
Abstract
Synapse loss is an early and invariant feature of Alzheimer's disease (AD) and there is a strong correlation between the extent of synapse loss and the severity of dementia. Accordingly, it has been proposed that synapse loss underlies the memory impairment evident in the early phase of AD and that since plasticity is important for neuronal viability, persistent disruption of plasticity may account for the frank cell loss typical of later phases of the disease. Extensive multi-disciplinary research has implicated the amyloid β-protein (Aβ) in the aetiology of AD and here we review the evidence that non-fibrillar soluble forms of Aβ are mediators of synaptic compromise. We also discuss the possible mechanisms of Aβ synaptotoxicity and potential targets for therapeutic intervention.
Similar content being viewed by others

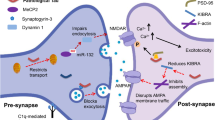

Introduction
Alzheimer's disease (AD) is an irreversible, progressive brain disorder that slowly destroys memory and cognitive skills. It is the most common human dementia and as such confers a huge emotional and economic burden on patients, caregivers and society [1]. Age is the most significant risk factor, with the chance for developing AD doubling every five years after 65 [2, 3]. However, the disease can also strike in mid-life and so-called early-onset AD (EOAD) is designated as dementia developing before 65 years old. Fortunately EOAD is rare with an estimated incidence of 4.2 per 100,000 persons in the 45-64 year age group [4]. The cognitive and pathological changes evident in EOAD and late onset AD (LOAD) are highly similar with the former apparently an accelerated form of the more common LOAD. Because many EOAD cases have a strong Mendelian inheritance pattern they have proved instructive in identifying key gene products involved in the disease process. Specifically, autosomal dominant mutations identified in familial AD all appear to converge on altering the processing of the amyloid precursor protein (APP) [5].
The precise onset of clinical AD is difficult to discern but is often manifested as subtle and intermittent deficits in episodic memory. After many months of gradually progressive impairment of first declarative and then also non-declarative memory, other cognitive symptoms appear and slowly advance. Over a further period of years a profound dementia develops that affects multiple cognitive and behavioral spheres [6]. Pathologically, the Alzheimer brain is characterized by atrophy and microscopically there are decreases in the numbers of neuronal cell bodies in the limbic and association cortices and in certain subcortical nuclei [7–9] and numerous amyloid plaques and neurofibrillary tangles litter the cerebrum [10–12]. Many studies have examined the relationship between cognitive impairment and plaque and tangle counts and while, in general, the number of neurofibrillary tangles correlates better with severity of dementia than the number of amyloid plaques, the most robust correlation in the staging of dementia and early AD is the magnitude of synapse loss [13–15].
Synaptic impairment is likely to be the basis of memory loss in AD
The notion that dementia could result from some sort of synaptic degeneration has been with us for over 100 years and was eloquently expressed by Santiago Ramon y Cajal when he suggested that "dementia could result when synapses between neurons are weakened as a result of a more or less pathological condition, that is, when processes atrophy and no longer form contacts, when cortical mnemonic or association areas suffer partial disorganization" [16]. Quantification using electron microscopy or immunohistochemical staining for synaptic markers has documented significant decreases in synaptic density in the association cortices and hippocampus of AD brain [13, 14, 17–21] and biochemical and immunohistochemical analysis has revealed a similar loss of both pre-synaptic and post-synaptic components [22]. Moreover, the initial decrease in synapse number and density seems disproportionate to the loss of neuronal cell bodies [13, 18, 23], suggesting that pruning of synaptic endings may precede frank neuronal loss. Indeed, synaptic degeneration appears to be an early event in pathogenesis with synapse loss evident in patients with early AD and mild cognitive impairment (MCI) [14, 15, 24].
An important feature of the memory impairment associated with AD and MCI is the selective vulnerability of the ability to consolidate new memories; whereas, the capacity to recall information from the distant past is preserved. This selective impairment of recent memory together with the rapid decay of newly acquired information is reminiscent of the deficits seen in the temporal lobe amnesias and in patients with bilateral hippocampal damage [25, 26]. Analogously, extensive experimentation has demonstrated that the memory deficits observed in aged rodents are highly similar to those in animals with bilateral lesioning of the hippocampus and that age-dependent memory impairment is associated with decreased hippocampal synaptic plasticity. Specifically, estimation of the number of axospinous synapses per neuron in the molecular layer of the dentate gyrus revealed that aged rats with impaired spatial memory had fewer simple and perforated synapses than did memory-intact aged or young rats [27–30]. Moreover, old animals with good spatial memory exhibited similar levels of synaptic enhancement as young animals, whereas memory-impaired old rats had impaired enhancement [31]. The prominent pathology evident in the perforant pathway in the early stages of AD [32] and the documented importance of the hippocampus in encoding the memory of recent events [33–35] argue strongly that disconnection of the hippocampal formation is likely to underlie the progressive memory disturbances in AD.
Irrespective of educational or social status few humans will escape the age-dependent memory impairment that is a frequent companion of old age, yet the majority of elderly will not suffer the ignominy of AD. So, what distinguishes MCI and the early periods of AD from non-pathogenic age-dependent memory loss? Clearly the spectrum from healthy ageing to AD is a very broad one, though not necessarily a continuum, with age-dependent deficits often having a strongly frontal involvement [36, 37]. However, it has been suggested that AD is an extreme and accelerated version of age-related memory loss [38], but that once this accelerated process is initiated it develops a pathogenic profile not seen in normal ageing. Here we propose that the amyloid β-protein (Aβ) is the molecular accelerant that hastens age-related memory decline and triggers AD pathogenesis.
Measures of synaptic plasticity provide a useful read-out to assess AD-related pathophysiology
Hebbs postulate describes a basic mechanism for synaptic plasticity wherein an increase in synaptic efficacy arises from the pre-synaptic cell's repeated and persistent stimulation of the post-synaptic cell [39]. Accordingly, long term potentiation (LTP) and long term depression (LTD) are widely used as models of learning and memory and such processes are believed to play important roles in neural circuits of the brain, with effects lasting for hours, days, or longer [34, 35, 40, 41]. Similar to LTP, the induction of hippocampal LTD requires activation of NMDA receptors and/or metabotropic glutamate receptors (mGluR), depending on the experimental conditions [42, 43]. Mechanistically, the balance between synapse potentiation versus depression is thought to depend on alterations in cytosolic Ca2+ concentration and the differential activation of certain kinases and phosphatases, such as calcium/calmodulin dependent kinase II (CamKII), calcineurin, and cyclic AMP response element binding protein (CREB) [42, 43]. Ultimately, this balance in intraneuronal signalling appears to regulate the stability of the post-synaptic density [44] and the post-synaptic content of AMPA receptors [45, 46]. Together, these structural changes dynamically modulate the magnitude of neurotransmission at the synaptic level.
Anatomical studies in normal rodents suggest that the induction of LTP is associated with spine formation and increased spine volume, whereas the induction of LTD results in decreased spine volume and spine elimination [47–49]. Similarly, processing of information for long-term storage requires specific patterns of activity that lead to modification of synapse structure and eventually to change in neural connectivity [50]. Increases in synapse density and stabilization of dendritic spines in the mouse barrel cortex have been described following whisker stimulation, providing insights into structural changes in synapses associated with learning and memory [51]. It is also well-established that synapses throughout the CNS integrate synaptic activity to maintain and regulate receptors and channels at synapses [40]. Without regulation of synapse activity, maintenance of synaptic proteins is lost, leading to excitotoxicity and degradation of stored memory [40]. Consequently, we believe that studies of synaptic plasticity employing both electrophysiological and morphological measures are essential for developing an understanding of how memory mechanisms are disrupted in disease.
Aβ exists in many different assembly forms
The molecular pathways leading to synapse loss and dysfunction in AD are not well understood, but substantial data indicate that Aβ may be responsible for these affects [52–54]. Over the past 17 years researchers have built up a detailed picture of the natural economy of brain Aβ. The steady state level of Aβ is controlled by its production, degradation and clearance [55, 56] and it is proposed that in disease a defect leading to over-production or decreased clearance causes an accumulation of Aβ. This in turn triggers a pathogenic cascade culminating in the cognitive deficits that characterize AD [57]. Like several other disease-associated proteins, Aβ has the ability to self-associate [58], and can form an array of different assembly forms ranging from dimers all the way to aggregates of fibrils [59]. Initially, it was assumed that toxicity was mediated by fibrillar Aβ similar to that present in amyloid plaques. However, the quantity and temporal progression of amyloid plaques do not correlate well with the clinical progression of the disease [20, 60, 61], thus raising the simple question: if Aβ causes AD, then why doesn't the amount of Aβ in the form of amyloid plaques relate to the severity of dementia?
