
Abstract
Alzheimer's disease (AD) is a progressive neurodegenerative disease characterized by insidious cognitive decline and memory dysfunction. Synapse loss is the best pathological correlate of cognitive decline in AD and mounting evidence suggests that AD is primarily a disease of synaptic dysfunction. Soluble oligomeric forms of amyloid beta (Aβ), the peptide that aggregates to form senile plaques in the brain of AD patients, have been shown to be toxic to neuronal synapses both in vitro and in vivo. Aβ oligomers inhibit long-term potentiation (LTP) and facilitate long-term depression (LTD), electrophysiological correlates of memory formation. Furthermore, oligomeric Aβ has also been shown to induce synapse loss and cognitive impairment in animals. The molecular underpinnings of these observations are now being elucidated, and may provide clear therapeutic targets for effectively treating the disease. Here, we review recent findings concerning AD pathogenesis with a particular focus on how Aβ impacts synapses.
Similar content being viewed by others

Background
First described by the German neuropathologist Alois Alzheimer in 1906, Alzheimer's disease (AD) is a progressive neurodegenerative disease characterized by insidious cognitive decline and loss of memory function [1, 2]. Over 35 million people are afflicted with AD worldwide, 5.5 million of them in the United States alone, and these numbers are expected to quadruple by 2050 [3]. AD is the sixth leading cause of death in the United States, and remains one of the only causes of death that increased by as much as 66% over the last decade [4]. No disease-modifying drug has been developed for treating AD, making it one of the most pressing public health problems in the world today. Tremendous progress has been made over the last few decades in understanding the underlying biology of the disease. Here we review pertinent research findings concerning AD pathogenesis with a particular focus on how neuronal synapses are impacted in disease progression. Understanding the molecular underpinnings of AD pathogenesis may aid in developing effective therapeutic approaches for combating it.
Neuropathology and Pathogenesis of Alzheimer's disease
AD is characterized pathologically by cortical atrophy, neuronal cell death, neuroinflammation, synapse loss, and the accumulation of two definitive pathological lesions: neurofibrillary tangles and senile plaques [5]. Neurofibrillary tangles (NFTs) deposit within neurons and are composed of hyperphosphoryated tau protein whereas senile plaques occur in the extracellular space and are made up largely of the 38-43 amino acid peptide amyloid-beta (Aβ) [6]. Aβ is believed to be a key trigger of AD pathogenesis, one that is upstream of NFTs. It is formed by the sequential cleavage of the amyloid precursor protein (APP) by β- and γ-secretase, after which Aβ is released into the extracellular space [6]. There, Aβ can assume a variety of conformational states ranging from monomers to soluble oligomers, protofibrils, and fibrils, which aggregate to form plaques [7–9].
Several lines of evidence support the hypothesis that alterations in amyloid processing can lead to AD. First, APP is located on chromosome 21, and Down syndrome patients who have trisomy of chromosome 21 invariably develop AD [10]. Further, individuals with trisomy 21 with a chromosome 21q break such that APP diploidy occurs in the setting of trisomy 21 do not develop clinical or neuropathological AD [11]. Conversely, a small cohort of patients who inherited an extra copy of APP due to microduplication of small portions of chromosome 21q containing the APP locus developed AD-like dementia with plaque deposition [12].
Second, most genetic mutations associated with rare familial early onset AD lead to increased production of Aβ or an increase in Aβ42-to-Aβ40 ratio, which increases the propensity for Aβ aggregation [13]. Mutations leading to early onset familial AD have been found in the APP gene on chromosome 21q [14], in the presenilin 1 gene (PSEN 1) on chromosome 14q, and the presenilin 2 gene (PSEN 2, a homolog of PSEN 1) located on chromosome 1q [13]. Presinilin forms the catalytic site of γ-secretase, which is one of the enzymes involved in the cleavage of APP to form Aβ [15–17] All of these mutations influence Aβ metabolism and production [18, 19].
Third, Aβ has been shown to be toxic to neurons in vitro and in vivo [6]. Injecting synthetic or naturally secreted Aβ, at concentrations akin to those seen in the brains of AD patients, into the brains of rodents induces behavioral deficits and tau hyperphosphorylation [5].
Fourth, transgenic mouse models overexpressing human APP and/or PSEN genes with known familial early onset AD mutations develop amyloid plaque deposition and some of the morphological changes of AD (e.g. synapses loss) [20–22]. While most of these transgenic mice do not develop the typical neuronal cell loss observed in AD, they manifest age-dependent memory impairments and cognitive deficits [20–22].
Finally, immunization of AD transgenic mice with Aβ or anti-Aβ antibodies reduces amyloid plaque deposition, clears existing plaques, and ameliorates cognitive deficits in transgenic mice [23, 24], indicating that removal of Aβ is beneficial to the brain.
Taken together, these findings suggest that Aβ is an essential element in the pathogenesis of AD. The mechanistic link between Aβ and neurodegeneration, however, remains elusive. Mounting evidence suggests that AD is primarily a disease of synaptic dysfunction [25] and it is becoming clear that Aβ, particularly in oligomeric form, is toxic to synapses. There is therefore a growing interest in understanding how oligomeric Aβ induces synaptic dysfunction in AD.
Aβ-mediated synaptic dysfunction in Alzheimer's disease
AD brains are characterized by dramatic synapses loss in mesiotemporal regions [26–29]. Significant synapse loss also occurs in patients with mild cognitive impairment, a harbinger for future AD [30]. In fact, synapse loss is the best pathological correlate of cognitive dysfunction in AD, suggesting that synaptic changes are crucial for AD pathogenesis [28, 31, 32]. Synapse loss is most prominent in the immediate vicinity of senile plaques, suggesting that plaques may be a reservoir of synaptotoxic molecules such as Aβ [33–36]. Indeed, recent studies using multiphoton in vivo imaging revealed a halo of oligomeric Aβ around plaques in the brain of AD transgenic mice suggesting that oligomeric Aβ may exist in equilibrium with plaques in AD [37].
