
Abstract
Background
Clinical features, complications and treatments of Gaucher’s disease (GD), a rare autosomal–recessive disorder due to a confirmed lysosomal enzyme (glucocerebrosidase) deficiency, are described.
Methods
All patients with known GD, living in France, with ≥1 consultations (1980–2010), were included in the French GD registry, yielding the following 4 groups: the entire cohort, with clinical description; and its subgroups: patients with ≥1 follow-up visits, to investigate complications; recently followed (2009–2010) patients; and patients treated during 2009–2010, to examine complications before and during treatment. Data are expressed as medians (range) for continuous variables and numbers (%) for categorical variables.
Results
Among the 562 registry patients, 265 (49.6%) were females; 454 (85.0%) had type 1, 22 (4.1%) type 2, 37 (6.9%) perinatal–lethal type and 21 (3.9%) type 3. Median ages at first GD symptoms and diagnosis, respectively, were 15 (0–77) and 22 (0–84) years for all types. The first symptom diagnosing GD was splenomegaly and/or thrombocytopenia (37.6% and 26.3%, respectively). Bone-marrow aspiration and/or biopsy yielded the diagnosis for 54.7% of the patients, with enzyme deficiency confirming GD for all patients. Birth incidence rate was estimated at 1/50,000 and prevalence at 1/136,000. For the 378 followed patients, median follow-up was 16.2 (0.1–67.6) years. Major clinical complications were bone events (BE; avascular necrosis, bone infarct or pathological fracture) for 109 patients, splenectomy for 104, and Parkinson’s disease for 14; 38 patients died (neurological complications for 15 type-2 and 3 type-3 patients, GD complications for 11 type-1 and another disease for 9 type-1 patients). Forty-six had monoclonal gammopathy. Among 283 recently followed patients, 36 were untreated and 247 had been treated during 2009–2010; 216 patients received treatment in December 2010 (126 with imiglucerase, 45 velaglucerase, 24 taliglucerase, 21 miglustat). BE occurred before (130 in 67 patients) and under treatment (60 in 41 patients) with respective estimated frequencies (95% CI) of first BE at 10 years of 20.3% (14.1%–26.5%) and 19.8% (13.5%–26.1%).
Conclusion
This registry enabled the epidemiological description of GD in France and showed that BE occur even during treatment.
Similar content being viewed by others

Background
Gaucher’s disease (GD), a rare autosomal–recessive disorder with an approximate prevalence of 1/75,000 live births worldwide, is due to the deficiency of a lysosomal enzyme (glucocerebrosidase, glucosylceramidase or glucosidase-β acid (EC 3.2.1.45))[1] or, more rarely, its activator (saposin C)[2, 3]. GD diagnosis is based on deficient glucocerebrosidase activity in peripheral leukocytes or cultured skin fibroblasts. Genotyping can sometimes provide prognostic information[4]. This lysosomal storage disease is characterized by liver and spleen enlargement, and bone manifestations (Erlenmeyer-flask deformity, osteoporosis, lytic lesions, pathological and vertebral compression fractures, bone infarcts and avascular necroses leading to degenerative arthropathy)[1, 5]. Based on the neurological signs, 3 clinical phenotypes are recognized: type 1, the classic form usually defined by the absence of central nervous system impairment, although an association between type-1 GD and Parkinsonism has been described[6]; types 2 and 3, both rare and severe, have neurological involvement[7]; and the perinatal–lethal GD type, with perinatal onset and death before 3 months of age[8]. GD signs usually appear after a symptom-free period, except in rare cases of fetal onset[9]. Thrombocytopenia and anemia are common, and several biomarkers (chitotriosidase, ferritin, angiotensin-converting enzyme [ACE] and tartrate-resistant acid phosphatase [TRAP]) are elevated during GD evolution[10–17].
Enzyme-replacement therapy (ERT; alglucerase [Ceredase©, Genzyme Corporation, available since 1991][18], followed by imiglucerase [Cerezyme©, Genzyme Corporation, available since 1996], velaglucerase [Vpriv©, Shire, available since 2010][19], and taliglucerase [Uplyso©, Pfizer, only authorized for temporary use][20]), is the reference treatment. Substrate-reduction therapy (SRT), namely miglustat (Zavesca©, Actelion, available since 2002)[21], is indicated for moderate GD when ERT is unsuitable. In June 2009, an acute imiglucerase shortage occurred because of viral contamination (Vesivirus 2117) of cell cultures and other production problems[22]. Since then, that ERT has been in short supply, which was further aggravated in August 2009.
Genzyme Corporation developed an international registry[23] and several countries, e.g., Spain[24, 25], Brazil[26] or Japan[27], identified GD cohorts and established exhaustive national registries. While the international registry conducted many important ancillary studies[28–34], its non-exhaustive cohort did not address public health issues in terms of incidence, prevalence and monitoring of care of GD patients. Since 2004, France has created referral centers dedicated to the clinical management of rare diseases, and assigned them several objectives, e.g., improving overall patient clinical care and professional practices, and collecting epidemiological data. In this context, a Referral Center for Lysosomal Diseases (RCLD) was established and a national GD-patient registry was created, in 2009, as a means to examine and meet some of those goals.
The main aims of this study were to describe the epidemiological profile of GD patients in France: GD demographic, clinical, biological and genetic features; complications in patients with follow-up (2009–2010); and treatments for those with recent (2009–2010) follow-up based on data collected since 1980 and available in the French Gaucher Disease Registry (FGDR).
Patients and methods
Registry design
The FGDR developed and maintains a designated RCLD since 2009. Its Evaluation of Gaucher-Disease–Treatment Committee (EGDTC) is a national scientific committee to monitor and optimize GD management in France. The French Data-Protection Commission’s (CNIL) approval of the FGDR required oral or written informed consent from patients or their parents. Data from patients who did not consent were not entered. The FGDR was finally certified in 2009 by the French Institute for Public Health Surveillance (InVS) and the French National Institute of Health and Medical Research (INSERM). All GD patients living in France and having ≥1 consultations (i.e., hospitalization or outpatient consultation with a GD specialist) since 1980 were included. For all patients, GD was diagnosed by demonstration of deficient glucocerebrosidase activity in leukocytes[35] or cultured skin fibroblasts. Exhaustive identification of cases was achieved through 3 sources. Only 3 diagnostic laboratories (all included in EGDTC) are accredited in France and, therefore, identify all patients with an enzymatic assay for GD. Based on our Reference Center’s expertise, the French national health insurance (Rare Disease Committee with EGDTC members) validates each GD diagnosis and authorizes coverage for its treatment. Once experts have validated the case, they can ask the treating physician to include the patient in our registry. The indications for treatment are well established to maximize efficacy and avoid unnecessary health insurance expenditures. Each treating physician contacted allowed access to the medical data entered in the FGDR, which is certified by InVS and INSERM.