Recent studies suggest soluble non-fibrillar Aβ assemblies, which go by names such as ADDLs, oligomers, paranuclei, and protofibrils (Table 1) [62–66] may provide the missing link, but as yet the specific form(s) of Aβ which causes injury to neurons in vivo has not been identified. Perhaps the most compelling argument in support of a role for non-fibrillar soluble Aβ comes from a common sense inspection of available data. Extensive multi-disciplinary research provides incontrovertible evidence that Aβ plays an important role in AD [57]. This together with the findings that monomer is innocuous and that amyloid plaques alone cannot account for disease has lead many to conclude, if it isn't fibrillar Aβ (akin to that found in amyloid plaques) and it isn't Aβ monomer then it must be some other form of Aβ. Similarly, it seems reasonable that the synaptic and neuronal compromise seen at sites distant from plaques is mediated by an Aβ species that can readily diffuse and access the space in and surrounding the synaptic cleft.
In recent years biochemical analysis of AD brain have revealed a robust correlation between soluble Aβ levels and the extent of synaptic loss and severity of cognitive impairment [67–70]. But what exactly constitutes soluble Aβ is as yet not well-understood. The term soluble Aβ is an operational definition, embracing all forms of Aβ that remain in aqueous solution following high speed centrifugation of brain extracts [68–71]. Moreover, the origin of soluble Aβ is ambiguous. Extraction of Aβ from brain invariably involves homogenization and consequent cell fracture thus the extracted pool will include truly soluble extracellular Aβ, extracellular Aβ loosely associated or in equilibrium with plaques and a portion of intracellular Aβ. To date, most studies of soluble cerebral Aβ have employed ELISA methods that cannot disclose the aggregation state of the species detected and for the most part appear to preferentially detect Aβ monomer [72–76]. Although the detection methods used provide little information about the assembly state of Aβ the fact that they are not sedimented by ultracentrifugation indicates that they are not mature amyloid fibrils. While a huge amount of data has been gathered concerning the primary sequence of cerebral Aβ only limited attempts have been made to assess the assembly forms of Aβ present in human brain. Using aqueous buffer free of detergents or chaotropes, Kuo and colleagues [71] isolated a range of non-fibrillar forms of Aβ from both AD and control brain. Both sample populations contained a continuous distribution of Aβ species from monomer up to oligomers in excess of 100 kDa, with the major contribution coming from low-n oligomers ranging from dimers to octamers. In agreement with these results we have found that aqueous extracts of AD brain contain a distribution of Aβ species which migrate on non-denaturing size exclusion chromatography (SEC) [77] and glycerol velocity gradient centrifugation (GVGC) over a broad molecular weight range (J. McDonald and DMW unpublished). However, given that Aβ can also bind to other proteins, the molecular weight distribution determined by ultrafiltration, SEC and GVGC cannot be definitively ascribed to homo-oligomers of Aβ.
In a complementary study, McLean and colleagues extracted samples of frontal cortex and putamen in PBS and centrifuged these at 175,000 g for 30 min [68]. Western blot analysis of the supernates from AD brain revealed the presence of variable proportions of monomeric, dimeric and trimeric Aβ species [68]. Such SDS-stable low-n oligomers have also been detected in human CSF by LC-MS [78] and appear to represent highly stable non-covalently associated dimers of Aβ1-40 and trimers of either Aβ6-42 or Aβ1-35. Higher molecular weight SDS-stable assemblies have not been reported in human CSF or soluble extracts of human brain, but whether the dimers and trimers detected represent true low-n oligomers or are breakdown products of larger SDS-unstable assemblies is unclear. Moreover, the presence of SDS-stable dimers and trimers in the soluble fraction of human brain and in extracts of amyloid plaques [68, 74, 75, 79] suggest that in addition to being the earliest mediators of neuronal dysfunction SDS-stable low-n oligomers of Aβ may be the fundamental building blocks of insoluble amyloid deposits.
Transgenic mouse models over-expressing human APP develop many facets of amyloid pathology [80, 81] and have been studied in an effort to identify toxic Aβ assemblies [82–87]. Particularly noteworthy evidence supporting soluble forms of Aβ as the principal mediators of neuronal compromise comes from a report using PDAPP mice in which Aβ-mediated deficits of memory were reversed by a single intraperitoneal injection of an anti-Aβ antibody [88]. In these acute (< 24 hr) experiments, brain amyloid burden was not decreased, suggesting that the antibody was acting on soluble, diffusible species of Aβ and that neutralization or clearing these small intermediates allowed rapid improvement in object recognition performance.
Using another well-characterized APP transgenic mouse model, Tg2576, Lesne and colleagues reported that in brain extracts from Tg2576 mice ~42 kDa (nonamer) and ~56 kDa (dodecamer) Aβ species were detected at an age that coincided with the first observed changes in spatial memory [86]. Aβ monomer, trimer and hexamer were seen at earlier time points and hence were not considered to be associated with a deleterious effect on cognition. Indeed, comparison of spatial memory and the levels of Aβ monomer, trimer, hexamer, nonamer and dodecamer revealed that only nonamer and dodecamer levels correlated with impairment of spatial memory. Importantly, ventricular injection of purified dodecamer into normal pre-trained wild-type rats dramatically perturbed the memory of learned behaviour [86, 89], thus demonstrating that a soluble, brain-derived form of Aβ can directly mediate brain dysfunction. However, it is unlikely that nonamer and dodecamer alone are the only Aβ assemblies capable of altering brain function. For instance, Kawarabayashi and colleagues reported that the appearance of SDS-stable dimers present in lipid rafts also coincided with impairment of spatial memory [85]. In addition, the same Tg2576 mice demonstrate poor performance in a hippocampus-dependent contextual fear conditioning assay, decreased spine density in the dentate gyrus, and impairment of long term potentiation (LTP) at ages long before the first detection of Aβ dodecamer [86, 90, 91]. Thus, while the appearance of dodecamers correlate with the impairment of spatial memory in Tg2576 mice, it does not correlate with changes in other forms of memory, nor do dodecamer levels correlate with changes in synaptic form and function. Indeed, very recent data support the presence of multiple bioactive Aβ species in APP transgenic mouse brain [92, 93]. In J20 APP transgenic mice, over-expression of neprilysin dramatically reduced total Aβ levels, but did not alter dodecamer and SDS-stable Aβ trimer levels nor did it recover spatial reference learning and memory impairments evident in J20 mice. In addition, changes in hippocampal Fos levels and hyperactivity were attributed to a third unidentified species [93]. Similarly, we found the presence of several different Aβ assemblies in the cerebrum of J20 mice, before, coincident with, and after the onset of detectable synapto-dendritic compromise [94] thus it seems likely that more than one Aβ species mediates the various synaptic deficits evident in APP transgenic mice.
SDS-stable low-n oligomers bearing some similarities to those detected in human and mouse brain have been detected in the conditioned medium (CM) and/or lysates of a variety of cell lines [73, 95–100]. Chinese hamster ovarian (CHO) cells that express mutant (V717F) human APP (referred to as 7PA2 cells) produce and secrete low nanomolar amounts of Aβ species that migrate in denaturing SDS-PAGE with molecular weights consistent for Aβ monomer, dimer and trimer [95, 101]. Because of the easy maintenance and fast growth rate of 7PA2 cells, 7PA2 CM has become the media of choice to investigate the biological activities of cell-derived SDS-stable low-n Aβ oligomers. 7PA2 medium containing Aβ low-n oligomers has a variety of plasticity and memory impairing effects. It can block LTP in vivo [102, 103] and in vitro [104, 105], reduce spine density in cultured neurons [100, 106] and decrease the density of synapses in vivo (Freir et al. Cell-derived Aβ oligomers inhibit synapse remodelling necessary for memory consolidation, submitted). When injected into the lateral ventricle 7PA2 CM or oligomer-containing fractions thereof impair performance on operant lever tasks such as the alternating lever cyclic ratio test [89, 107], disrupt working memory in rats tested in a radial-arm maze [99] and show a time-dependent interference with consolidation of avoidance learning (Freir et al. Cell-derived Aβ oligomers inhibit synapse remodelling necessary for memory consolidation, submitted).