Aβ oligomerizes via an unknown mechanism, adopting several higher order conformations such as soluble dimers, trimers, dodecamers, higher order oligomers (also named Aβ-derived diffusible ligands (ADDL)), protofibrils, and fibrils [38–42]. Most of these higher order Aβ structures have been found to be toxic to neurons. Synthetic Aβ oligomers or natural soluble oligomeric Aβ purified from the media of cultured cells expressing mutant human APP (hAPP) or extracted directly from the brains of AD patients have potent synaptic effects. Sodium dodecyl sulfate (SDS) stable Aβ oligomers, ADDLs and protofibrils [43–47] have all been shown to induce synaptic dysfunction [43–48]. Specifically, oligomeric Aβ inhibits the induction of long-term potentiation (LTP), an electrophysiological correlate of memory formation [41, 44, 49–53]. Biophysical methods such as size exclusion chromatography (SEC) and mass spectroscopy have been used to show that Aβ dimers and trimers are most potent at inhibiting LTP [50, 51]. Inhibitors of Aβ oligomerization rescue impairment of LTP induced by Aβ containing media, suggesting that monomeric Aβ is not a potent inhibitor of LTP [54]. Complementing its effects on LTP inhibition, oligomeric Aβ has also been shown to facilitate the induction of long-term depression (LTD) in hippocampal synapses [52, 55, 56]. Impairments in LTP and facilitation of LTD culminate in synaptic depression and impairments in neuronal networks [57].
Molecular basis of oligomeric Aβ mediated synaptic depression
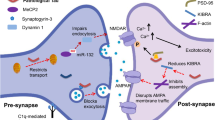
The molecular mechanisms underlying oligomeric Aβ-mediated synapse dysfunction is very complex. Oligomeric Aβ can induce calcium dyshomeostasis, trigger activation of caspases and calcineurin, and modulate the activity of synaptic excitatory receptors and receptor tyrosine kinases, instigating a cascade of molecular events that culminate in the inhibition of LTP, facilitation of LTD, and synapse loss (Figure 1).

Molecular pathways of oligomeric Aβ mediated synaptic dysfunction. Oligomeric Aβ (oAβ) can induce calcium dyshomeostasis, trigger activation of caspase 3, or modulate the activity of NMDARs either directly or through intermediate molecules (shown as X) involved in the trafficking of NMDAR (e.g. EphB2). Activation of different subtypes of NMDA receptors may trigger different intrasynaptic pathways. Activation of NR2A containing NMDARs may lead to high changes in synaptic calcium concentration ([Ca2+]), which triggers downstream events involving CaMKII and pCREB (not shown), facilitating the induction of LTP, which promotes dendritic spine enlargement. Alternatively, activation of NR2B containing NMDAR may trigger a low rise in intrasynaptic calcium, which is favored by oAβ interactions with synapses (away from dotted line pathway), leading to calcineurin (CaN) activation; oAβ-dependent active caspase 3 can also activate CaN. Activated CaN dephosphorylates GluR subunits of AMPARs promoting internalization of AMPARs from the surface of synapses leading to LTD, which leads to dendritic spine shrinkage. Furthermore, active CaN dephosphorylates cofilin rendering it active to depolymerize dendritic spine actin, which leads to dendritic spine collapse and synapse loss.
Physiologically, LTP and LTD depend on calcium influx through N-methyl-D-aspartate (NMDA) receptors and/or activation of metabotropic glutamate receptors (mGluRs) [58–62]. Synapse potentiation or depression depends on the rate of influx of calcium as well as the level of cytosolic calcium. LTP occurs when rapid and high levels of calcium influx occur through NMDA receptors, whereas LTD is favored when low level calcium influx through NMDA receptors occurs [63]. LTP requires the activation of NR2A containing NMDA receptors, whereas LTD requires activation of NR2B containing NMDA receptors [64]. These different subclasses of NMDA receptors have distinct calcium influx kinetics [65, 66] and modulate distinct postsynaptic signaling pathways [67, 68]. LTP is associated with dendritic spine enlargement and increase in synapse density, whereas LTD leads to dendritic spine shrinkage and synapse collapse [69–72]. Several protein kinases such as p38 mitogen-activated protein kinase (MAPK), calcium calmodoulin-dependent protein kinase II (CaMKII), glycogen synthase kinase 3-beta (GSK3β), and ephrin receptor B2 (EphB2) have all been shown to modulate LTP induction in the brain [73, 74]. Phosphatases and proteases such as calcineurin (protein phosphatase 2B [PP2B]) and caspases play key intracellular roles in the induction of LTD [58, 62, 75]. Transcription factors such as the cyclic AMP response element binding protein (CREB) are crucial for the induction of continuous LTP, by increasing the expression of several genes including those encoding brain derive neurotrophic factor (BDNF) and nitric oxide synthase [76, 77].
Oligomeric Aβ has been shown to inhibit LTP and enhance LTD by modulating the activity of all of the above molecular pathways. Oligomeric Aβ-induced loss of excitatory synapses in the hippocampus requires functional NMDA receptors [51]. Several studies have shown that oligomeric Aβ induces partial blockade of NMDA receptor currents, which leads to reduction of calcium influx into spines promoting LTD over LTP [78, 79]. Aβ binds to 7α-nicotinic acetylcholine receptors (nAchR) [80], triggering a series of events that leads to internalization of NMDA receptors via a mechanism requiring calcineurin activation [81]. Reduced calcium influx through NMDA receptors induced by Aβ limits CAMKII function, LTP, and spine enlargement [82]. In fact, oligomeric Aβ-mediated LTP impairment is believed to involve a decrease in the activation of MAPK, CaMKII and Akt/protein kinase B, but not protein kinases A and C [53, 83, 84]. Aβ has also been shown to induce synaptic depression by activating mGluRs, which triggers a series of downstream molecular events involving MAPK and calcineurin, ultimately promoting internalization of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors and synapse collapse [73, 85]. In vivo studies suggest that Aβ indirectly modulates calcineurin activation by causing calcium dysregulation [86–88]. Calcineurin activation promotes the induction of LTD by decreasing surface expression of NMDA receptors and increasing internalization of AMPA receptors via dynamin-mediated endocytosis [79, 89]. Indeed, Aβ-mediated internalization of AMPA [85] and NMDA receptors [81], loss of dendritic spines [85], and cognitive decline [90] can all be rescued by inhibiting calcineurin activation [91–93], indicating that calcineurin plays a crucial role in Aβ-dependent modulation of synaptic plasticity. Further, oligomeric Aβ activation of calcineurin has been shown to induce dendritic simplification, spine loss, and neuritic dystrophies at least in part by activating NFAT (nuclear factor of activated T-cells) pathways both in vitro and in vivo [91]. Oligomeric Aβ has also been shown to activate other synaptic phosphatases such as STEP (striatal-enriched tyrosine phosphatase), which function to dephosphorylate NR2B subunits of NMDA receptors and promote their endocytosis, thereby inducing synaptic depression [94–96].