Each patient’s data were also collected by RCLD physicians or clinical research assistants. The FGDR director controlled data quality. Dr D. Hamroun developed the original Internet software for the FGDR, using 4th Dimension language from 4D (http://www.4D.com). Data were collected retrospectively between 2009 and 2010, and as of 2011, all data have been recorded prospectively.
A standardized case-report form was used to collect the following information: initial data (age at diagnosis, sex, history related or unrelated to GD, symptoms leading to diagnosis and first symptoms, first diagnostic exam, phenotype, genotype and affected family members); clinical information during the first consultation, at diagnosis and throughout follow-up; body mass index expressed according to the World Health Organization classification; organomegaly (liver and/or spleen, ultrasound measurement of the largest diameter); biological findings initially and throughout follow-up (hemoglobin level, platelet count, leukocyte count, chitotriosidase, ferritin, ACE, TRAP, gammaglobulin (with respective normal values of >12 g/dL, >150×103/mm3, >4 × 103/mm3, <100 nmol/mL/h, <250 ng/L, <45 IU/L, <7 IU/L and <13 g/L). Plasma chitotriosidase activity was determined using the fluorescent substrate 4-methylumbelliferylβ-D-N N′,N′′-triacetylchitotriose[10]; ACE, TRAP, ferritin and other markers were measured in the appropriate local laboratories. Bone findings (X-rays, magnetic resonance imaging and, for some patients, scintigraphy and dual-energy X-ray absorptiometry) were recorded during follow-up, with identification of intercurrent events, particularly bone complications. Bone events (BE) were defined clinically, using the bone indications for treatment recommended by the French National Health Authority[36]: avascular necrosis of an epiphysis, bone infarct, pathological and/or vertebral compression fracture(s). Each BE caused a clinical manifestation and was confirmed radiologically. Bone pain alone was not considered a BE without radiological confirmation. Acute bone pain defined a bone crisis. Bone crisis was included in BE only when a bone infarct was identified. Any event, GD-related or not, occurring during follow-up and monitoring of GD-specific therapy was also recorded.
Study design
This investigation was undertaken to describe and analyze clinical, biological, radiological and therapeutic data recorded in the FGDR for all patients from diagnosis until 31 December 2010, the closing date. The local Institutional Review Board of Northern Paris Hospitals, Paris–Diderot University, AP–HP (Ethics Committee) reviewed and approved the research project.
To simplify the description, we defined 4 groups: the entire cohort and its subgroups. Data from the entire cohort of patients entered in the FGDR described, when available, diagnosis characteristics for these patients, the GD-incidence rate (defined as total number of cases diagnosed between 1980 and 2010 divided by the total French population during the same period), birth incidence rate (defined as the total number of cases diagnosed between 1980 and 2010, divided by the total number of live births during the same period) and prevalence were estimated for the French population. For patients with ≥1 follow-up visits in addition to the initial assessment form, we investigated their GD complications (splenectomy, Parkinson’s disease (PD), monoclonal gammopathy (MG), BE, first treatment and deaths). Recently followed patients had consulted in 2009–2010: a map showing the locations of hospitals monitoring them was drawn. For patients seen and treated in 2009–2010, BE were analyzed, before and under (ERT and/or SRT (ERT/SRT)). Clinical, biological and radiological monitoring of these recently treated patients was also investigated. Specific GD ERT (imiglucerase, velaglucerase, taliglucerase, miglustat) was studied, particularly during the period of imiglucerase shortage (June 2009–December 2010). Patients were distinguished according to their age on 1 June 2009 (≤15 or >15 years) for the description of the shortage that began at that time. That age was chosen because it defines the limit between adult medicine and pediatrics.
Statistical analyses
All statistical analyses were computed with SAS software (version 9.2; SAS Institute Inc; Cary, NC). Data are expressed as medians ((range) or interquartile range [IQR; Q1;Q3]) for continuous variables and numbers (%) for categorical variables. Because this was a retrospective study, some data were missing, particularly at the onset of follow-up (during the diagnosis phase) or at treatment onset. Given the demonstrated relatively stable clinical and laboratory parameters of untreated patients after GD diagnosis[37], the biological data during the next 2 years changed only minimally and were considered similar to those at diagnosis. Likewise, data for the previous 2 years under ERT/SRT were stable compared to those at treatment onset. Under treatment data were the last values before the end of therapy or at the closing date. When ERT/SRT was interrupted for <6 months, patients were always considered to be on treatment.
Non-parametric tests were used to compare categorical variables (Fisher’s exact test) across patient subgroups. A two-sided p<0.05 was considered significant. For recently treated patients, time to first BE was estimated with the Kaplan–Meier method for 2 periods: diagnosis to treatment onset (before ERT) and first ERT to closing date (under ERT), with only the first BE occurring during each period being considered. Data were censored when no BE occurred prior to ERT start for the first analysis, and until the closing date or treatment discontinuation for the second. We aimed to study a risk effect of BE. First, the impact of splenectomy, time to treatment onset (< or ≥2 years after diagnosis, based on Mistry et al.’s demonstration of lower BE risk after the latter[38]) or age at diagnosis (≤ or >15 years) on BE occurrence was tested using the log-rank test; second a Cox model was used to derive a predictive model. When several univariate model covariates were significant, a multivariate Cox model with backward selection was used to retain only significant ones. For BE under ERT, the impact of BE occurrence before treatment was also tested.