Importantly, biologically active non-fibrillar, non-monomeric assemblies of synthetic Aβ have also been identified and such species of Aβ have been shown to exert disease relevant toxicities [63, 64, 94, 100, 108–112]. Preparations containing toxicity mediating forms of Aβ are frequently referred to as oligomers, however, by and large, the assembly state of these have been poorly defined and it is very difficult to compare Aβ preparations used in different studies. Here we will review some of the most commonly encountered and better characterized Aβ preparations. Protofibrils (PFs) have been characterized using a combination of size exclusion chromatography, quasi-elastic light scattering, electron microscopy (EM) and atomic force microscopy (AFM) and derive their name due to the fact that these structures share some physical similarities with amyloid fibrils but appear prior to the detection of fibrils [62, 63]. In solution PFs are highly polydisperse and by EM or AFM range in size from spheres ~5 nm in diameter to curvilinear structures up to 200 nm in length. Their formation is dependent on concentration, pH and ionic strength [113] and they appear to behave as true fibril intermediates in that they can both form fibrils and dissociate to low molecular weight species of Aβ [63, 113]. Annular PFs with external and internal diameters of ~8 and ~2 nm have been detected and seem to be particularly well-populated in preparations of Aβ peptide bearing the Arctic mutation (E22G) [114]. Under conditions where there appeared to be little conversion of PFs to fibrils, addition of PFs to rat cortical cultures caused a time-dependent decrease in neuronal viability as measured by LDH release or an increase in Hoechst staining [108, 115]. Moreover, PFs caused a dose-dependent inhibition of MTT reduction by primary neuronal cultures that was detectable after only 2 hours of incubation with cells [63], an almost immediate enhancement of electrical activity of neurons [108] and a robust block of LTP [116]. It has also been demonstrated that that certain naturally occurring lipids can cause disassembly of fibrils leading to the formation of protofibrils and that when these so-called "backward PFs" are injected into the ventricle can spread rapidly throughout the brain and alter learned behaviour [117].
Aβ-derived diffusible ligands (ADDLs) which are sometimes simply referred to as Aβ oligomers were first detected and studied by Mary Lambert, Bill Klein and colleagues [64]. Until relatively recently there was little physical definition of ADDLs. The original report simply documented the detection by AFM of imperfect spheres ~5-6 nm in diameter which appeared highly similar to the smallest PF species detected by AFM and EM [62, 118, 119]. In contrast solution state analysis of ADDLs using SEC, light scattering and analytical ultracentrifugation indicate that the classic "Klein ADDL preparation" contain a mixture of different sized species and include Aβ assemblies which are not well-detected by EM or AFM [120, 121]. ADDL preparations contain a huge array of species ranging from monomer to large assemblies with molecular weights approaching a million Daltons [120], however, the relative abundance of the high molecular weight species and monomer appear to vary depending on the precise incubation conditions. For instance, Hepler and colleagues found monomer to be the major species in their ADDL preparation [120], whereas Lauren and colleagues found the high molecular weight species to predominant [121].
ADDLs have been shown to cause neuronal death, block LTP [64, 109] and inhibit reduction of MTT by neural cell lines [109, 122, 123]. When incubated with organotypic mouse brain slice cultures for a 24 h period low concentrations of ADDLs (5 nM) caused ~20% loss in cell number whereas at higher concentrations (500 nM) and brief incubation periods (45-60 minutes) cell loss was not evident but a near complete abrogation of LTP was observed [64, 109]. Consistent with their synaptotoxic activity, ADDLs have been shown to avidly bind and decorate dendritic arbors of certain cultured neurons [121, 124, 125] and to mediate dendritic spine loss [111]. The in vivo relevance of ADDLs is suggested by the finding that antibodies (M93 and M94) to synthetic ADDLs prevented Aβ-induced toxicity and that both synthetic Aβ1-42 and some forms of brain-derived Aβ bound specifically to the surface of cultured hippocampal neurons [125, 126]. These antibodies only weakly reacted with monomer and preferentially recognized assembled forms of Aβ [126], and dot blot analysis of soluble extracts from human AD brain revealed a dramatic increase in M93 immunoreactivity compared to control brain [125]. Similarly, soluble brain extracts from old Tg2576 mice displayed significant anti-ADDL immunoreactivity [127]. However, given the highly heterogeneous nature of the synthetic ADDL immunogen used to generate M93 it is unclear exactly what species of Aβ this antibody recognizes.
Besides the use of ADDLs and PFs a number of studies have employed a variety of other non-fibrillar preparations and found these also to be toxic to cultured neurons [65, 128–132]. For example, when Deshpande and colleagues examined the effect of 3 distinct assembly forms of synthetic Aβ, they found that all preparations tested were toxic to primary human cortical neurons, but that the extent and mechanism of toxicity differed [133]. The forms of Aβ investigated were, high molecular weight oligomers (formed as described by [134]) ADDLs and fibrillar Aβ. Low micromolar concentrations (5 μM) of high molecular weight synthetic oligomers caused widespread death within 24 h, whereas similar concentrations of ADDLs took 5-times longer to cause cell loss, and 4-fold higher concentrations of fibrillar Aβ took 10 days to induce only modest cell death. Both high molecular weight oligomers and ADDLs bound rapidly and avidly to synaptic contacts. High molecular weight oligomers caused activation of the mitochondrial death pathway, but activation of this pathway also occurred when sub-lethal levels of the same oligomers were used, suggesting that such changes may underlie defective synaptic activity in neurons that are still viable. In contrast, Wogulis and colleagues found that overt neuronal loss required the presence of both unaggregated soluble Aβ and fibrillar Aβ [135]. The apparent conflict between this report and those ascribing toxicity to a specific Aβ species likely results because of the use of relatively long incubation conditions. During prolonged incubation with neurons intermediates have the potential to further associate and transition to higher ordered aggregates and fibrils have the ability to dissociate Thus such experimental formats render it difficult to unambiguously ascribe cytopathological activity to a discrete species. Other studies have reported that the application of sub-lethal concentrations of various non-fibrillar Aβ assemblies can alter neuronal architecture, cause perturbations in axonal transport and reduced cell surface levels of NMDA receptors [130, 131, 136–139].
The role of low-n oligomers of Aβ in the range of dimer to tetramer is supported by results from studies using peptides bearing disease-associated and design mutations. A mutation associated with EOAD which results in the deletion of glutamic acid 22 produces Aβ which in vitro exhibits accelerated oligomerization without fibrillation and which potently blocks LTP [140]. In contrast, design mutations substituting glycine for leucine within the GxxxG repeat motif of Aβ show a greater propensity for aggregation and decrease the abundance of mass spec-detected dimers and trimers relative to wild type Aβ1-42. Accordingly, such mutants exhibit reduced toxicity when incubated with cultured neurons [141]. Using a similar approach Harmeier and colleagues found that Aβ1-42 peptides containing G33A or G29/33A substitutions formed reduced amounts of low-n oligomers, and that the low-n oligomers formed did not block LTP. This latter finding indicates that aggregation size alone is not the sole determinant of synaptotoxicity, but that the solution structure of assemblies is also critical. Interestingly, in comparison to wild type Aβ, over-expression of these peptides in drosophila resulted in lower cell loss and reduced eye degeneration [142].
In summary, studies using synthetic Aβ peptides, Aβ-containing cell culture medium, APP transgenic mouse and human brain demonstrate that Aβ toxicity is a complex and multifaceted phenomenon that may be induced by multiple assembly forms of Aβ and which can result in a variety of effects ranging from reversible changes in synaptic form and function all the way to frank neuronal loss.
Mechanisms of Aβ-mediated synaptic dysfunction
How Aβ mediates its effects on synaptic plasticity may take many years to fully understand, but already we know that it is likely to involve three different levels. The first and most important mechanism impugns a toxic gain of function for Aβ which results due to self-association and attainment of new structures capable of novel interactions that lead to impaired plasticity. The other two scenarios predicate that Aβ has a normal physiological role. On the one hand insufficient Aβ could lead to a loss of normal function, whereas excess Aβ may precipitate dysfunction.