Oligomeric Aβ can also directly interact with synaptic surface receptor tyrosine kinases that play key roles in LTP and LTD modulation. For instance, it has been shown that oligomeric Aβ binds to the fibronectin domain of EphB2, a receptor tyrosine kinase known to modulate NMDA receptor trafficking and downstream transcription factors such as Fos, which plays a critical role in the induction of LTP [97–100]. Oligomeric Aβ binding to EphB2 promotes its degradation in the proteasome, impairing the induction of LTP [101]. Indeed, EphB2 is depleted in the brains of transgenic hAPP mice and AD patients [102], and replacement of EphB2 reverses cognitive impairment in hAPP mice [101].
Other studies have shown that Aβ facilitates hippocampal LTD via a mechanism that depends on both NMDAR and mGluR activity. Exogenous extracellular glutamate scavengers reverse oligomeric Aβ mediated facilitation of LTD, whereas inhibitors of glutamate reuptake mimic oligomeric Aβ-mediated LTD facilitation, suggesting that the effects of oligomeric Aβ-mediated LTD facilitation may occur as a result of impaired glutamate reuptake at the synapse, leading to post-synaptic NMDA receptor desensitization [55]. Metabotropic glutamate receptor activity, GSK-3β signaling, and protein phosphatase 2B activity are all necessary for oligomeric-Aβ mediated LTD enhancement [55, 73].
Caspase-3 activity has also been found to be crucial for oligomeric Aβ-mediated facilitation of LTD. Soluble Aβ induces caspase-3 activation at a low level that is not sufficient to induce apoptosis [84]. Mitochondria-dependent caspase-3 activation is necessary for physiologic LTD via a mechanism involving Akt proteolysis [75]. Soluble Aβ activates caspase-3, which leads to LTD via a mechanism involving activation of different protein phosphatases that dephosphorylate AMPA receptors and promote their endocytosis from synaptic surfaces, suggesting that prevention of caspase-3 activation may be a viable therapeutic approach for treating AD [84]. Acute inhibition of caspase-3 activity is beneficial, but unfortunately, chronic inhibition of caspase-3 activation beyond the baseline did not reverse cognitive decline in hAPP mice, but instead exacerbated cognitive impairment, possibly due to a requirement for caspase-3 activity in normal synaptic function [84]. Aβ also influences CREB activation, which is crucial for the maintenance of LTP, insofar as CREB regulates the expression of genes necessary for LTP. One study showed that Aβ decreases the activity of CREB and thus reduces expression of genes encoding proteins that are essential for LTP [103]. Another study found that excessive activation of extrasynaptic NR2B-containing NMDA receptors, which leads to downregulation of CREB underlies oligomeric Aβ-mediated LTP inhibition [104].
Oligomeric Aβ causes synapse shrinkage in Alzheimer's disease
The acute effects of Aβ on synaptic physiology appear to translate into structural changes in synaptic morphology because enhanced LTD leads to dendritic spine shrinkage whereas inhibition of LTP limits spine enlargement [69–72]. Exposure of cultured neurons or rat hippocampal slices to oligomeric Aβ induces dendritic spine shrinkage and collapse, a phenomenon that can be reversed by treatment with Aβ antibodies [51, 105]. APP transgenic mice have significant synapse loss and neutralization of oligomeric Aβ with anti-Aβ antibodies leads to reversal of synapse collapse [106–108]. Furthermore, increased concentration of Aβ may reduce glutamatergic transmission and leads to synapse loss in hAPP transgenic mice even before plaque formation [21, 109, 110]. Oligomeric Aβ-mediated inhibition of LTP and enhancement of LTD lead to dendritic spine loss as a result of F-actin remodeling [105]. LTD accompanied by shrinkage of dendritic spines occurs via a mechanism involving cofilin-mediated depolymerization of actin [71]. Specifically, Aβ indirectly stimulates cofilin binding to actin and induction of actin depolymerization in neuronal cytoskeleton. Binding of cofilin to actin is promoted by dephosphorylation at Ser3 by phosphatase Slingshot, and inhibited by phosphorylation by LIM kinase 1, a process that is modulated by oligomeric Aβ [105]. Indeed, in addition to dendritic spine protein loss, increased amounts of dephosphorylated cofilin have been found in the brain of AD patients [111, 112].
Oligomeric Aβ induces cognitive impairments
The electrochemical and structural effects of oligomeric Aβ on synapses described above may lead to potent behavioral and cognitive deficits in animals. Intra-cerebral injection of synthetic or naturally secreted oligomeric Aβ impairs complex behavior including memory and cognitive function in animals [113–115]. APP transgenic mice with increased soluble Aβ in the brain display dramatic cognitive impairments even before the onset of plaque deposition [21]. Neutralization of soluble oligomeric Aβ with anti-Aβ antibodies reverses behavioral deficits seen in different AD transgenic mice [116–118], suggesting that behavioral deficits in AD transgenic mice are caused by soluble Aβ. Inhibition of oligomeric Aβ formation decreases both histopathological and behavioral AD phenotypes in APP transgenic mice [119], implicating higher order Aβ structures such as soluble oligomeric Aβ, but not Aβ monomers, in AD pathogenesis. Levels of soluble oligomeric Aβ, but not senile plaques, in the brain correlates with severity of memory loss in human AD patients, however, the precise contribution of different Aβ species to cognitive decline is not clear [120].
While it is now well-established that increased oligomeric Aβ levels in the brain leads to synaptic dysfunction, it should be noted that at physiologic levels, Aβ might play a normal role in modulating synaptic activity, which likely becomes deranged in the setting of excess Aβ production or accumulation, leading ultimately to the clinical manifestation of cognitive impairment. Indeed, there is a small but growing body of evidence suggesting that Aβ at low concentrations actually promotes LTP and normal synaptic function [121–124]. Thus, therapeutic approaches aimed at improving cognition by counteracting the toxic effects of Aβ will have to be tailored to target only the toxic function of oligomeric Aβ. Nonspecific total inhibition of Aβ may lead to negative effects on synaptic function and cognition.