Results
Entire registry cohort
The FGDR contained 562 patients with confirmed GD, living in France and having ≥1 consultations between 1980 and 2011. Patient characteristics at diagnosis are reported in Table 1. The male/female ratio was 1.0151 (49.6% female), while median age at first symptoms was 15 (0–77) years. During the 31 years (1980–2010), 474 patients were diagnosed in France, with an annual median of 15 (9–32) patients. The incidence rate was estimated at 0.26 patients/106 person-years and birth incidence at 1/50,000. The prevalence was 1/136,000. The most frequent first symptom leading to diagnosis (available for 232 patients) was splenomegaly and/or thrombocytopenia, although others were found, including anemia in 14, 14 splenectomies, 10 severe hemorrhages, 6 neurological symptoms, 3 avascular necroses, 1 vertebral collapse and 1 bone infarct. Although the most common diagnostic test yielding the diagnosis was bone-marrow aspiration, all patients’ diagnoses were confirmed by enzymatic assay. Genotypes were determined for 261 patients, with 203 having homo- (39 patients) or heterozygous p.N370S mutations (164 patients, including 41 with p.L444P/p.N370S mutations). Genotype distributions differed significantly (p<0.0001): L444P/L444P or L444P/other were found in 14 (6.4%) phenotype-1, 13 (100%) phenotype-2 and 12 (100%) phenotype-3 patients, while the genotypes N370S/L444P, N370S/other or N370S/N370S were identified in 203 (93.6%) phenotype-1, no phenotype-2 and no phenotype-3 patients. Other mutations found (associated or not with p.N370S or p.L444P) were: p.R48W, p.C16W, p.G193R, p.K303I, p.W312S, p.G377S, p.W393R, p.V394L, p.D409H, p.M416I, p.A446P, p.R463C, p.1416, 1417delAG and p.RecNcil. Patients were predominantly (84.7%) type 1. Among 161 patients with an affected family member, all but 3 (1 mother, 1 uncle, 1 cousin) were siblings. Ninety-seven patients died: 37 perinatal deaths (28 fetuses and 9 newborns <3 months old) and 60 later (33 type 1, 22 type 2 and 5 type 3).
Followed patients
A total of 378 patients, predominantly type 1, had ≥1 follow-up visits after their initial evaluations. The median follow-up duration was 16.2 (0.1–67.6) years. Their characteristics at diagnosis are reported in Table 1. During follow-up, 225 complained of chronic bone pain or clinical bone crisis, 231 had splenomegaly and 163 had hepatomegaly at least once.
Splenectomy
Surgery was performed on 104 patients: 17 before diagnosis, 46 at diagnosis and 41 >1 year postdiagnosis. Among the 41 patients splenectomized after diagnosis, 27 had the surgery before 1991 and only 14 thereafter. Among the 14 splenectomies after 1991, indications were: 2 for refractory idiopathic thrombocytopenic purpura, 2 for splenic rupture, 2 for major hypersplenism, 2 for severe splenic infarct and 2 for splenic hematoma but 4 had wrong or unknown indications. Splenectomy was more frequent in type 1 (29.3%) than type 3 patients (2, 13.3%) (p=0.009). Median age at splenectomy was 24.6 (3–76) years. Only 3 patients were taking ERT/SRT when splenectomy was performed for splenic complications.
BE
One hundred and nine patients experienced BE during follow-up, with a median of 1 (1–8) BE per patient and a total of 223 BE: 89 avascular necroses, 40 bone infarcts, 67 pathological fractures and 27 vertebral collapses. Sites were known for 188 BE: 56 avascular necroses affected the femur, 8 the humerus, 5 the tibia and 7 other bones (astragal, iliac crest, calcaneus), with 13 unknown sites; 15 bone infarcts involved the femur, 5 the tibia, 5 the iliac crest and 2 other bones, with 13 sites unknown; 11 pathological fractures concerned the wrists, 9 the ribs, 8 the femur, 8 the humerus and 22 other bones, with 9 localizations unknown. Type-1 patients had 107 (30.7%) BE, type-3 patients had 2 (13.3%), while type-2 patients had no BE (p=0.009). Median age at first BE was 34.1 (0–75.9) years. The first BE occurred in 88 patients without treatment and in 22 under ERT/SRT.
MG
Forty-six patients, all type 1, developed MG during follow-up. Median age at MG diagnosis was 48.2 (24.3–81) years. The median GD-diagnosis-to-MG-diagnosis interval was 1.3 (0–47.5) years. MG was not significantly more frequent in any given genotype (p=0.37): 1 (4%) p.N370S/p.N370S, 15 (60%) p.N370S/other, 6 (24%) p.N370S/p.L444P, 1 (4%) p.L444P/other and 2 (8%) in other/other; 21 patients with MG had no genotype determination. Comparing the p.L444P allele to the others, no significant difference was found (p=1). Thirteen patients were on ERT/SRT when MG was diagnosed (but no pretreatment immunofixation was available), 23 were treated thereafter and 10 had never been treated at closing date, despite MG.
PD
Neurologist-confirmed PD was diagnosed in 14 patients, all had type-1 GD; 6 had dementia. Median age at PD diagnosis was 60.4 (38.2–77.1) years. The median age at GD diagnosis of patients who developed PD was 46.2 (4.4–70.7) years. PD was significantly more frequent in patients with the p.L444P/p.N370S mutation versus others (p<0.0001): PD was diagnosed in 1 (2.9%) p.N370S/p.N370S patient, 2 (1.7%) p.N370S/other and 5 (13.5%) p.N370S/p.L444P; 6 PD patients had no genotype determination. Five patients were on ERT/SRT at PD diagnosis, 7 were treated thereafter and 2 had never been treated at closing date, despite PD. Treatment (imiglucerase or miglustat) did not have any apparent effect on PD signs. Two patients received miglustat and both stopped it after 1 year because of PD progression or dementia.
Treatments
Among the 378 patients with follow-up, 298 (78.8%) received treatment. The first ERT was alglucerase for 62 (20.8%) patients, imiglucerase for 224 (75.2%), miglustat for 7 (2.3%), velaglucerase for 4 (13.4%) and taliglucerase in 1 (0.3%). The median ERT dose (known for 228 patients) was 120 (30–284) IU/kg/month, with 204 patients receiving ≥120 IU/kg/month. Sixty-two patients were given miglustat during follow-up, with a median treatment duration of 0.7 (0.1–6.7) years. Ten patients had an indwelling catheter and 85 received ≥1 ERT administrations at home. The median diagnosis-to-first-treatment interval was 9.1 (0–61.4) years for all 298 patients, but only 1.4 (0–16) years for the 145 patients diagnosed after 1991.