A growing body of literature supports a physiological role for Aβ in normal synapse function. For instance, in organotypic hippocampal slices, β-secretase activity is increased by synaptic activity and the resulting Aβ peptides depress excitatory transmission through AMPA and NMDA receptors, suggesting a role for Aβ in homeostatic plasticity [143]. Indeed in APP transgenic mouse brain there is a strong positive correlation between synaptic activity and the concentration of Aβ in the interstitial fluid [56] and in humans cerebral Aβ concentration increases as neuronal function and mental status recover in patients with traumatic brain injury [144]. Aβ may also have a role in regulating well-described forms of synaptic plasticity. Specifically, exogenous application of Aβ partially occludes mGluR-dependent LTD, suggesting shared pathways that elicit and modulate, this form of endogenous synaptic plasticity [145]. It has also been suggested that Aβ is important for neuronal survival [146] and it has been shown that picomolar concentrations of human Aβ1-42 increase the magnitude of LTP generated in hippocampus by recruiting an α7-acetylcholine receptors pathway [147]. While a physiologic role for Aβ can be surmised from such studies, the Aβ assembly form responsible for these effects is not known. However, it seems likely that monomeric Aβ would mediate these effects, not least since monomer would be the predominant form of Aβ present in freshly reconstituted synthetic peptide or cortical Aβ. Consequently, pathologic effects on synapse physiology may not only arise from the appearance of higher order Aβ assemblies, which assume a toxic gain of function, but also rising level of monomeric Aβ. Thus increased concentrations of Aβ could lead to synaptic dysregulation mediate by abnormally high levels of monomer and the formation of toxic oligomers. For instance it seems plausible that the increase in non-convulsive seizures observed in APP transgenic mice result due to an Aβ-dependent imbalance of excitatory and inhibitory activity [148]. In this scenario Aβ promotes neuronal over-excitability, which results in GABAergic sprouting of inhibitory synapses as a compensatory mechanism. Similarly, using multiphoton imaging of intraneuronal calcium fluctuations high focal levels of Aβ were shown to increase heterogeneity in the excitability of neurons within 60 μm of amyloid plaques [149]. Moreover, dendritic spine loss observed within 20 μm of amyloid plaques in the Tg2576 APP transgenic mouse provides a structural correlate to the physiologic findings [150].
Interactions between Aβ and various receptors have been shown through biochemical and pharmacologic techniques. Given the profound loss of cholinergic transmission in AD nicotinic and muscarinic acetylcholine receptors have drawn considerable attention. Synthetic Aβ has been shown to bind the calcium permeable α7 nicotinic acetylcholine receptors with high affinity [151]. Functionally, this interaction has been proposed to account for the internalization of NMDA receptors through a calcineurin dependent pathway [137, 152]. Because these studies focus on post-synaptic cholinergic transmission, it is unclear whether interactions with acetylcholine receptor signalling directly account for the disruption of pre-synaptic cholinergic projections in AD, such as those extending from the nucleus basalis of Meynert.
Hebbian mechanisms of synapse modulation often implicate involvement of NMDA receptors, which transduce the level of synaptic activity into a calcium signal that can initiate an array of signalling pathways. The current-voltage relationship that is generated by the magnesium block of this receptor is a key determinant in the amount of calcium that enters upon glutamate binding. Whether NMDA receptor activation will result in LTP or LTD appears to be dependent on the amplitude and kinetics of the calcium transient through the channel [153, 154]. A number of studies have reported that the effects of Aβ on the viability, morphology and physiology of neurons are dependent on NMDA receptor activation [106, 111, 155]. Memantine, an activity dependent NMDA receptor antagonist is used for the treatment of AD. Initially it was proposed to mitigate glutamate excitotoxicity, but it may also block more subtle effects of NMDA receptor activation that lead to synaptic depression and loss.
Activation of metabotropic glutamate receptors (mGluR) recruits a number of signalling pathways (such as p38 MAP kinase or ERK), stimulates release of intracellular calcium stores through generation of inositol triphosphate, or modulates associated ionic channels. These effects may result in post-synaptic AMPA receptor endocytosis [156] and decreased pre-synaptic neurotransmitter release probability [157], both of which decrease synaptic strength. Understanding the contribution of these receptors to Aβ-mediated synaptic depression is difficult because of the high variability in mechanisms linked to mGluRs across brain regions and developmental time periods. However, various groups have reported that Aβ mediates synaptic depression and loss through activation of group I mGluRs with p38 MAP kinase and calcineurin as downstream effectors [77, 94, 104, 145, 152].
We have found that Aβ oligomers from a variety of sources can facilitate LTD through both mGluRs and NMDA receptors and that this appears to involve dysregulation of the neuronal glutamate transporter [94]. A role for aberrant glutamate transport is supported by in vivo microdialysis experiments in which micro-injection of oligomer-containing 7PA2 CM caused a rapid and massive increase in hippocampal interstitial fluid glutamate [158].
In addition to receptors well-known for their involvement in synaptic plasticity, Aβ also appears to engage with other synapse proteins. For example, ADDLs appear to perturb insulin signalling by inducing preferential somatic distribution of the receptor via an NMDAR-dependent pathway [159]. Similarly, non-infectious conformations of the cellular prion protein (PrP) have also been demonstrated to bind ADDLs and the role of PrP as a disease-relevant receptor for Aβ is supported by the finding that knock-out of PrP rescues the block of LTP mediated by ADDLs [121].
But how can the myriad interactions demonstrated between Aβ and several classes of receptors be explained? One possibility is that these reports are all correct, but that Aβ does not directly target a single receptor per se, but rather it interacts with synaptic membranes and influences the expression and distribution of receptors thus mimicking the same effect as if Aβ had directly bound and antagonized the effected receptors. Another possibility lies in the diversity of the different assembly forms and conformations of Aβ used. It is quite possible that different Aβ assemblies or conformation have different targets thus depending on the form of Aβ used one group can report activation of the insulin receptor and another antagonism of α7 nicotinic acetylcholine receptors.
Therapeutic targeting of synaptotoxic Aβ oligomers
Although our understanding of how and what forms of Aβ mediate synaptic dysfunction is incomplete, there is a growing consensus that soluble non-fibrillar Aβ interacts either directly or indirectly with one or more receptors initiating transduction mechanisms that result in decreased plasticity (see appendix 1). If correct, this information already provides for the rational design of disease-modifying therapeutics. Plausible approaches include neutralizing one or more forms of synaptotoxic, non-fibrillar Aβ, and preventing Aβ-mediated perturbations of receptors. As our understanding of the mechanisms contributing to synapse dysfunction continues to develop so too additional therapeutic targets are likely be revealed. In this regard the application of unbiased paradigms in which synaptic alterations are delineated following exposure to well defined soluble Aβ assembly forms should yield novel insights on the pathogenesis of synapse loss (see appendix 2). This information may not only assist in drug design to halt neurodegeneration in AD, but may also promote synapse regeneration. Thus the dream of curing AD may be realized not by just preventing neuronal death, but by actively promoting synaptic connectivity.
Appendix 1
Key observations
-
1.
Synaptic loss is an early and invariant feature of AD the extent of which correlates closely with severity of dementia.
-
2.
MCI and early stage AD have a strong amnestic presentation implicating dysfunction of hippocampal and medial temporal lobe circuitry.
-
3.
Soluble forms of Aβ present in human brain correlate well with synaptic loss and dementia status.
-
4.
Synaptic plasticity as measured by changes in LTP and spine density are perturbed in transgenic animals models of AD and are associated with impaired spatial learning and memory.
-
5.
Changes in plasticity and memory often occur at intervals before transgenic animals have detectable amyloid deposits and can be reversed by acute treatment with anti-Aβ antibodies.
-
6.
Synthetic, cell culture- and brain-derived Aβ inhibits LTP, facilitates LTD, reduces spine and synaptic densities and impairs the memory of learned behaviour.
Appendix 2
Critical next steps
-
1.
Employ sophisticated biophysical techniques to determine the size and structure of synaptotoxic human brain-derived Aβ.
-
2.
Identify the cellular and molecular targets of Aβ oligomers.
-
3.
Elucidate the mechanism by which Aβ mediates synaptic compromise: delineating whether these effects involve disruption of receptors secondary to membrane destabilization or are a result of direct Aβ receptor binding.
-
4.
Determine if and how Aβ oligomers alter tau aggregation and/or phosphorylation and how this is linked to neuronal loss.
References
Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y, Jorm A, Mathers C, Menezes PR, Rimmer E, Scazufca M: Global prevalence of dementia: a Delphi consensus study. Lancet. 2005, 366: 2112-2117. 10.1016/S0140-6736(05)67889-0.
Kawas CH: Clinical practice. Early Alzheimer'sdisease. N Engl J Med. 2003, 349: 1056-1063. 10.1056/NEJMcp022295.
Nussbaum RL, Ellis CE: Alzheimer's disease and Parkinson's disease. N Engl J Med. 2003, 348: 1356-1364. 10.1056/NEJM2003ra020003.
Mercy L, Hodges JR, Dawson K, Barker RA, Brayne C: Incidence of early-onset dementias in Cambridgeshire, United Kingdom. Neurology. 2008, 71: 1496-1499. 10.1212/01.wnl.0000334277.16896.fa.
Selkoe DJ: Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001, 81: 741-766.
Romanelli MF, Morris JC, Ashkin K, Coben LA: Advanced Alzheimer's disease is a risk factor for late-onset seizures. Arch Neurol. 1990, 47: 847-850.
Khachaturian ZS: Diagnosis of Alzheimer's disease. Arch Neurol. 1985, 42: 1097-1105.
Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT: Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997, 41: 17-24. 10.1002/ana.410410106.
Uylings HB, de Brabander JM: Neuronal changes in normal human aging and Alzheimer's disease. Brain Cogn. 2002, 49: 268-276. 10.1006/brcg.2001.1500.
Alzheimer A: Über einen eigenartigen schweren Erkrankungsprozeβ der Hirnrinde. Neurologisches Centralblatt. 1906, 23: 1129-1136.
Terry RD: The fine structure of neurofibrillary tangles in Alzheimer's disease. J Neuropathol Exp Neurol. 1963, 22: 629-642. 10.1097/00005072-196310000-00005.
Kidd M: Alzheimer's disease - An electron microscopical study. Brain. 1964, 87: 307-320. 10.1093/brain/87.2.307.
Davies CA, Mann DM, Sumpter PQ, Yates PO: A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer's disease. J Neurol Sci. 1987, 78: 151-164. 10.1016/0022-510X(87)90057-8.
Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW, Morris JC: Altered expression of synaptic proteins occurs early during progression of Alzheimer's disease. Neurology. 2001, 56: 127-129.
Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ: Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007, 68: 1501-1508. 10.1212/01.wnl.0000260698.46517.8f.
Cajal Ry: Degeneration and regeneration of the nervous system. 1928, Oxford Press, London
Bertoni-Freddari C, Fattoretti P, Meier-Ruge W, Ulrich J: Computer-assisted morphometry of synaptic plasticity during aging and dementia. Pathol Res Pract. 1989, 185: 799-802.
DeKosky ST, Scheff SW: Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990, 27: 457-464. 10.1002/ana.410270502.
Masliah E, Cole G, Shimohama S, Hansen L, DeTeresa R, Terry RD, Saitoh T: Differential involvement of protein kinase C isozymes in Alzheimer's disease. J Neurosci. 1990, 10: 2113-2124.
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R: Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991, 30: 572-580. 10.1002/ana.410300410.
Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ: Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol. 1997, 56: 933-944. 10.1097/00005072-199708000-00011.
Reddy PH, Mani G, Park BS, Jacques J, Murdoch G, Whetsell W, Kaye J, Manczak M: Differential loss of synaptic proteins in Alzheimer's disease: implications for synaptic dysfunction. J Alzheimers Dis. 2005, 7: 103-117. discussion 173-180.
Bertoni-Freddari C, Fattoretti P, Casoli T, Caselli U, Meier-Ruge W: Deterioration threshold of synaptic morphology in aging and senile dementia of Alzheimer's type. Anal Quant Cytol Histol. 1996, 18: 209-213.
Scheff SW, Price DA, Schmitt FA, Mufson EJ: Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiol Aging. 2006, 27: 1372-1384. 10.1016/j.neurobiolaging.2005.09.012.
Scoville WB, Milner B: Loss of recent memory after bilateral hippocampal lesions. J Neurol Neurosurg Psychiatry. 1957, 20: 11-21. 10.1136/jnnp.20.1.11.
Zola-Morgan S, Squire LR, Amaral DG: Human amnesia and the medial temporal region: enduring memory impairment following a bilateral lesion limited to field CA1 of the hippocampus. J Neurosci. 1986, 6: 2950-2967.
deToledo-Morrell L, Geinisman Y, Morrell F: Age-dependent alterations in hippocampal synaptic plasticity: relation to memory disorders. Neurobiol Aging. 1988, 9: 581-590. 10.1016/S0197-4580(88)80117-9.
Geinisman Y, de Toledo-Morrell L, Morrell F: Loss of perforated synapses in the dentate gyrus: morphological substrate of memory deficit in aged rats. Proc Natl Acad Sci USA. 1986, 83: 3027-3031. 10.1073/pnas.83.9.3027.
Geinisman Y, de Toledo-Morrell L, Morrell F: Aged rats need a preserved complement of perforated axospinous synapses per hippocampal neuron to maintain good spatial memory. Brain Res. 1986, 398: 266-275. 10.1016/0006-8993(86)91486-1.
Geinisman Y, deToledo-Morrell L, Morrell F, Persina IS, Rossi M: Age-related loss of axospinous synapses formed by two afferent systems in the rat dentate gyrus as revealed by the unbiased stereological dissector technique. Hippocampus. 1992, 2: 437-444. 10.1002/hipo.450020411.
Barnes CA: Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol. 1979, 93: 74-104. 10.1037/h0077579.
Hyman BT, Van Hoesen GW, Kromer LJ, Damasio AR: Perforant pathway changes and the memory impairment of Alzheimer's disease. Ann Neurol. 1986, 472-481. 10.1002/ana.410200406.
Geinisman Y: Age-related decline in memory function: is it associated with a loss of synapses?. Neurobiol Aging. 1999, 20: 353-356. 10.1016/S0197-4580(99)00072-X. discussion 359-360.
Morris RG: Long-term potentiation and memory. Philos Trans R Soc Lond B Biol Sci. 2003, 358: 643-647. 10.1098/rstb.2002.1230.
Lynch MA: Long-term potentiation and memory. Physiol Rev. 2004, 84: 87-136. 10.1152/physrev.00014.2003.
Buckner RL: Memory and executive function in aging and AD: multiple factors that cause decline and reserve factors that compensate. Neuron. 2004, 44: 195-208. 10.1016/j.neuron.2004.09.006.
Persson J, Nyberg L, Lind J, Larsson A, Nilsson LG, Ingvar M, Buckner RL: Structure-function correlates of cognitive decline in aging. Cereb Cortex. 2006, 16: 907-915. 10.1093/cercor/bhj036.
Mesulam MM: Neuroplasticity failure in Alzheimer's disease: bridging the gap between plaques and tangles. Neuron. 1999, 24: 521-529. 10.1016/S0896-6273(00)81109-5.
Hebb DO: A Neuropsychological Theory. 1949, Lawrence Erlbaum Associates, Mahwah N, J
Malenka RC, Bear MF: LTP and LTD: an embarrassment of riches. Neuron. 2004, 44: 5-21. 10.1016/j.neuron.2004.09.012.
Whitlock JR, Heynen AJ, Shuler MG, Bear MF: Learning induces long-term potentiation in the hippocampus. Science. 2006, 313: 1093-1097. 10.1126/science.1128134.
Kemp N, Bashir ZI: Long-term depression: a cascade of induction and expression mechanisms. Prog Neurobiol. 2001, 65: 339-365. 10.1016/S0301-0082(01)00013-2.
Citri A, Malenka RC: Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. 2008, 33: 18-41. 10.1038/sj.npp.1301559.
Colbran RJ: Targeting of calcium/calmodulin-dependent protein kinase II. Biochem J. 2004, 378: 1-16. 10.1042/BJ20031547.
Kessels HW, Kopec CD, Klein ME, Malinow R: Roles of stargazin and phosphorylation in the control of AMPA receptor subcellular distribution. Nat Neurosci. 2009, 12: 888-896. 10.1038/nn.2340.
Kessels HW, Malinow R: Synaptic AMPA receptor plasticity and behavior. Neuron. 2009, 61: 340-350. 10.1016/j.neuron.2009.01.015.
Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H: Structural basis of long-term potentiation in single dendritic spines. Nature. 2004, 429: 761-766. 10.1038/nature02617.
Zhou Q, Homma KJ, Poo MM: Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004, 44: 749-757. 10.1016/j.neuron.2004.11.011.
Bastrikova N, Gardner GA, Reece JM, Jeromin A, Dudek SM: Synapse elimination accompanies functional plasticity in hippocampal neurons. Proc Natl Acad Sci USA. 2008, 105: 3123-3127. 10.1073/pnas.0800027105.
Lamprecht R, LeDoux J: Structural plasticityand memory. Nat Rev Neurosci. 2004, 5: 45-54. 10.1038/nrn1301.
Holtmaat A, Wilbrecht L, Knott GW, Welker E, Svoboda K: Experience-dependent and cell-type-specific spine growth in the neocortex. Nature. 2006, 441: 979-983. 10.1038/nature04783.
Klein WL, Krafft GA, Finch CE: Targeting small Abeta oligomers: the solution to an Alzheimer's disease conundrum?. Trends Neurosci. 2001, 24: 219-224. 10.1016/S0166-2236(00)01749-5.
Selkoe DJ: Alzheimer's disease is a synaptic failure. Science. 2002, 298: 789-791. 10.1126/science.1074069.
Walsh DM, Selkoe DJ: Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron. 2004, 44: 181-193. 10.1016/j.neuron.2004.09.010.
Selkoe DJ: Clearing the brain's amyloid cobwebs. Neuron. 2001, 32: 177-180. 10.1016/S0896-6273(01)00475-5.
Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM: Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005, 48: 913-922. 10.1016/j.neuron.2005.10.028.
Hardy J, Selkoe DJ: The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002, 297: 353-356. 10.1126/science.1072994.
Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE: Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009, 78: 959-991. 10.1146/annurev.biochem.052308.114844.
Walsh DM, Selkoe DJ: A beta oligomers - a decade of discovery. J Neurochem. 2007, 101: 1172-1184. 10.1111/j.1471-4159.2006.04426.x.
Katzman R: Alzheimer's disease. N Engl J Med. 1986, 314: 964-973.
Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P: Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging. 1995, 16: 285-298. 10.1016/0197-4580(95)00013-5. discussion 298-304.
Harper JD, Wong SS, Lieber CM, Lansbury PT: Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem & Biol. 1997, 4: 119-125. 10.1016/S1074-5521(97)90255-6.
Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB: Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J Biol Chem. 1999, 274: 25945-25952. 10.1074/jbc.274.36.25945.
Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, iosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL: Diffusible, nonfribrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998, 95: 6448-6453. 10.1073/pnas.95.11.6448.
Hoshi M, Sato M, Matsumoto S, Noguchi A, Yasutake K, Yoshida N, Sato K: Spherical aggregates of beta-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3beta. Proc Natl Acad Sci USA. 2003, 100: 6370-6375. 10.1073/pnas.1237107100.
Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB: Amyloid β-protein (Aβ) assembly: Aβ 40 and Aβ 42 oligomerize through distinct pathways. Proc Natl Acad Sci USA. 2003, 100: 330-335. 10.1073/pnas.222681699.
Lemere CA, Spooner ET, Leverone JF, Mori C, Clements JD: Intranasal immunotherapy for the treatment of Alzheimer's disease: Escherichia coli LT and LT(R192G) as mucosal adjuvants. Neurobiol Aging. 2002, 23: 991-1000. 10.1016/S0197-4580(02)00127-6.
McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL: Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999, 46: 860-866. 10.1002/1531-8249(199912)46:6<860::AID-ANA8>3.0.CO;2-M.
Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J: Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999, 155: 853-862.
Wang J, Dickson DW, Trojanowski JQ, Lee VM: The levels of soluble versus insoluble brain Abeta distinguish Alzheimer's disease from normal and pathologic aging. Exp Neurol. 1999, 158: 328-337. 10.1006/exnr.1999.7085.
Kuo YM, Emmerling MR, Vigo-Pelfrey C, Kasunic TC, Kirkpatrick JB, Murdoch GH, Ball MJ, Roher AE: Water-soluble Abeta (N-40, N-42) oligomers in normal and Alzheimer disease brains. J Biol Chem. 1996, 271: 4077-4081. 10.1074/jbc.271.8.4077.
Stenh C, Englund H, Lord A, Johansson AS, Almeida CG, Gellerfors P, Greengard P, Gouras GK, Lannfelt L, Nilsson LN: Amyloid-beta oligomers are inefficiently measured by enzyme-linked immunosorbent assay. Ann Neurol. 2005, 58: 147-150. 10.1002/ana.20524.
Morishima-Kawashima M, Ihara Y: The presence of amyloid β-protein in the detergent-insoluble membrane compartment of human neuroblastoma cells. Biochem. 1998, 37: 15247-15253. 10.1021/bi981843u.
Enya M, Morishima-Kawashima M, Yoshimura M, Shinkai Y, Kusui K, Khan K, Games D, Schenk D, Sugihara S, Yamaguchi H, Ihara Y: Appearance of sodium dodecyl sulfate-stable amyloid beta-protein (Abeta) dimer in the cortex during aging. Am J Pathol. 1999, 154: 271-279.
Funato H, Enya M, Yoshimura M, Morishima-Kawashima M, Ihara Y: Presence of sodium dodecyl sulfate-stable amyloid beta-protein dimers in the hippocampus CA1 not exhibiting neurofibrillary tangle formation. Am J Pathol. 1999, 155: 23-28.
Englund H, Degerman Gunnarsson M, Brundin RM, Hedlund M, Kilander L, Lannfelt L, Ekholm Pettersson F: Oligomerization Partially Explains the Lowering of Abeta42 in Alzheimer's Disease Cerebrospinal Fluid. Neurodegener Dis. 2009, 6: 139-147. 10.1159/000225376.
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ: Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008, 14: 837-842. 10.1038/nm1782.
Vigo-Pelfrey C, Lee D, Keim PS, Lieberburg I, Schenk D: Characterization of beta-amyloid peptide from human cerebrospinal fluid. Journal Neurochemistry. 1993, 61: 1965-1968. 10.1111/j.1471-4159.1993.tb09841.x.
Roher A, Wolfe D, Palutke M, KuKuruga D: Purification, ultrastructure, and chemical analyses of Alzheimer's disease amyloid plaque core protein. Proc Natl Acad Sci USA. 1986, 83: 2662-2666. 10.1073/pnas.83.8.2662.
Ashe KH: Learning and memory in transgenic mice modeling Alzheimer's disease. Learn Mem. 2001, 8: 301-308. 10.1101/lm.43701.
Games D, Buttini M, Kobayashi D, Schenk D, Seubert P: Mice as models: transgenic approaches and Alzheimer's disease. J Alzheimers Dis. 2006, 9: 133-149.
Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L: High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000, 20: 4050-4058.
Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L: Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA. 1999, 96: 3228-3233. 10.1073/pnas.96.6.3228.
Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, Ashe KH: The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2002, 22: 1858-1867.
Kawarabayashi T, Shoji M, Younkin LH, Wen-Lang L, Dickson DW, Murakami T, Matsubara E, Abe K, Ashe KH, Younkin SG: Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer's disease. J Neurosci. 2004, 24: 3801-3809. 10.1523/JNEUROSCI.5543-03.2004.
Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH: A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006, 440: 352-357. 10.1038/nature04533.
Cheng IH, Scearce-Levie K, Legleiter J, Palop JJ, Gerstein H, Bien-Ly N, Puolivali J, Lesne S, Ashe KH, Muchowski PJ, Mucke L: Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem. 2007, 282: 23818-23828. 10.1074/jbc.M701078200.
Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM: Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat Neurosci. 2002, 5: 452-457.
Reed MN, Hofmeister JJ, Jungbauer L, Welzel AT, Yu C, Lesné S, LaDu MJ, Walsh DM, Ashe KH, Cleary JP: Cognitive Effects of Naturally and Synthetically Derived A®Oligomers. Neurobiology of Aging. 2009,
Dineley KT, Xia XF, Bui D, Sweatt JD, Zheng H: Accelerated plaque accumulation, associative learning deficits, and up-regulation of alpha 7 nicotinic receptor protein in transgenic mice co-expressing mutant human presenilin 1 and amyloid precursor proteins. Journal of Biological Chemistry. 2002, 277: 22768-22780. 10.1074/jbc.M200164200.
Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, Bloom FE: Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2006, 103: 5161-5166. 10.1073/pnas.0600948103.
Shankar GM, Leissring MA, Adame A, Sun X, Spooner E, Masliah E, Selkoe DJ, Lemere CA, Walsh DM: Biochemical and immunohistochemical analysis of an Alzheimer's disease mouse model reveals the presence of multiple cerebral Abeta assembly forms throughout life. Neurobiol Dis. 2009, 36: 293-302. 10.1016/j.nbd.2009.07.021.
Meilandt WJ, Cisse M, Ho K, Wu T, Esposito LA, Scearce-Levie K, Cheng IH, Yu GQ, Mucke L: Neprilysin overexpression inhibits plaque formation but fails to reduce pathogenic Abeta oligomers and associated cognitive deficits in human amyloid precursor protein transgenic mice. J Neurosci. 2009, 29: 1977-1986. 10.1523/JNEUROSCI.2984-08.2009.
Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D: Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009, 62: 788-801. 10.1016/j.neuron.2009.05.012.
Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, Selkoe DJ: Aggregation of secreted amyloid beta-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J Biol Chem. 1995, 270: 9564-9570. 10.1074/jbc.270.16.9564.
Xia W, Zhang J, Kholodenko D, Citron M, Podlisny MB, Teplow DB, Haass C, Seubert P, Koo EH, Selkoe DJ: Enhanced production and oligomerization of the 42-residue amyloid beta-protein by Chinese hamster ovary cells stably expressing mutant presenilins. Journal Biological Chemistry. 1997, 272: 7977-7982. 10.1074/jbc.272.12.7977.
Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ: The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry. 2000, 39: 10831-10839. 10.1021/bi001048s.
Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ: Effects of secreted oligomers of amyloid {beta}-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006, 572: 477-492. 10.1113/jphysiol.2005.103754.