Seeing Aβ in action at synapses
Collectively, all of the above evidence suggests that soluble oligomeric Aβ is a potent mediator of cognitive impairment in AD. Oligomeric Aβ inhibits the induction of LTP, lowers the threshold for inducing LTD, and causes synapse collapse, which may ultimately lead to cognitive decline resulting from disrupted neuronal network connectivity [57]. For several years, limitations in the resolution of conventional microscopy techniques made it difficult to ascertain whether oligomeric Aβ directly associates with neuronal synapses and plays a role in their shrinkage and collapse in vivo. Recent advances in high-resolution microscopy techniques have made it possible to address these questions. For example development of array tomography [125, 126], an ultra-high resolution fluorescence imaging technique that allow direct simultaneous visualization of several thousand small structures such as synapses and peptides in tissue has allowed determination of whether oligomeric Aβ plays a direct role in synapse loss in AD. Using array tomography and a conformation specific antibody (NAB61) [127], we demonstrated that oligomeric Aβ in the brain of APP/PS1 transgenic mice directly colocalizes with a subset of synapses and is associated with their shrinkage and collapse [37] (Figure 2), suggesting that the in vitro effects of Aβ oligomers observed using cell based assays likely also occur in vivo, supporting the notion that oligomeric Aβ adversely impacts synapses. High-resolution techniques such as array tomography could be extended to study the effects of oligomeric Aβ on synapses in the brain of AD patients. Furthermore, it will be important to determine whether Aβ oligomers are targeted to synapses by specific carrier proteins or whether they are produced locally at synapses. A number of studies have suggested that production of Aβ (at least in monomeric form) is regulated by activity [110, 128–130] and Aβ appears to play a negative feedback function on synaptic activity [110, 131, 132]. Mechanistically, synaptic activity-dependent production of Aβ requires clathrin-mediated endocytosis of APP, which is then cleaved by β- and γ-secretase in late endosomes at synapses to form Aβ [129]. Nonetheless, it is also possible that Aβ binding proteins like apolipoprotein E, which also play a role at the synapse, may stabilize Aβ oligomers [133] in the extracellular space and deliver them to synaptic sites.

Oligomeric Aβ associates with a subset of synapses in the brain of Alzheimer's disease transgenic mice. A) Array tomograms showing oligomeric Aβ (oAβ) localized to synaptic sites near senile plaques in APP/PS1 mice. B) A higher magnification view of the outlined square in panel A showing multiple pre- and post-synaptic elements colocalized with oAβ (arrows) stained with an oAβ specific antibody (NAB61). Scale bar is 10 μm in A and 2 μm in B.
It is presently unclear whether Aβ oligomers interact directly with specific high affinity receptors at the synapse to induce synapse dysfunction. A number of recent studies have reported high affinity binding of oligomeric Aβ to cellular prion protein (PrPC), which was necessary for Aβ to mediate acute synaptic depression, synapse loss, and cognitive impairment in vivo [134, 135]. Subsequent studies, however, could not reproduce these findings [136–138]. This is likely because of differences in experimental paradigms used in the subsequent studies. Single particle tracking of Aβ oligomers labeled with quantum dots exposed to hippocampal neurons in culture have nonetheless demonstrated that the diffusion of Aβ oligomers is dramatically limited upon binding to synaptic sites, suggesting that high affinity oligomeric Aβ receptors may be present at synapses [139]. Identifying these high affinity receptors could aid in designing drugs capable of blocking the deleterious effects of oligomeric Aβ on neuronal synapses.
Concluding remarks
Based on the evidence discussed here, we postulate that AD begins as a disease of synaptic dysfunction and synapse loss then progresses to include widespread neuronal loss and neuronal network failure. Findings from recent experiments continue to provide insight into the complicated molecular underpinnings of synapse dysfunction in AD with mounting evidence pointing to soluble oligomeric Aβ as a key player in the induction of synaptic failure. Oligomeric Aβ activates a variety of molecular cascades that culminate in synapse dysfunction, shrinkage, collapse and loss (Figure 1). These pathological Aβ-triggered molecular events, however, may become independent of Aβ as the disease progresses, with downstream tau effects causing overt neuronal loss, exacerbating the loss of connectivity between neurons [140]. If this is correct, at least two main therapeutic approaches could be taken to combat the disease effectively: 1) early interventions that prevent the initiation of Aβ-triggered pathological events; or 2) inhibition of specific downstream pathways activated by Aβ. The failure of previous therapeutic approaches aimed at removing toxic Aβ species from the brain (e.g. active immunization with Aβ peptide) in clinical trials may be because they were given to the wrong cohort of patients (i.e. patients with advanced AD, whose Aβ-triggered neuronal events may have become independent of Aβ) [140]. Perhaps, a more effective approach will be to initiate such anti-Aβ therapeutic regimens at very early stages of the disease. For this approach to be successful, highly sensitive and specific biomarkers for diagnosing AD need to be developed to identify AD patients at the very early stages of the disease. For patients who have progressed into symptomatic AD, it will likely be necessary to target pathways downstream of Aβ, including tau hyperphosphorylation and accumulation in the soma, which are linked to neuronal death [141, 142]. In conclusion, Aβ-mediated synaptic dysfunction appears to be an important driving factor in AD pathogenesis and understanding the molecular underpinnings may provide effective therapeutic targets for combating the disease.
References
Alzheimer A: Ubereine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift fur Psychiatrie und Psychisch-Gerichtliche Medizin. 1907, 64: 146-148.
Goedert M, Spillantini MG: A century of Alzheimer's disease. Science. 2006, 314 (5800): 777-781. 10.1126/science.1132814.
Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA: Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol. 2003, 60 (8): 1119-1122. 10.1001/archneur.60.8.1119.
Alzheimer's A, Thies W, Bleiler L: 2011 Alzheimer's disease facts and figures. Alzheimers Dement. 2011, 7 (2): 208-244.
Holtzman DM, Morris JC, Goate AM: Alzheimer's disease: the challenge of the second century. Sci Transl Med. 2011, 3 (77): 77sr71-
Hardy J, Selkoe DJ: The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002, 297 (5580): 353-356. 10.1126/science.1072994.
Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG: Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003, 300 (5618): 486-489. 10.1126/science.1079469.
Lansbury PT: Evolution of amyloid: what normal protein folding may tell us about fibrillogenesis and disease. Proc Natl Acad Sci USA. 1999, 96 (7): 3342-3344. 10.1073/pnas.96.7.3342.
Lansbury PT: In pursuit of the molecular structure of amyloid plaque: new technology provides unexpected and critical information. Biochemistry. 1992, 31 (30): 6865-6870. 10.1021/bi00145a001.
Tanzi RE, Bertram L: Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005, 120 (4): 545-555. 10.1016/j.cell.2005.02.008.
Prasher VP, Farrer MJ, Kessling AM, Fisher EM, West RJ, Barber PC, Butler AC: Molecular mapping of Alzheimer-type dementia in Down's syndrome. Ann Neurol. 1998, 43 (3): 380-383. 10.1002/ana.410430316.
Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, et al: APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006, 38 (1): 24-26. 10.1038/ng1718.
Bertram L, Lill CM, Tanzi RE: The genetics of Alzheimer disease: back to the future. Neuron. 2010, 68 (2): 270-281. 10.1016/j.neuron.2010.10.013.
Bertram L, Tanzi RE: Thirty years of Alzheimer's disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci. 2008, 9 (10): 768-778. 10.1038/nrn2494.