Deaths
Thirty-eight patients died during follow-up: type 2 was fatal for all 15 patients compared to 20 (5.7%) type 1 or 3 (20%) type 3 (p<0.001). Age at death was known for 28 patients: median of 8.4 (0.3–83.4) years for all deceased, but 64.5 (38.4–83.4) years for 13 type-1 patients, 1 (0.3–2.2) year for the 13 type-2 patients and 8.4 (7.2–9.6) years for the 2 type-3 patients. Type-2 and −3 patients died of neurological impairment, whereas 11 type-1 patients’ deaths were attributed to: 2 lymphomas, 3 PD, 1 myeloma, 1 osteosarcoma, 2 with anemia or thrombocytopenia complications (1 each with myocardial infarct with anemia or pancytopenia) and 2 had pulmonary hypertension. Mortality was significantly more frequent (p<0.0001) for patients whose genotypes carried the p.L444P mutation; most of them had neurological impairment, as reported above: 1 (2.9%) with p.N370S/p.N370S, 3 (2.6%) p.N370S/other, 4 (10.8%) p.N370S/p.L444P, 5 (31.2%) p.L444P/other and 6 (54.5%) p.L444P/p.L444P; 17 deceased patients had no genotype determination. Fifteen patients were on ERT/SRT when they died, while all others had never been treated (including all type-2 patients).
Recently followed patients
Among followed patients, 95 had no known data during 2009–2010. Median time since the last hospital consultation for these patients was 10.7 (2.4–31.6) years. Thus, 283 patients had consulted during 2009–2010: 36 had never been treated and 247 patients had received ERT/SRT during that period. Their characteristics at diagnosis are reported in Table 1.
Hospital locations of followed patients
Figure 1 shows where the hospitals caring for the 283 patients with recent follow-up are located. Ninety-five patients were followed in Paris, including 73 (76.8%) in the RCLD. Follow-up was done in an internal medicine department for 152 (54%) patients, hematology for 65 (23%), pediatrics for 30 (11%) and other departments (7 gastroenterology, 6 neurology, 10 rheumatology, 13 others) for 36.

Map showing the locations of the hospitals monitoring the 283 patients with recent follow-up (2009–2010).
Patients without treatment
Thirty-six patients had never been treated: median diagnosis-to-closing-date interval was 7.8 (0.4–39.9) years, for 35 type-1 patients and 1 with type 2; 14 patients had familial GD (all siblings) and 3 patients, who developed BE (2 avascular necroses, 1 pathological fracture and 1 vertebral collapse) during follow-up, refused treatment.
Recently seen and treated patients
During 2009–2010, 247 patients (239 type 1 and 8 type 3) received treatment, with median follow-up at 19.3 (0.2–66.2) years. Among these recently treated patients, all 5 type-1 patients died at a median age of 65.8 (56.8–83.4) years. Table 2 provides clinical, biological and bone data at diagnosis, treatment onset and closing date. Clinical findings, biological values and bone findings tended to improve under ERT/SRT.
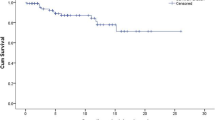
During follow-up, 190 BE (73 avascular necroses, 36 bone infarcts, 58 pathological fractures, 23 vertebral compressions) occurred in 86 patients, with a median of 1.5 (1–8) BE/patient. Figure 2 shows Kaplan–Meier estimates of the time to the first BE for the 247 recently seen and treated patients, between diagnosis and ERT/SRT onset (9.2 years of follow-up), and between the latter and the closing date (7.8 years of follow-up). Treatment at the time of BE was imiglucerase for 56 patients, alglucerase for 5 and miglustat for 4. The median imiglucerase/alglucerase dose when BE occurred in 52 patients was 120 (43.5–240) IU/kg/month; 42 patients had doses ≥120 IU/kg/month. The probabilities (95% confidence interval [CI]) of BE occurring by 10 years before and during treatment were estimated at 20.3% (14.1%–26.1%) and 19.8% (13.5%–26.1%), respectively. Before treatment, 67 patients developed 128 BE: 35 patients with 1 BE, 17 with 2 BE and 15 with ≥3 BE; whereas under treatment, 41 developed 62 BE: 28 patients with 1 BE, 8 with 2 BE and 5 with ≥3 BE, including 22 patients with BE before and under ERT/SRT.

Time to the first bone event (BE) in 247 GD patients receiving ERT/SRT. The dashed lines represent the curve’s 95% CI; the estimated probability of BE occurrence after 10 years is reported on the y-axis. (A) Between diagnosis and first treatment during the first 30 years of follow-up. (B) Between first treatment and end of treatment or closing date during 15 years of follow-up. No. at risk represents the number of patients followed at the indicated time; No. with BE represents the number of patients with a BE.
The probabilities (95% CI) of BE occurring by 10 years before ERT/SRT were 11.5% (3.1%–19.8%) versus 24.9% (16.6%–33.2%) for age at diagnosis ≤15 years and > 15 years, respectively (p=0.047). No other covariates were found to influence BE occurrence before ERT/SRT. During treatment, the probabilities (95% CI) of experiencing BE by 10 years were: 11.8% (5.9%–17.6%) and 35.9% (22.2%–49.5%) for patients without or with BE (Figure 3) before ERT/SRT, respectively, with an HR of 9.8 (5.9–16.3) (p<0.001); 29.1% (16.1%–42.1%) versus 14.7% (8.3%–21.1%) with and without splenectomy, respectively, with an HR of 2.1 (1.5–2.9) (p=0.005); 34.9% (29.8%–40.1%) vs 50.3% (46.1%-54.4%) for age at diagnosis ≤15 years and > 15 years, respectively, with an HR of 0.7 (0.5–0.98) (p=0.04); and 22.3% (15.1%–29.5%) versus 8.1% (0%–17.5%) for diagnosis-to-treatment interval >2 and ≤2 years, respectively, with an HR of 0.5 (0.3–0.9) (p=0.01). Age at diagnosis >15 years was not significantly associated with BE under treatment (p=0.54). In the multivariate analysis with backward elimination, BE before treatment was the only significant risk factor retained.

