochem Pharmacol. 2004, 68: 621-629. 10.1016/j.bcp.2004.05.012.
Hirano T, Higa S, Arimitsu J, Naka T, Ogata A, Shima Y, Fujimoto M, Yamadori T, Ohkawara T, Kuwabara Y, Kawai M, Matsuda H, Yoshikawa M, Maezaki N, Tanaka T, Kawase I, Tanaka T: Luteolin, a flavonoid, inhibits AP-1 activation by basophils. Biochem Biophys Res Commun. 2006, 340: 1-7.
Hirano T, Higa S, Arimitsu J, Naka T, Shima Y, Ohshima S, Fujimoto M, Yamadori T, Kawase I, Tanaka T: Flavonoids such as luteolin, fisetin and apigenin are inhibitors of interleukin-4 and interleukin-13 production by activated human basophils. Int Arch Allergy Immunol. 2004, 134: 135-140. 10.1159/000078498.
Hirata Y, Masuda Y, Kakutani H, Higuchi T, Takada K, Ito A, Nakagawa Y, Ishii H: Sp1 is an essential transcription factor for LPS-induced tissue factor expression in THP-1 monocytic cells, and nobiletin represses the expression through inhibition of NF-kappaB, AP-1, and Sp1 activation. Biochem Pharmacol. 2008, 75: 1504-1514. 10.1016/j.bcp.2007.12.019.
Zheng LT, Ock J, Kwon BM, Suk K: Suppressive effects of flavonoid fisetin on lipopolysaccharide-induced microglial activation and neurotoxicity. Int Immunopharmacol. 2008, 8: 484-494. 10.1016/j.intimp.2007.12.012.
Kim JS, Jobin C: The flavonoid luteolin prevents lipopolysaccharide-induced NF-kappaB signalling and gene expression by blocking IkappaB kinase activity in intestinal epithelial cells and bone-marrow derived dendritic cells. Immunology. 2005, 115: 375-387. 10.1111/j.1365-2567.2005.02156.x.
Goyarzu P, Malin DH, Lau FC, Taglialatela G, Moon WD, Jennings R, Moy E, Moy D, Lippold S, Shukitt-Hale B, Joseph JA: Blueberry supplemented diet: Effects on object recognition memory and nuclear factor-kappa B levels in aged rats. Nutr Neurosci. 2004, 7: 75-83. 10.1080/10284150410001710410.
Joseph JA, Denisova NA, Arendash G, Gordon M, Diamond D, Shukitt-Hale B, Morgan D: Blueberry supplementation enhances signaling and prevents behavioral deficits in an alzheimer disease model. Nutr Neurosci. 2003, 6: 153-162. 10.1080/1028415031000111282.
Obregon DF, Rezai-Zadeh K, Bai Y, Sun N, Hou H, Ehrhart J, Zeng J, Mori T, Arendash GW, Shytle D, Town T, Tan J: ADAM10 activation is required for green tea (-)-epigallocatechin-3-gallate-induced alpha-secretase cleavage of amyloid precursor protein. J Biol Chem. 2006, 281: 16419-16427. 10.1074/jbc.M600617200.
Joseph JA, Shukitt-Hale B, Denisova NA, Bielinski D, Martin A, McEwen JJ, Bickford PC: Reversals of age-related declines in neuronal signal transduction, cognitive, and motor behavioral deficits with blueberry, spinach, or strawberry dietary supplementation. J Neurosci. 1999, 19: 8114-8121.
Laurin D, Masaki KH, Foley DJ, White LR, Launer LJ: Midlife dietary intake of antioxidants and risk of late-life incident dementia: The honolulu-asia aging study. Am J Epidemiol. 2004, 159: 959-967. 10.1093/aje/kwh124.
White LR, Petrovitch H, Ross GW, Masaki K, Hardman J, Nelson J, Davis D, Markesbery W: Brain aging and midlife tofu consumption. J Am Coll Nutr. 2000, 19: 242-255.
Engelhart MJ, Geerlings MI, Ruitenberg A, van Swieten JC, Hofman A, Witteman JC, Breteler MM: Dietary intake of antioxidants and risk of alzheimer disease. JAMA. 2002, 287: 3223-3229. 10.1001/jama.287.24.3223.
Commenges D, Scotet V, Renaud S, Jacqmin-Gadda H, Barberger-Gateau P, Dartigues JF: Intake of flavonoids and risk of dementia. Eur J Epidemiol. 2000, 16: 357-363. 10.1023/A:1007614613771.
Acknowledgements
BG is supported by an NIMH K08 Clinical Scientist Career Development Award (1K08MH082642-01A1). TT is supported by an Alzheimer's Association grant and a "Pathway to Independence" award from the NIH/NIA (4R00AG029726-02).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
The authors made the following contributions. BG did the majority of the writing and compilation of the literature. DO wrote the Abeta vaccination section. AT provided editing of the entire manuscript as did EO. FF provided clinical consultion. TT edited the entire manuscript and provided consultation on microglial activation and flavonoids. Finally, JT wrote the section on HUCBC and also edited the entire manuscript.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Giunta, B., Fernandez, F., Nikolic, W.V. et al. Inflammaging as a prodrome to Alzheimer's disease. J Neuroinflammation 5, 51 (2008). https://doi.org/10.1186/1742-2094-5-51
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-2094-5-51