Poling A, Morgan-Paisley K, Panos JJ, Kim EM, O'Hare E, Cleary JP, Lesne S, Ashe KH, Porritt M, Baker LE: Oligomers of the amyloid-beta protein disrupt working memory: confirmation with two behavioral procedures. Behav Brain Res. 2008, 193: 230-234. 10.1016/j.bbr.2008.06.001.
Calabrese B, Shaked GM, Tabarean IV, Braga J, Koo EH, Halpain S: Rapid, concurrent alterations in pre-and postsynaptic structure induced by naturally-secreted amyloid-beta protein. Mol Cell Neurosci. 2007, 35: 183-193. 10.1016/j.mcn.2007.02.006.
Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ: Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans. 2002, 30: 552-557. 10.1042/BST0300552.
Walsh D, Klyubin I, Fadeeva J, William K, Cullen W, Anwyl R, Wolfe M, Rowan M, Selkoe D: Naturally secreted oligomers of the Alzheimer amyloid beta-protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002, 416: 535-539. 10.1038/416535a.
Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, Lemere CA, Cullen WK, Peng Y, Wisniewski T, Selkoe DJ, Anwyl R, Walsh DM, Rowan MJ: Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008, 28: 4231-4237. 10.1523/JNEUROSCI.5161-07.2008.
Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R: Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004, 24: 3370-3378. 10.1523/JNEUROSCI.1633-03.2004.
Walsh DM, Townsend M, Podlisny MB, Shankar GM, Fadeeva JV, Agnaf OE, Hartley DM, Selkoe DJ: Certain inhibitors of synthetic amyloid beta-peptide (Abeta) fibrillogenesis block oligomerization of natural Abeta and thereby rescue long-term potentiation. J Neurosci. 2005, 25: 2455-2462. 10.1523/JNEUROSCI.4391-04.2005.
Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL: Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007, 27: 2866-2875. 10.1523/JNEUROSCI.4970-06.2007.
Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH: Natural oligomers, but not monomers, of the amyloid β-protein specifically disrupt the memory of learned behavior. Nature Neuroscience. 2005, 8: 79-84. 10.1038/nn1372.
Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ: Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999, 19: 8876-8884.
Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL: Soluble oligomers of beta-amyloid(1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Research. 2002, 924: 133-140. 10.1016/S0006-8993(01)03058-X.
Shrestha BR, Vitolo OV, Joshi P, Lordkipanidze T, Shelanski M, Dunaevsky A: Amyloid beta peptide adversely affects spine number and motility in hippocampal neurons. Mol Cell Neurosci. 2006, 33: 274-282. 10.1016/j.mcn.2006.07.011.
Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL: Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007, 27: 796-807. 10.1523/JNEUROSCI.3501-06.2007.
Walsh DM, Thulin E, Minogue AM, Gustavsson N, Pang E, Teplow DB, Linse S: A facile method for expression and purification of the Alzheimer's disease-associated amyloid beta-peptide. FEBS J. 2009, 276: 1266-1281. 10.1111/j.1742-4658.2008.06862.x.
Harper JD, Wong SS, Lieber CM, Lansbury PT: Assembly of Aβ amyloid protofibrils: An in vitro model for a possible early event in Alzheimer's disease. Biochemistry. 1999, 38: 8972-8980. 10.1021/bi9904149.
Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT: Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002, 418: 291-10.1038/418291a.
Isaacs AM, Senn DB, Yuan M, Shine JP, Yankner BA: Acceleration of amyloid beta-peptide aggregation by physiological concentrations of calcium. J Biol Chem. 2006, 281: 27916-27923. 10.1074/jbc.M602061200.
Hartley DM, Zhao C, Speier AC, Woodard GA, Li S, Li Z, Walz T: Transglutaminase induces protofibril-like amyloid beta-protein assemblies that are protease-resistant and inhibit long-term potentiation. J Biol Chem. 2008, 283: 16790-16800. 10.1074/jbc.M802215200.
Martins IC, Kuperstein I, Wilkinson H, Maes E, Vanbrabant M, Jonckheere W, Van Gelder P, Hartmann D, D'Hooge R, De Strooper B, Schymkowitz J, Rousseau F: Lipids revert inert Abeta amyloid fibrils to neurotoxic protofibrils that affect learning in mice. EMBO J. 2008, 27: 224-233. 10.1038/sj.emboj.7601953.
Walsh DM, Lomakin A, Benedek GB, Maggio JE, Condron MM, Teplow DB: Amyloid β-protein fibrillogenesis: Detection of a protofibrillar intermediate. J Biol Chem. 1997, 272: 22364-22374. 10.1074/jbc.272.35.22364.
Nybo M, Svehag SE, Holm Nielsen E: An ultrastructural study of amyloid intermediates in A beta1-42 fibrillogenesis. Scand J Immunol. 1999, 49: 219-223. 10.1046/j.1365-3083.1999.00526.x.
Hepler RW, Grimm KM, Nahas DD, Breese R, Dodson EC, Acton P, Keller PM, Yeager M, Wang H, Shughrue P, Kinney G, Joyce JG: Solution state characterization of amyloid beta-derived diffusible ligands. Biochemistry. 2006, 45: 15157-15167. 10.1021/bi061850f.
Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM: Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009, 457: 1128-1132. 10.1038/nature07761.
Dahlgren KN, Manelli AM, Stine WB, Baker LK, Krafft GA, LaDu MJ: Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J Biol Chem. 2002, 277: 32046-32053. 10.1074/jbc.M201750200.
Kim HJ, Chae SC, Lee DK, Chromy B, Lee SC, Park YC, Klein WL, Krafft GA, Hong ST: Selective neuronal degeneration induced by soluble oligomeric amyloid beta protein. Faseb J. 2003, 17: 118-120.
Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL: Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J Neurosci. 2004, 24: 10191-10200. 10.1523/JNEUROSCI.3432-04.2004.
Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL: Alzheimer's disease-affected brain: presence of oligomeric Abeta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proceedings of the National Academy of Sciences (USA). 2003, 100: 10417-10422. 10.1073/pnas.1834302100.
Lambert MP, Viola KL, Chromy BA, Chang L, Morgan TE, Yu J, Venton DL, Krafft GA, Finch CE, Klein WL: Vaccination with soluble Abeta oligomers generates toxicity-neutralizing antibodies. J Neurochem. 2001, 79: 595-605. 10.1046/j.1471-4159.2001.00592.x.
Chang L, Bakhos L, Wang ZG, Venton DL, Klein WL: Femtomole Immunodetection of synthetic and endogenous amyloid-b oligomers and its application to Alzheimer's disease drug candidate screening. Journal of Molecular Neuroscience. 2003, 20: 305-313. 10.1385/JMN:20:3:305.
Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG: Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003, 300: 486-489. 10.1126/science.1079469.
Whalen BM, Selkoe DJ, Hartley DM: Small non-fibrillar assemblies of amyloid beta-protein bearing the Arctic mutation induce rapid neuritic degeneration. Neurobiol Dis. 2005, 20: 254-266. 10.1016/j.nbd.2005.03.007.
Maloney MT, Minamide LS, Kinley AW, Boyle JA, Bamburg JR: Beta-secretase-cleaved amyloid precursor protein accumulates at actin inclusions induced in neurons by stress or amyloid beta: a feedforward mechanism for Alzheimer's disease. J Neurosci. 2005, 25: 11313-11321. 10.1523/JNEUROSCI.3711-05.2005.
Kelly BL, Ferreira A: beta-Amyloid-induced dynamin 1 degradation is mediated by N-methyl-D-aspartate receptors in hippocampal neurons. J Biol Chem. 2006, 281: 28079-28089. 10.1074/jbc.M605081200.
Barghorn S, Zheng-Fischhofer Q, Ackmann M, Biernat J, von Bergen M, Mandelkow E: Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias [In Process Citation]. Biochemistry. 2000, 39: 11714-11721. 10.1021/bi000850r.
Deshpande A, Mina E, Glabe C, Busciglio J: Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci. 2006, 26: 6011-6018. 10.1523/JNEUROSCI.1189-06.2006.
Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG: Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem. 2005, 280: 17294-17300. 10.1074/jbc.M500997200.
Wogulis M, Wright S, Cunningham D, Chilcote T, Powell K, Rydel RE: Nucleation-dependent polymerization is an essential component of amyloid-mediated neuronal cell death. J Neurosci. 2005, 25: 1071-1080. 10.1523/JNEUROSCI.2381-04.2005.
White AR, Reyes R, Mercer JF, Camakaris J, Zheng H, Bush AI, Multhaup G, Beyreuther K, Masters CL, Cappai R: Copper levels are increased in the cerebral cortex and liver of APP and APLP2 knockout mice. Brain Research. 1999, 842: 439-444. 10.1016/S0006-8993(99)01861-2.
Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P: Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005, 8: 1051-1058. 10.1038/nn1503.
Tamagno E, Bardini P, Guglielmotto M, Danni O, Tabaton M: The various aggregation states of beta-amyloid 1-42 mediate different effects on oxidative stress, neurodegeneration, and BACE-1 expression. Free Radic Biol Med. 2006, 41: 202-212. 10.1016/j.freeradbiomed.2006.01.021.
Zhao L, Ma QL, Calon F, Harris-White ME, Yang F, Lim GP, Morihara T, Ubeda OJ, Ambegaokar S, Hansen JE, Weisbart RH, Teter B, Frautschy SA, Cole GM: Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nat Neurosci. 2006, 9: 234-242. 10.1038/nn1630.
Tomiyama T, Nagata T, Shimada H, Teraoka R, Fukushima A, Kanemitsu H, Takuma H, Kuwano R, Imagawa M, Ataka S, Wada Y, Yoshioka E, Nishizaki T, Watanabe Y, Mori H: A new amyloid beta variant favoring oligomerization in Alzheimer's-type dementia. Ann Neurol. 2008, 63: 377-387. 10.1002/ana.21321.
Hung LW, Ciccotosto GD, Giannakis E, Tew DJ, Perez K, Masters CL, Cappai R, Wade JD, Barnham KJ: Amyloid-beta peptide (Abeta) neurotoxicity is modulated by the rate of peptide aggregation: Abeta dimers and trimers correlate with neurotoxicity. J Neurosci. 2008, 28: 11950-11958. 10.1523/JNEUROSCI.3916-08.2008.
Harmeier A, Wozny C, Rost BR, Munter LM, Hua H, Georgiev O, Beyermann M, Hildebrand PW, Weise C, Schaffner W, Schmitz D, Multhaup G: Role of amyloid-beta glycine 33 in oligomerization, toxicity, and neuronal plasticity. J Neurosci. 2009, 29: 7582-7590. 10.1523/JNEUROSCI.1336-09.2009.
Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R: APP processing and synaptic function. Neuron. 2003, 37: 925-937. 10.1016/S0896-6273(03)00124-7.
Brody DL, Magnoni S, Schwetye KE, Spinner ML, Esparza TJ, Stocchetti N, Zipfel GJ, Holtzman DM: Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science. 2008, 321: 1221-1224. 10.1126/science.1161591.
Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R: AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006, 52: 831-843. 10.1016/j.neuron.2006.10.035.
Plant LD, Boyle JP, Smith IF, Peers C, Pearson HA: The production of amyloid beta peptide is a critical requirement for the viability of central neurons. J Neurosci. 2003, 23: 5531-5535.
Puzzo D, Privitera L, Leznik E, Fa M, Staniszewski A, Palmeri A, Arancio O: Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008, 28: 14537-14545. 10.1523/JNEUROSCI.2692-08.2008.
Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L: Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007, 55: 697-711. 10.1016/j.neuron.2007.07.025.
Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O: Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008, 321: 1686-1689. 10.1126/science.1162844.
Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT: Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005, 25: 7278-7287. 10.1523/JNEUROSCI.1879-05.2005.
Wang HY, Lee DH, Davis CB, Shank RP: Amyloid peptide Abeta(1-42) binds selectively and with picomolar affinity to alpha7 nicotinic acetylcholine receptors. J Neurochem. 2000, 75: 1155-1161. 10.1046/j.1471-4159.2000.0751155.x.
Dewachter I, Filipkowski RK, Priller C, Ris L, Neyton J, Croes S, Terwel D, Gysemans M, Devijver H, Borghgraef P, Godaux E, Kaczmarek L, Herms J, Van Leuven F: Deregulation of NMDA-receptor function and down-stream signaling in APP[V717I] transgenic mice. Neurobiol Aging. 2009, 30: 241-256. 10.1016/j.neurobiolaging.2007.06.011.
Yang SN, Tang YG, Zucker RS: Selective induction of LTP and LTD by postsynaptic [Ca2+]i elevation. J Neurophysiol. 1999, 81: 781-787.
Neveu D, Zucker RS: Postsynaptic levels of [Ca2+]i needed to trigger LTD and LTP. Neuron. 1996, 16: 619-629. 10.1016/S0896-6273(00)80081-1.
Ye C, Walsh DM, Selkoe DJ, Hartley DM: Amyloid beta-protein induced electrophysiological changes are dependent on aggregation state: N-methyl-D-aspartate (NMDA) versus non-NMDA receptor/channel activation. Neurosci Lett. 2004, 366: 320-325. 10.1016/j.neulet.2004.05.060.
Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF: Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci. 2001, 4: 1079-1085. 10.1038/nn746.
Zakharenko SS, Zablow L, Siegelbaum SA: Altered presynaptic vesicle release and cycling during mGluR-dependent LTD. Neuron. 2002, 35: 1099-1110. 10.1016/S0896-6273(02)00898-X.
O'Shea SD, Smith IM, McCabe OM, Walsh DM, O'Connor WT: Intracerebroventricular administration of amyloid β-protein oligomers selectively increases dorsal hippocampal dialysate glutamate levels in the awake rat. Sensors. 2008, 8: 7428-7437. 10.3390/s8117428.
Zhao WQ, De Felice FG, Fernandez S, Chen H, Lambert MP, Quon MJ, Krafft GA, Klein WL: Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008, 22: 246-260. 10.1096/fj.06-7703com.
Lashuel HA, Hartley DM, Petre BM, Wall JS, Simon MN, Walz T, Lansbury PT: Mixtures of wild-type and a pathogenic (E22G) form of Abeta40 in vitro accumulate protofibrils, including amyloid pores. J Mol Biol. 2003, 332: 795-808. 10.1016/S0022-2836(03)00927-6.
Yu L, Edalji R, Harlan JE, Holzman TF, Lopez AP, Labkovsky B, Hillen H, Barghorn S, Ebert U, Richardson PL, Miesbauer L, Solomon L, Bartley D, Walter K, Johnson RW, Hajduk PJ, Olejniczak ET: Structural characterization of a soluble amyloid beta-peptide oligomer. Biochemistry. 2009, 48: 1870-1877. 10.1021/bi802046n.
Barghorn S, Nimmrich V, Striebinger A, Krantz C, Keller P, Janson B, Bahr M, Schmidt M, Bitner RS, Harlan J, Barlow E, Ebert U, Hillen H: Globular amyloid beta-peptide oligomer-a homogenous and stable neuropathological protein in Alzheimer's disease. J Neurochem. 2005, 95: 834-847. 10.1111/j.1471-4159.2005.03407.x.
Hermann D, Both M, Ebert U, Gross G, Schoemaker H, Draguhn A, Wicke K, Nimmrich V: Synaptic transmission is impaired prior to plaque formation in amyloid precursor protein-overexpressing mice without altering behaviorally-correlated sharp wave-ripple complexes. Neuroscience. 2009, 162: 1081-1090. 10.1016/j.neuroscience.2009.05.044.
Noguchi A, Matsumura S, Dezawa M, Tada M, Yanazawa M, Ito A, Akioka M, Kikuchi S, Sato M, Ideno S, Noda M, Fukunari A, Muramatsu SI, Itokazu Y, Sato K, Takahashi H, Teplow DB, Nabeshima YI, Kakita A, Imahori K, Hoshi M: Isolation and characterization of patient-derived, toxic, high-mass amyloid {beta}-protein (A{beta}) assembly from Alzheimer's disease brains. J Biol Chem. 2009, 32895-905. 10.1074/jbc.M109.000208. 47
Hu NW, Smith IM, Walsh DM, Rowan MJ: Soluble amyloid-beta peptides potently disrupt hippocampal synaptic plasticity in the absence of cerebrovascular dysfunction in vivo. Brain. 2008, 131: 2414-2424. 10.1093/brain/awn174.
Acknowledgements
DMW thanks Prof. Michael Kidd for first directing him to the importance of synaptic loss in AD and for setting him on a decade long odyssey of discovery. We also thank Profs. Dennis Selkoe and Ciaran Regan and members of the Walsh lab for useful discussions. This work was supported by funding from the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement No. 200611 (DMW) and by the NIH (1R01AG027443, DMW).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
DMW is a shareholder and member of the scientific advisory board of Senexis, plc.
Authors' contributions
DMW designed the layout and content of the review and DMW and GMS co-wrote the text.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Shankar, G.M., Walsh, D.M. Alzheimer's disease: synaptic dysfunction and Aβ. Mol Neurodegeneration 4, 48 (2009). https://doi.org/10.1186/1750-1326-4-48
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1750-1326-4-48