Impact of BE before treatment on BE occurrence under ERT/SRT for 247 treated GD patients. The solid bold grey line represents patients without BE before treatment; the solid bold black line represents the times to first BE. Dashed lines represent the 95% CI of those curves; the estimated probability of BE occurrence after 10 years reported on the y-axis.
Table 3 reports ERT/SRT prescribed according to the supply-shortage dates for the 247 recently treated patients. The first treatment prescribed was imiglucerase for 185 patients, alglucerase for 50, miglustat for 7, velaglucerase for 4 patients and taliglucerase for 1. The median diagnosis-to-treatment interval was 9.2 (0.0–47) years for all patients, but only 1.5 (0.0–16) years for patients diagnosed after 1991. The median treatment duration was 7.8 (0–18.3) years. During the supply shortages (June–August 2009), 106 patients discontinued their treatment, 9 switched to miglustat and 46 reduced their imiglucerase doses. Two patients received a combination of miglustat and imiglucerase, which was continued during the shortage (counted only under imiglucerase in Table 3).
Discussion
To date, no other publication has analyzed the comprehensive data entered in the FGDR for 562 patients, minus 3 who refused to participate and 97 who died, leaving 465 patients (among 65.8 million inhabitants), yielding prevalence of 1/140,000 inhabitants in France, a number that is probably underestimated. Concerning the entire cohort, although type 1 predominated (85%), types 2 and type 3 represented 4% each, along with 37 (6.9%) perinatal–lethal type. Moreover, the type-2 incidence was the same as that of type 3 but its prevalence was low because of its associated early mortality. The recent publication on the exhaustive Spanish registry[26] reported data similar to ours, with 88.3% type 1, 6.7% type 2 and 5% type 3. Our birth incidence (1/50,000) was higher than previously reported for the GD frequency in non-Jewish populations from EU countries[25, 39, 40], with a prevalence (1/136,000), close to that of the Spanish registry (1/149,000)[26].
Bone-marrow aspiration (or biopsy) remained the most common laboratory test (57%) providing the GD diagnosis. It is usually the first-line analysis when thrombocytopenia is associated (or not) with splenomegaly and there is no reason to think of immune thrombocytopenia purpura. It is not mandatory and should not be done if the GD diagnosis has been established by enzymatic assay or is already strongly suspected (e.g., possible family history). Rarely, bone-marrow aspiration was considered “normal” but another sample contained the characteristic GD cells.
Fourteen (3.7%) of our 378 followed patients had PD, reaching a prevalence comparable to that reported by Bultron et al.[41]. MG and polyclonal gamma globulinemia occur frequently in GD[42–45]. Among the 378 followed patients, 46 (12.2%) had MG, a rate within the previously reported range (1%[42] to 35%[45]), and median gamma globulinemia at ERT/SRT onset in recently treated patients was 21.7 g/L. Usually, MG is unaffected by ERT[43, 44]. However, for patients whose MG was diagnosed under treatment, no pretreatment evaluation was available, and MG had probably been present at treatment onset.
Before 1991, splenectomy was the only available treatment but, since then, it should not have been performed (albeit with exceptions) as a GD treatment. However, it has been used sometimes as a diagnostic tool when splenomegaly and thrombocytopenia coexisted, but should no longer be. Fourteen splenectomies were done after 1991 and after GD diagnosis, usually for patients with splenic complications (splenic infarcts, spleen rupture or large fibrous splenomegaly not amenable to ERT) or a mistaken indication.
BE are the most serious GD complications. They are usually prevented by ERT/SRT, with substantial attenuation of bone pain, bone crises and bone-mineral density[46], although the BE decrease is difficult to evaluate without randomized placebo-controlled trials. In addition, the definition of BE is not homogeneous across studies. Apparently, ERT/SRT does not prevent all BE, as indicated by the estimated respective probabilities of BE occurring by 10 years before and during treatment of 20.3% and 19.8%. It is likely that patients on ERT/SRT would probably have had more complications had they not been treated. Furthermore, we showed that BE before treatment increased the risk of BE under ERT/SRT and was the only factor retained in our multivariate analysis. Note that, as reported by Mistry et al.[38], our univariate analyses also found splenectomy and treatment >2 years after GD diagnosis to increase that risk, while sex and age at diagnosis ≤15 years were associated with increased risk of BE before but not under ERT/SRT. Thus, BE persist as a problem that is not fully resolved by treatment. The continuing challenges remain how to identify patients at risk before and under ERT/SRT, and then to decide whether or not these patients would benefit from earlier treatment onset and/or dose intensification.
In summary, the FGDR strong points are its comprehensiveness, independence, accreditation and/or certification by the various health authorities and cooperation generated among the different French centers. This registry also had to manage the imiglucerase shortage, when more severe GD and children were accorded priority treatment. The FGDR also enabled, during that shortage, nationwide management of the ERT/SRT stock and selection of those patients most in need of therapy (velaglucerase and taliglucerase). In France, GD-patient management is organized so that patients receive treatment near their homes, which improves their quality of life. Even though monitoring is not centralized, the FGDR identification and tracking of patients should contribute to improving their specific care management.
Abbreviations
- ACE:
-
Angiotensin-converting enzyme
- BE:
-
Bone events
- CI:
-
Confidence interval
- EGDTC:
-
Evaluation of Gaucher-Disease Treatment Committee
- ERT:
-
Enzyme-replacement therapy
- FGDR:
-
French Gaucher Disease Registry
- GD:
-
Gaucher’s disease
- HR:
-
Hazard ratio
- IQR:
-
Interquartile range
- MG:
-
Monoclonal gammopathy
- PD:
-
Parkinson’s disease
- RCLD:
-
Referral Center for Lysosomal Diseases
- SRT:
-
Substrate-reduction therapy
- TRAP:
-
Tartrate-resistant acid phosphatase.
References
Brady RO, Kanfer JN, Shapiro D: Metabolism of glucocerebrosides. II. Evidence of an enzymatic deficiency in Gaucher's disease. Biochem Biophys Res Commun. 1965, 18: 221-225. 10.1016/0006-291X(65)90743-6.
Staretz-Chacham O, Lang TC, LaMarca ME, Krasnewich D, Sidransky E: Lysosomal storage disorders in the newborn. Pediatrics. 2009, 123: 1191-1207. 10.1542/peds.2008-0635.