Osenkowski P, Ye W, Wang R, Wolfe MS, Selkoe DJ: Direct and potent regulation of gamma-secretase by its lipid microenvironment. J Biol Chem. 2008, 283 (33): 22529-22540. 10.1074/jbc.M801925200.
Selkoe DJ, Wolfe MS: In search of gamma-secretase: presenilin at the cutting edge. Proc Natl Acad Sci USA. 2000, 97 (11): 5690-5692. 10.1073/pnas.97.11.5690.
Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ: Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999, 398 (6727): 513-517. 10.1038/19077.
Murakami K, Irie K, Morimoto A, Ohigashi H, Shindo M, Nagao M, Shimizu T, Shirasawa T: Neurotoxicity and physicochemical properties of Abeta mutant peptides from cerebral amyloid angiopathy: implication for the pathogenesis of cerebral amyloid angiopathy and Alzheimer's disease. J Biol Chem. 2003, 278 (46): 46179-46187. 10.1074/jbc.M301874200.
Tsubuki S, Takaki Y, Saido TC: Dutch, Flemish, Italian, and Arctic mutations of APP and resistance of Abeta to physiologically relevant proteolytic degradation. Lancet. 2003, 361 (9373): 1957-1958. 10.1016/S0140-6736(03)13555-6.
Games D, Adams D, Alessandrini R, Barbour R, Borthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, et al: Alzheimer-type neuropathology in transgenic mice overexpressing V717F [beta]-amyloid precursor protein. Nature. 1995, 373 (6514): 523-527. 10.1038/373523a0.
Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L: High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000, 20 (11): 4050-4058.
Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH: A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006, 440 (7082): 352-357. 10.1038/nature04533.
Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM: Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004, 43 (3): 321-332. 10.1016/j.neuron.2004.07.003.
Lemere CA, Masliah E: Can Alzheimer disease be prevented by amyloid-beta immunotherapy?. Nat Rev Neurol. 2010, 6 (2): 108-119. 10.1038/nrneurol.2009.219.
Selkoe DJ: Alzheimer's disease is a synaptic failure. Science. 2002, 298 (5594): 789-791. 10.1126/science.1074069.
Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW, Morris JC: Altered expression of synaptic proteins occurs early during progression of Alzheimer's disease. Neurology. 2001, 56 (1): 127-129.
DeKosky ST, Scheff SW: Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990, 27 (5): 457-464. 10.1002/ana.410270502.
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R: Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991, 30 (4): 572-580. 10.1002/ana.410300410.
Davies CA, Mann DM, Sumpter PQ, Yates PO: A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer's disease. J Neurol Sci. 1987, 78 (2): 151-164. 10.1016/0022-510X(87)90057-8.
Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ: Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007, 68 (18): 1501-1508. 10.1212/01.wnl.0000260698.46517.8f.
DeKosky ST, Scheff SW, Styren SD: Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration. 1996, 5 (4): 417-421. 10.1006/neur.1996.0056.
Coleman PD, Yao PJ: Synaptic slaughter in Alzheimer's disease. Neurobiol Aging. 2003, 24 (8): 1023-1027. 10.1016/j.neurobiolaging.2003.09.001.
Lanz TA, Carter DB, Merchant KM: Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol Dis. 2003, 13 (3): 246-253. 10.1016/S0969-9961(03)00079-2.
Moolman DL, Vitolo OV, Vonsattel JP, Shelanski ML: Dendrite and dendritic spine alterations in Alzheimer models. J Neurocytol. 2004, 33 (3): 377-387.
Tsai J, Grutzendler J, Duff K, Gan WB: Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci. 2004, 7 (11): 1181-1183. 10.1038/nn1335.
Spires TL, Hannan AJ: Nature, nurture and neurology: gene-environment interactions in neurodegenerative disease. FEBS Anniversary Prize Lecture delivered on 27 June 2004 at the 29th FEBS Congress in Warsaw. FEBS J. 2005, 272 (10): 2347-2361. 10.1111/j.1742-4658.2005.04677.x.
Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, et al: Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci USA. 2009, 106 (10): 4012-4017. 10.1073/pnas.0811698106.
Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ: Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999, 19 (20): 8876-8884.
Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB: Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J Biol Chem. 1999, 274 (36): 25945-25952. 10.1074/jbc.274.36.25945.
Harper JD, Wong SS, Lieber CM, Lansbury PT: Observation of metastable Abeta amyloid protofibrils by atomic force microscopy. Chem Biol. 1997, 4 (2): 119-125. 10.1016/S1074-5521(97)90255-6.
Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, et al: Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998, 95 (11): 6448-6453. 10.1073/pnas.95.11.6448.
Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT: Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002, 418 (6895): 291-
Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ: The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry. 2000, 39 (35): 10831-10839. 10.1021/bi001048s.
Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ: Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002, 416 (6880): 535-539. 10.1038/416535a.
Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH: A specific amyloid-[beta] protein assembly in the brain impairs memory. 2006, 440 (7082): 352-357.
Enya M, Morishima-Kawashima M, Yoshimura M, Shinkai Y, Kusui K, Khan K, Games D, Schenk D, Sugihara S, Yamaguchi H, et al: Appearance of sodium dodecyl sulfate-stable amyloid beta-protein (Abeta) dimer in the cortex during aging. Am J Pathol. 1999, 154 (1): 271-279. 10.1016/S0002-9440(10)65273-X.
Funato H, Enya M, Yoshimura M, Morishima-Kawashima M, Ihara Y: Presence of sodium dodecyl sulfate-stable amyloid beta-protein dimers in the hippocampus CA1 not exhibiting neurofibrillary tangle formation. Am J Pathol. 1999, 155 (1): 23-28. 10.1016/S0002-9440(10)65094-8.
Roher AE, Chaney MO, Kuo YM, Webster SD, Stine WB, Haverkamp LJ, Woods AS, Cotter RJ, Tuohy JM, Krafft GA, et al: Morphology and toxicity of Abeta-(1-42) dimer derived from neuritic and vascular amyloid deposits of Alzheimer's disease. J Biol Chem. 1996, 271 (34): 20631-20635. 10.1074/jbc.271.34.20631.
Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, et al: Soluble oligomers of beta amyloid (1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002, 924 (2): 133-140. 10.1016/S0006-8993(01)03058-X.
Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ: Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006, 572 (Pt 2): 477-492.
Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL: Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007, 27 (11): 2866-2875. 10.1523/JNEUROSCI.4970-06.2007.
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, et al: Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008, 14 (8): 837-842. 10.1038/nm1782.
Townsend M, Mehta T, Selkoe DJ: Soluble Abeta inhibits specific signal transduction cascades common to the insulin receptor pathway. J Biol Chem. 2007, 282 (46): 33305-33312. 10.1074/jbc.M610390200.