Christomanou H, Aignesberger A, Linke RP: Immunochemical characterization of two activator proteins stimulating enzymic sphingomyelin degradation in vitro. Absence of one of them in a human Gaucher disease variant. Biol Chem Hoppe Seyler. 1986, 367: 879-890. 10.1515/bchm3.1986.367.2.879.
Qi X, Grabowski GA: Molecular and cell biology of acid beta-glucosidase and prosaposin. Prog Nucleic Acid Res Mol Biol. 2001, 66: 203-239.
Grabowski GA: Recent clinical progress in Gaucher disease. Curr Opin Pediatr. 2005, 17: 519-524. 10.1097/01.mop.0000172702.33128.19.
Beutler E: Gaucher disease. Arch Intern Med. 1999, 159: 881-882. 10.1001/archinte.159.8.881.
Kraoua I, Stirnemann J, Ribeiro MJ, Rouaud T, Verin M, Annic A, Rose C, Defebvre L, Remenieras L, Schupbach M, et al: Parkinsonism in Gaucher's disease type 1: ten new cases and a review of the literature. Mov Disord. 2009, 24: 1524-1530. 10.1002/mds.22593.
Mignot C, Doummar D, Maire I, de Villemeur TB: Type 2 Gaucher disease: 15 new cases and review of the literature. Brain Dev. 2006, 28: 39-48. 10.1016/j.braindev.2005.04.005.
Eblan MJ, Goker-Alpan O, Sidransky E: Perinatal lethal Gaucher disease: a distinct phenotype along the neuronopathic continuum. Fetal Pediatr Pathol. 2005, 24: 205-222. 10.1080/15227950500405296.
Mignot C, Gelot A, Bessieres B, Daffos F, Voyer M, Menez F, Fallet Bianco C, Odent S, Le Duff D, Loget P, et al: Perinatal-lethal Gaucher disease. Am J Med Genet A. 2003, 120: 338-344.
Hollak CE, van Weely S, van Oers MH, Aerts JM: Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J Clin Invest. 1994, 93: 1288-1292. 10.1172/JCI117084.
Deegan PB, Moran MT, McFarlane I, Schofield JP, Boot RG, Aerts JM, Cox TM: Clinical evaluation of chemokine and enzymatic biomarkers of Gaucher disease. Blood Cells Mol Dis. 2005, 35: 259-267. 10.1016/j.bcmd.2005.05.005.
Lieberman J, Beutler E: Elevation of serum angiotensin-converting enzyme in Gaucher's disease. N Engl J Med. 1976, 294: 1442-1444. 10.1056/NEJM197606242942609.
Zimran A, Kay A, Gelbart T, Garver P, Thurston D, Saven A, Beutler E: Gaucher disease. Clinical, laboratory, radiologic, and genetic features of 53 patients. Medicine (Baltimore). 1992, 71: 337-353. 10.1097/00005792-199211000-00002.
Beutler E: Gaucher disease. Blood Rev. 1988, 2: 59-70. 10.1016/0268-960X(88)90009-4.
Morgan MA, Hoffbrand AV, Laulicht M, Luck W, Knowles S: Serum ferritin concentration in Gaucher's disease. Br Med J (Clin Res Ed). 1983, 286: 1864-10.1136/bmj.286.6381.1864.
Tuchman LR, Suna H, Carr JJ: Elevation of serum acid phosphatase in Gaucher's disease. Journal of Mount Sinai Hospital. 1956, 23: 227-229.
Troy K, Cuttner J, Reilly M, Grabowski G, Desnick R: Tartrate-resistant acid phosphatase staining of monocytes in Gaucher disease. Am J Hematol. 1985, 19: 237-244. 10.1002/ajh.2830190305.
Barton NW, Brady RO, Dambrosia JM, Di Bisceglie AM, Doppelt SH, Hill SC, Mankin HJ, Murray GJ, Parker RI, Argoff CE, et al: Replacement therapy for inherited enzyme deficiency – macrophage-targeted glucocerebrosidase for Gaucher's disease. N Engl J Med. 1991, 324: 1464-1470. 10.1056/NEJM199105233242104.
Zimran A, Altarescu G, Philips M, Attias D, Jmoudiak M, Deeb M, Wang N, Bhirangi K, Cohn GM, Elstein D: Phase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48-month experience. Blood. 2010, 115: 4651-4656. 10.1182/blood-2010-02-268649.
Shaaltiel Y, Bartfeld D, Hashmueli S, Baum G, Brill-Almon E, Galili G, Dym O, Boldin-Adamsky SA, Silman I, Sussman JL, et al: Production of glucocerebrosidase with terminal mannose glycans for enzyme replacement therapy of Gaucher's disease using a plant cell system. Plant Biotechnol J. 2007, 5: 579-590. 10.1111/j.1467-7652.2007.00263.x.
Aerts JM, Hollak CE, Boot RG, Groener JE, Maas M: Substrate reduction therapy of glycosphingolipid storage disorders. J Inherit Metab Dis. 2006, 29: 449-456. 10.1007/s10545-006-0272-5.
Hollak CE, Vom Dahl S, Aerts JM, Belmatoug N, Bembi B, Cohen Y, Collin-Histed T, Deegan P, van Dussen L, Giraldo P, et al: Force majeure: therapeutic measures in response to restricted supply of imiglucerase (Cerezyme) for patients with Gaucher disease. Blood Cells Mol Dis. 2010, 44: 41-47. 10.1016/j.bcmd.2009.09.006.
Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, Rosenbloom BE, Scott CR, Wappner RS, Weinreb NJ, Zimran A: The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med. 2000, 160: 2835-2843. 10.1001/archinte.160.18.2835.
Giraldo P, Pocovi M, Perez-Calvo J, Rubio-Felix D, Giralt M: Report of the Spanish Gaucher's disease registry: clinical and genetic characteristics. Haematologica. 2000, 85: 792-799.
Giraldo P, Alfonso P, Irun P, Gort L, Chabas A, Vilageliu L, Grinberg D, Sa Miranda CM, Pocovi M: Mapping the genetic and clinical characteristics of Gaucher disease in the Iberian Peninsula. Orphanet J Rare Dis. 2012, 7: 17-10.1186/1750-1172-7-17.
Sobreira E, Pires RF, Cizmarik M, Grabowski GA: Phenotypic and genotypic heterogeneity in Gaucher disease type 1: a comparison between Brazil and the rest of the world. Mol Genet Metab. 2007, 90: 81-86. 10.1016/j.ymgme.2006.08.009.
Eto Y, Ida H: Clinical and molecular characteristics of Japanese Gaucher disease. Neurochem Res. 1999, 24: 207-211. 10.1023/A:1022553819241.
Mistry PK, Weinreb NJ, Kaplan P, Cole JA, Gwosdow AR, Hangartner T: Osteopenia in Gaucher disease develops early in life: response to imiglucerase enzyme therapy in children, adolescents and adults. Blood Cells Mol Dis. 2011, 46: 66-72. 10.1016/j.bcmd.2010.10.011.
Rosenbloom B, Balwani M, Bronstein JM, Kolodny E, Sathe S, Gwosdow AR, Taylor JS, Cole JA, Zimran A, Weinreb NJ: The incidence of parkinsonism in patients with type 1 gaucher disease: data from the ICGG gaucher registry. Blood Cells Mol Dis. 2011, 46: 95-102. 10.1016/j.bcmd.2010.10.006.
Tylki-Szymanska A, Vellodi A, El-Beshlawy A, Cole JA, Kolodny E: Neuronopathic gaucher disease: demographic and clinical features of 131 patients enrolled in the international collaborative gaucher group neurological outcomes subregistry. J Inherit Metab Dis. 2010, 33: 339-346. 10.1007/s10545-009-9009-6.
Weinreb NJ, Deegan P, Kacena KA, Mistry P, Pastores GM, Velentgas P, Vom Dahl S: Life expectancy in Gaucher disease type 1. Am J Hematol. 2008, 83: 896-900. 10.1002/ajh.21305.
Fairley C, Zimran A, Phillips M, Cizmarik M, Yee J, Weinreb N, Packman S: Phenotypic heterogeneity of N370S homozygotes with type I Gaucher disease: an analysis of 798 patients from the ICGG Gaucher Registry. J Inherit Metab Dis. 2008, 31: 738-744. 10.1007/s10545-008-0868-z.
Weinreb NJ, Taylor J, Cox T, Yee J, Vom Dahl S: A benchmark analysis of the achievement of therapeutic goals for type 1 Gaucher disease patients treated with imiglucerase. Am J Hematol. 2008, 83: 890-895. 10.1002/ajh.21280.
Wenstrup RJ, Kacena KA, Kaplan P, Pastores GM, Prakash-Cheng A, Zimran A, Hangartner TN: Effect of enzyme replacement therapy with imiglucerase on BMD in type 1 Gaucher disease. J Bone Miner Res. 2007, 22: 119-126.
Daniels LB, Glew RH: Beta-glucosidase assays in the diagnosis of Gaucher's disease. Clin Chem. 1982, 28: 569-577.
Stirnemann J, Belmatoug N, Vincent C, Fain O, Fantin B, Mentre F: Bone events and evolution of biologic markers in Gaucher disease before and during treatment. Arthritis Res Ther. 2010, 12: R156-10.1186/ar3111.
Mistry PK, Deegan P, Vellodi A, Cole JA, Yeh M, Weinreb NJ: Timing of initiation of enzyme replacement therapy after diagnosis of type 1 Gaucher disease: effect on incidence of avascular necrosis. Br J Haematol. 2009, 147: 561-570. 10.1111/j.1365-2141.2009.07872.x.
Cox TM, Schofield JP: Gaucher's disease: clinical features and natural history. Baillieres Clin Haematol. 1997, 10: 657-689. 10.1016/S0950-3536(97)80033-9.
Lacerda L, Amaral O, Pinto R, Aerts J, Sa Miranda MC: The N370S mutation in the glucocerebrosidase gene of Portuguese type 1 Gaucher patients: linkage to the PvuII polymorphism. J Inherit Metab Dis. 1994, 17: 85-88. 10.1007/BF00735401.
Bultron G, Kacena K, Pearson D, Boxer M, Yang R, Sathe S, Pastores G, Mistry PK: The risk of Parkinson's disease in type 1 Gaucher disease. J Inherit Metab Dis. 2010, 33: 167-173. 10.1007/s10545-010-9055-0.
Brautbar A, Elstein D, Pines G, Abrahamov A, Zimran A: Effect of enzyme replacement therapy on gammopathies in Gaucher disease. Blood Cells Mol Dis. 2004, 32: 214-217. 10.1016/j.bcmd.2003.10.007.
Grosbois B, Rose C, Noel E, de Roux Serratrice C, Dobbelaere D, Gressin V, Cherin P, Hartmann A, Javier RM, Clerson P, et al: Gaucher disease and monoclonal gammopathy: a report of 17 cases and impact of therapy. Blood Cells Mol Dis. 2009, 43: 138-139. 10.1016/j.bcmd.2009.04.002.
De Fost M, Out TA, de Wilde FA, Tjin EP, Pals ST, van Oers MH, Boot RG, Aerts JF, Maas M, Vom Dahl S, Hollak CE: Immunoglobulin and free light chain abnormalities in Gaucher disease type I: data from an adult cohort of 63 patients and review of the literature. Ann Hematol. 2008, 87: 439-449. 10.1007/s00277-008-0441-8.
Marti GE, Ryan ET, Papadopoulos NM, Filling-Katz M, Barton N, Fleischer TA, Rick M, Gralnick HR: Polyclonal B-cell lymphocytosis and hypergammaglobulinemia in patients with Gaucher disease. Am J Hematol. 1988, 29: 189-194. 10.1002/ajh.2830290403.
Sims KB, Pastores GM, Weinreb NJ, Barranger J, Rosenbloom BE, Packman S, Kaplan P, Mankin H, Xavier R, Angell J, et al: Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet. 2008, 73: 430-440. 10.1111/j.1399-0004.2008.00978.x.