Walsh DM, Townsend M, Podlisny MB, Shankar GM, Fadeeva JV, El Agnaf O, Hartley DM, Selkoe DJ: Certain inhibitors of synthetic amyloid beta-peptide (Abeta) fibrillogenesis block oligomerization of natural Abeta and thereby rescue long-term potentiation. J Neurosci. 2005, 25 (10): 2455-2462. 10.1523/JNEUROSCI.4391-04.2005.
Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D: Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009, 62 (6): 788-801. 10.1016/j.neuron.2009.05.012.
Kim JH, Anwyl R, Suh YH, Djamgoz MB, Rowan MJ: Use-dependent effects of amyloidogenic fragments of (beta)-amyloid precursor protein on synaptic plasticity in rat hippocampus in vivo. J Neurosci. 2001, 21 (4): 1327-1333.
Palop JJ, Mucke L: Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci. 2010, 13 (7): 812-818. 10.1038/nn.2583.
Kemp N, Bashir ZI: Long-term depression: a cascade of induction and expression mechanisms. Prog Neurobiol. 2001, 65 (4): 339-365. 10.1016/S0301-0082(01)00013-2.
Wu J, Rowan MJ, Anwyl R: Long-term potentiation is mediated by multiple kinase cascades involving CaMKII or either PKA or p42/44 MAPK in the adult rat dentate gyrus in vitro. J Neurophysiol. 2006, 95 (6): 3519-3527. 10.1152/jn.01235.2005.
Harney SC, Rowan M, Anwyl R: Long-term depression of NMDA receptor-mediated synaptic transmission is dependent on activation of metabotropic glutamate receptors and is altered to long-term potentiation by low intracellular calcium buffering. J Neurosci. 2006, 26 (4): 1128-1132. 10.1523/JNEUROSCI.2753-05.2006.
Anwyl R: Induction and expression mechanisms of postsynaptic NMDA receptor-independent homosynaptic long-term depression. Prog Neurobiol. 2006, 78 (1): 17-37. 10.1016/j.pneurobio.2005.12.001.
Citri A, Malenka RC: Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. 2008, 33 (1): 18-41. 10.1038/sj.npp.1301559.
Kullmann DM, Lamsa KP: Long-term synaptic plasticity in hippocampal interneurons. Nat Rev Neurosci. 2007, 8 (9): 687-699. 10.1038/nrn2207.
Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT: Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004, 304 (5673): 1021-1024. 10.1126/science.1096615.
Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY: Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994, 368 (6467): 144-147. 10.1038/368144a0.
Chen N, Luo T, Raymond LA: Subtype-dependence of NMDA receptor channel open probability. J Neurosci. 1999, 19 (16): 6844-6854.
Sprengel R, Suchanek B, Amico C, Brusa R, Burnashev N, Rozov A, Hvalby O, Jensen V, Paulsen O, Andersen P, et al: Importance of the intracellular domain of NR2 subunits for NMDA receptor function in vivo. Cell. 1998, 92 (2): 279-289. 10.1016/S0092-8674(00)80921-6.
Sheng M, Pak DT: Ligand-gated ion channel interactions with cytoskeletal and signaling proteins. Annu Rev Physiol. 2000, 62: 755-778. 10.1146/annurev.physiol.62.1.755.
Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H: Structural basis of long-term potentiation in single dendritic spines. Nature. 2004, 429 (6993): 761-766. 10.1038/nature02617.
Nagerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T: Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004, 44 (5): 759-767. 10.1016/j.neuron.2004.11.016.
Zhou Q, Homma KJ, Poo MM: Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004, 44 (5): 749-757. 10.1016/j.neuron.2004.11.011.
Bastrikova N, Gardner GA, Reece JM, Jeromin A, Dudek SM: Synapse elimination accompanies functional plasticity in hippocampal neurons. Proc Natl Acad Sci USA. 2008, 105 (8): 3123-3127. 10.1073/pnas.0800027105.
Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R: Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004, 24 (13): 3370-3378. 10.1523/JNEUROSCI.1633-03.2004.
Tackenberg C, Brandt R: Divergent pathways mediate spine alterations and cell death induced by amyloid-beta, wild-type tau, and R406W tau. J Neurosci. 2009, 29 (46): 14439-14450. 10.1523/JNEUROSCI.3590-09.2009.
Li Z, Jo J, Jia JM, Lo SC, Whitcomb DJ, Jiao S, Cho K, Sheng M: Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell. 2010, 141 (5): 859-871. 10.1016/j.cell.2010.03.053.
Mayr BM, Canettieri G, Montminy MR: Distinct effects of cAMP and mitogenic signals on CREB-binding protein recruitment impart specificity to target gene activation via CREB. Proc Natl Acad Sci USA. 2001, 98 (19): 10936-10941. 10.1073/pnas.191152098.
Mayr B, Montminy M: Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001, 2 (8): 599-609. 10.1038/35085068.
Cummings BJ, Pike CJ, Shankle R, Cotman CW: Beta-amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer's disease. Neurobiol Aging. 1996, 17 (6): 921-933. 10.1016/S0197-4580(96)00170-4.
Mulkey RM, Endo S, Shenolikar S, Malenka RC: Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994, 369 (6480): 486-488. 10.1038/369486a0.
Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB: beta-Amyloid(1-42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J Biol Chem. 2000, 275 (8): 5626-5632. 10.1074/jbc.275.8.5626.
Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, et al: Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005, 8 (8): 1051-1058. 10.1038/nn1503.
Nicoll RA, Malenka RC: Expression mechanisms underlying NMDA receptor-dependent long-term potentiation. Ann N Y Acad Sci. 1999, 868: 515-525. 10.1111/j.1749-6632.1999.tb11320.x.
Jo J, Whitcomb DJ, Olsen KM, Kerrigan TL, Lo SC, Bru-Mercier G, Dickinson B, Scullion S, Sheng M, Collingridge G, et al: Abeta(1-42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3beta. Nat Neurosci. 2011, 14 (5): 545-547. 10.1038/nn.2785.
D'Amelio M, Cavallucci V, Middei S, Marchetti C, Pacioni S, Ferri A, Diamantini A, De Zio D, Carrara P, Battistini L, et al: Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer's disease. Nat Neurosci. 2011, 14 (1): 69-76. 10.1038/nn.2709.
Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R: AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006, 52 (5): 831-843. 10.1016/j.neuron.2006.10.035.
Abdul HM, Sama MA, Furman JL, Mathis DM, Beckett TL, Weidner AM, Patel ES, Baig I, Murphy MP, LeVine H, et al: Cognitive decline in Alzheimer's disease is associated with selective changes in calcineurin/NFAT signaling. J Neurosci. 2009, 29 (41): 12957-12969. 10.1523/JNEUROSCI.1064-09.2009.
Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ: Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008, 59 (2): 214-225. 10.1016/j.neuron.2008.06.008.
Wen Z, Guirland C, Ming GL, Zheng JQ: A CaMKII/calcineurin switch controls the direction of Ca(2+)-dependent growth cone guidance. Neuron. 2004, 43 (6): 835-846. 10.1016/j.neuron.2004.08.037.
Slepnev VI, Ochoa GC, Butler MH, Grabs D, De Camilli P: Role of phosphorylation in regulation of the assembly of endocytic coat complexes. Science. 1998, 281 (5378): 821-824.
Dineley KT, Hogan D, Zhang WR, Taglialatela G: Acute inhibition of calcineurin restores associative learning and memory in Tg2576 APP transgenic mice. Neurobiol Learn Mem. 2007, 88 (2): 217-224. 10.1016/j.nlm.2007.03.010.
Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL, et al: Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. 2010, 30 (7): 2636-2649. 10.1523/JNEUROSCI.4456-09.2010.
Rozkalne A, Hyman BT, Spires-Jones TL: Calcineurin inhibition with FK506 ameliorates dendritic spine density deficits in plaque-bearing Alzheimer model mice. Neurobiol Dis. 2011, 41 (3): 650-654. 10.1016/j.nbd.2010.11.014.
Spires-Jones TL, Kay K, Matsouka R, Rozkalne A, Betensky RA, Hyman BT: Calcineurin inhibition with systemic FK506 treatment increases dendritic branching and dendritic spine density in healthy adult mouse brain. Neurosci Lett. 2011, 487 (3): 260-263. 10.1016/j.neulet.2010.10.033.
Zhang Y, Kurup P, Xu J, Carty N, Fernandez SM, Nygaard HB, Pittenger C, Greengard P, Strittmatter SM, Nairn AC, et al: Genetic reduction of striatal-enriched tyrosine phosphatase (STEP) reverses cognitive and cellular deficits in an Alzheimer's disease mouse model. Proc Natl Acad Sci USA. 2010, 107 (44): 19014-19019. 10.1073/pnas.1013543107.
Kurup P, Zhang Y, Venkitaramani DV, Xu J, Lombroso PJ: The role of STEP in Alzheimer's disease. Channels (Austin). 2010, 4 (5): 347-350.
Kurup P, Zhang Y, Xu J, Venkitaramani DV, Haroutunian V, Greengard P, Nairn AC, Lombroso PJ: Abeta-mediated NMDA receptor endocytosis in Alzheimer's disease involves ubiquitination of the tyrosine phosphatase STEP61. J Neurosci. 2010, 30 (17): 5948-5957. 10.1523/JNEUROSCI.0157-10.2010.
Takasu MA, Dalva MB, Zigmond RE, Greenberg ME: Modulation of NMDA receptor-dependent calcium influx and gene expression through EphB receptors. Science. 2002, 295 (5554): 491-495. 10.1126/science.1065983.
Henderson JT, Georgiou J, Jia Z, Robertson J, Elowe S, Roder JC, Pawson T: The receptor tyrosine kinase EphB2 regulates NMDA-dependent synaptic function. Neuron. 2001, 32 (6): 1041-1056. 10.1016/S0896-6273(01)00553-0.
Dalva MB, Takasu MA, Lin MZ, Shamah SM, Hu L, Gale NW, Greenberg ME: EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell. 2000, 103 (6): 945-956. 10.1016/S0092-8674(00)00197-5.
Chen Y, Fu AK, Ip NY: Bidirectional signaling of ErbB and Eph receptors at synapses. Neuron Glia Biol. 2008, 4 (3): 211-221. 10.1017/S1740925X09990287.
Cisse M, Halabisky B, Harris J, Devidze N, Dubal DB, Sun B, Orr A, Lotz G, Kim DH, Hamto P, et al: Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature. 2011, 469 (7328): 47-52. 10.1038/nature09635.
Simon AM, de Maturana RL, Ricobaraza A, Escribano L, Schiapparelli L, Cuadrado-Tejedor M, Perez-Mediavilla A, Avila J, Del Rio J, Frechilla D: Early changes in hippocampal Eph receptors precede the onset of memory decline in mouse models of Alzheimer's disease. J Alzheimers Dis. 2009, 17 (4): 773-786.
Vitolo OV, Sant'Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M: Amyloid beta -peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci USA. 2002, 99 (20): 13217-13221. 10.1073/pnas.172504199.
Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ: Soluble A{beta} Oligomers Inhibit Long-Term Potentiation through a Mechanism Involving Excessive Activation of Extrasynaptic NR2B-Containing NMDA Receptors. J Neurosci. 2011, 31 (18): 6627-6638. 10.1523/JNEUROSCI.0203-11.2011.
Selkoe DJ: Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008, 192 (1): 106-113. 10.1016/j.bbr.2008.02.016.
Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT: Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005, 25 (31): 7278-7287. 10.1523/JNEUROSCI.1879-05.2005.
Rozkalne A, Spires-Jones TL, Stern EA, Hyman BT: A single dose of passive immunotherapy has extended benefits on synapses and neurites in an Alzheimer's disease mouse model. Brain Res. 2009, 1280: 178-185.
Spires-Jones TL, Mielke ML, Rozkalne A, Meyer-Luehmann M, de Calignon A, Bacskai BJ, Schenk D, Hyman BT: Passive immunotherapy rapidly increases structural plasticity in a mouse model of Alzheimer disease. Neurobiol Dis. 2009, 33 (2): 213-220. 10.1016/j.nbd.2008.10.011.
Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L: Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA. 1999, 96 (6): 3228-3233. 10.1073/pnas.96.6.3228.
Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R: APP processing and synaptic function. Neuron. 2003, 37 (6): 925-937. 10.1016/S0896-6273(03)00124-7.
Ferrer I, Gullotta F: Down's syndrome and Alzheimer's disease: dendritic spine counts in the hippocampus. Acta Neuropathol. 1990, 79 (6): 680-685. 10.1007/BF00294247.
Minamide LS, Striegl AM, Boyle JA, Meberg PJ, Bamburg JR: Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat Cell Biol. 2000, 2 (9): 628-636. 10.1038/35023579.
O'Hare E, Weldon DT, Mantyh PW, Ghilardi JR, Finke MP, Kuskowski MA, Maggio JE, Shephard RA, Cleary J: Delayed behavioral effects following intrahippocampal injection of aggregated A beta (1-42). Brain Res. 1999, 815 (1): 1-10. 10.1016/S0006-8993(98)01002-6.
Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH: Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005, 8 (1): 79-84. 10.1038/nn1372.
Cleary J, Hittner JM, Semotuk M, Mantyh P, O'Hare E: Beta-amyloid(1-40) effects on behavior and memory. Brain Res. 1995, 682 (1-2): 69-74. 10.1016/0006-8993(95)00323-I.
Kotilinek LA, Bacskai B, Westerman M, Kawarabayashi T, Younkin L, Hyman BT, Younkin S, Ashe KH: Reversible memory loss in a mouse transgenic model of Alzheimer's disease. J Neurosci. 2002, 22 (15): 6331-6335.
Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, et al: Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat Neurosci. 2002, 5 (5): 452-457.
Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, Spooner ET, Jiang L, Anwyl R, Selkoe DJ, et al: Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat Med. 2005, 11 (5): 556-561. 10.1038/nm1234.
McLaurin J, Kierstead ME, Brown ME, Hawkes CA, Lambermon MH, Phinney AL, Darabie AA, Cousins JE, French JE, Lan MF, et al: Cyclohexanehexol inhibitors of Abeta aggregation prevent and reverse Alzheimer phenotype in a mouse model. Nat Med. 2006, 12 (7): 801-808. 10.1038/nm1423.
Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL: Alzheimer's disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci USA. 2003, 100 (18): 10417-10422. 10.1073/pnas.1834302100.
Puzzo D, Privitera L, Fa M, Staniszewski A, Hashimoto G, Aziz F, Sakurai M, Ribe EM, Troy CM, Mercken M, et al: Endogenous amyloid-beta is necessary for hippocampal synaptic plasticity and memory. Ann Neurol. 2011, 69 (5): 819-830. 10.1002/ana.22313.
Puzzo D, Privitera L, Leznik E, Fa M, Staniszewski A, Palmeri A, Arancio O: Picomolar Amyloid-{beta} Positively Modulates Synaptic Plasticity and Memory in Hippocampus. 10.1523/JNEUROSCI.2692-08.2008. J Neurosci. 2008, 28 (53): 14537-14545. 10.1523/JNEUROSCI.2692-08.2008.
Venkitaramani DV, Chin J, Netzer WJ, Gouras GK, Lesne S, Malinow R, Lombroso PJ: Beta-amyloid modulation of synaptic transmission and plasticity. J Neurosci. 2007, 27 (44): 11832-11837. 10.1523/JNEUROSCI.3478-07.2007.
Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I: Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci. 2009, 12 (12): 1567-1576. 10.1038/nn.2433.
Micheva KD, O'Rourke N, Busse B, Smith SJ: Array tomography: production of arrays. Cold Spring Harb Protoc. 2010, 2010 (11): pdb prot 5524
Micheva KD, Smith SJ: Array tomography: a new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 2007, 55 (1): 25-36. 10.1016/j.neuron.2007.06.014.
Lee EB, Leng LZ, Zhang B, Kwong L, Trojanowski JQ, Abel T, Lee VM: Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J Biol Chem. 2006, 281 (7): 4292-4299. 10.1074/jbc.M511018200.
Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM: Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005, 48 (6): 913-922. 10.1016/j.neuron.2005.10.028.
Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, Holtzman DM: Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008, 58 (1): 42-51. 10.1016/j.neuron.2008.02.003.
Cirrito JR, May PC, O'Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, et al: In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci. 2003, 23 (26): 8844-8853.
Ting JT, Kelley BG, Lambert TJ, Cook DG, Sullivan JM: Amyloid precursor protein overexpression depresses excitatory transmission through both presynaptic and postsynaptic mechanisms. Proc Natl Acad Sci USA. 2007, 104 (1): 353-358. 10.1073/pnas.0608807104.
Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J: Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci. 2006, 26 (27): 7212-7221. 10.1523/JNEUROSCI.1450-06.2006.
Cerf E, Gustot A, Goormaghtigh E, Ruysschaert JM, Raussens V: High ability of apolipoprotein E4 to stabilize amyloid-{beta} peptide oligomers, the pathological entities responsible for Alzheimer's disease. FASEB J. 2011, 25 (5): 1585-1595. 10.1096/fj.10-175976.
Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM: Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009, 457 (7233): 1128-1132. 10.1038/nature07761.
Barry AE, Klyubin I, Mc Donald JM, Mably AJ, Farrell MA, Scott M, Walsh DM, Rowan MJ: Alzheimer's Disease Brain-Derived Amyloid-{beta}-Mediated Inhibition of LTP In Vivo Is Prevented by Immunotargeting Cellular Prion Protein. J Neurosci. 2011, 31 (20): 7259-7263. 10.1523/JNEUROSCI.6500-10.2011.
Kessels HW, Nguyen LN, Nabavi S, Malinow R: The prion protein as a receptor for amyloid-beta. Nature. 2010, 466 (7308): E3-4; discussion E4-5. 10.1038/nature09217.
Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, et al: Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci USA. 2010, 107 (5): 2295-2300. 10.1073/pnas.0911829107.
Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, Mansuy IM, Aguzzi A: Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol Med. 2010, 2 (8): 306-314. 10.1002/emmm.201000082.
Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, Triller A: Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron. 2010, 66 (5): 739-754. 10.1016/j.neuron.2010.04.029.
Hyman BT: Amyloid-Dependent and Amyloid-Independent Stages of Alzheimer Disease. Arch Neurol. 2011
Spires-Jones TL, Kopeikina KJ, Koffie RM, de Calignon A, Hyman BT: Are Tangles as Toxic as They Look?. J Mol Neurosci. 2011
Spires-Jones TL, de Calignon A, Meyer-Luehmann M, Bacskai BJ, Hyman BT: Monitoring protein aggregation and toxicity in Alzheimer's disease mouse models using in vivo imaging. Methods. 2011, 53 (3): 201-207. 10.1016/j.ymeth.2010.12.009.
Acknowledgements
This work was supported by K99 AG033670-01A1, Alzheimer's disease Drug Discovery Foundation/Association for Frontotemporal Dementias, P50 AG005134, AG12406, and AG08487. RMK is supported by the Harvard Biophysics and Medical Scientist Training Programs (NIH T32 GM07753) and the Paul and Daisy Soros Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
RMK designed the layout and content of the review; RMK collected illustrative data, RMK, BTH, and TLS-J wrote the text; all authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Koffie, R.M., Hyman, B.T. & Spires-Jones, T.L. Alzheimer's disease: synapses gone cold. Mol Neurodegeneration 6, 63 (2011). https://doi.org/10.1186/1750-1326-6-63
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1750-1326-6-63