Acknowledgments
The authors wish to thank Janet Jacobson for editorial assistance, the RCLD secretary, Samira Zebiche, GD patients and their physicians, who contributed to the FDGR (listed in alphabetic order followed by their city): Agape Philippe (Saint-Denis), Allanore Yannick (Paris), Amir Abdeslem (Vendôme), Amsallem Daniel (Besançon), Bauduer Fréderic (Bayonne), Benbouker Lotfi (Tours), Bergelin-Besacon Anne (Le Mans), Bergère Alain (La Flèche), Bernard Olivier (Paris), Beyler Constance (Paris), Bismuth Mickael (Montpellier), Boiffard Olivier (Saintes), Bonnet Brigitte (Le Havre), Bordessoule Dominique (Limoges), Bouheddou Nadia (Moulins), Bouteiller Gilles (Auch), Brissot Pierre (Rennes), Brottier-Mancini Elisabeth (La Rochelle), Carreiro Miguel (Montauban), Cassan-Faux Nelly (Châtellerault), Cathebras Pascal (Saint-Priest-en-Jarez), Ceccaldi Joël (Libourne), Chabrol Brigitte (Marseille), Cohen-de Lara André (Paris), Cohen-Valensi Rolande (Martigues), Colnot Fabrice (Épinal), Constantini Denis (Corbeil–Éssonne), Costello Régis (Marseille), Dalbies Florence (Brest), Danne Odile (Cergy), Deconinck Éric (Besançon), Delahaye Florence (Évreux), Delattre Pierre (Cayenne), Delieu Fabienne (Nice), Demeocq François (Clermont-Ferrand), Denis Jacques (Évry), Descos Bruno (Nice), Desmurs-Clavel Hélène (Lyon), Diallo Boubacar (Remiremont), Djerad Hama (Nevers), Dobbelaere Dries (Lilles), Dreyfus Marie (Le Kremlin–Bicêtre), Dupriez Brigitte (Lens), Durieu Isabelle (Lyon), Edan Christine (Rennes), Fabre Sylvie (Montpellier), Feillet François (Vandœuvre-les-Nancy), Ferry Régine (Charleville-Mézières), Flodrops Hugues (Saint-Pierre, La Réunion), Fontaine Bertrand (Paris), Gaches Francis (Toulouse), Gay Claire (Saint-Étienne), Germain Dominique (Garches), Gil Helder (Besançon), Granier Françoise (Mantes-la-Jolie), Guérin Jacques (Arpajon), Guillaumat Cécile (Corbeil–Éssonne), Harle Robert (Marseille), Herbrecht Raoul (Strasbourg), Hutin Pascal (Quimper), Jarnouen de Villartay Philippe (Pontoise), Jourdan Éric (Nîmes), Kpati Agbo (Lagny-sur-Marne), Kuster Alice (Nantes), Lackmy-Port-Lis Marylin (Pointe-à-Pitre), Lamagnère Jean-Pierre (Tours), Lavigne Christian (Angers), Le Bideau Marc (Saint-Nazaire), Le Coz Marie-Françoise (Lorient), Le Henaff Catherine (Morlaix), Le Lorier Bernard (Melun), Le Niger Catherine (Brest), Lefebvre Vincent (Rodez), Leguy-Seguin Vanessa (Dijon), Lejars Odile (Tours), Lèone Jean (Reims), Leporrier Michel (Caen), Lescoeur Brigitte (Paris), Lèvy Marc (Nanterre), Lidove Olivier (Paris), Linassier Claude (Tours), Lioure Bruno (Strasbourg), Macro Margaret (Caen), Maillot François (Tours), Marie Isabelle (Rouen), Mathieu Sophie (Paris), Mazodier Karine (Marseille), Morel Pierre (Lens), Navarro Robert (Montpellier), Ninet Jacques (Lyon), Noël Esther (Strasbourg), Oksenhendler Éric (Paris), Orzechowski Christine (Bry-sur-Marne), Oudot Caroline (Paris), Pan-Petesch Brigitte (Brest), Pellegrino Béatrice (Saint-Germain), Perreti Delphine (Le Kremlin–Bicêtre), Pers Yves-Marie (Montpellier), Plantier Isabelle (Roubaix), Plouvier Emmanuel (Besançon), Pujazon Marie-Christine (Toulouse), Quinsat Denis (Antibes), Rhorlich Pierre-Simon (Besançon), Rivera Serge (Bayonne), Roche Jean (Roanne), Ruivard Marc (Clermont-Ferrand), Savoye Guillaume (Rouen), Sedel Frédéric (Paris), Simon Anne (Paris), Solary Éric (Dijon), Steiger Jean-Marie (Bourges), Tchamgoue Serge (Libourne), Themelin Pascal (Beauvais), Tieule Nathalie (Nice), Trab Albert (Nice), Varet Bruno (Paris), Vives Laurent (Saint-Gaudens), Zenone Thierry (Valence), Zunic Patricia (Saint-Pierre). The salaries of a statistician and a clinical research associate who participated in this study were funded, in part, by a grant from Genzyme France. Genzyme played no role in designing the study; in collecting, analyzing and interpreting the data; writing the paper; or the decision to submit the manuscript for publication. Publication of this article was not contingent upon approval of the study’s sponsors. The development of the original software for the French Gaucher Disease Registry (FGDR) was funded by a grant from the association VML (Vaincre les Maladies Lysosomales). J. Stirnemann’s work was funded, in part, by a grant from INSERM (Institut National de la Santé et de la Recherche Médicale). The FGDR was funded, in part, by INSERM and InVS (Institut national de Veille Sanitaire).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
Research grants from Genzyme France to University Paris–Diderot to fund statistical analyses and to AP–HP to finance data acquisition. Research grant from Shire France to APRIMI (Beaujon Hospital’s association) to finance data acquisition. C. Serratrice, L. Rossi-Semerano and B. Grosbois, received consulting fees, speaking fees, and/or honoraria from Genzyme (less than $10,000). C. Serratrice, D. Heraoui, F. Camou, A. Masseau and B. Grosbois received consulting fees, speaking fees and/or honoraria from Actelion (less than $10,000). C. Serratrice, F. Camou and B. Grosbois received consulting fees, speaking fees, and/or honoraria from Shire (less than $10,000).
Authors’ contributions
JS, FM, MV, NB, OF and BF designed research analyzed and interpreted data. All authors, except for FM, MV and DH were involved in treating patients and collecting data. JS had full access to all of the study data and takes responsibility for their integrity and the accuracy of the data analysis. DH developed the original software for the FGDR. JS, MV, NB and FM wrote the draft of the paper, which was then corrected and approved by all authors. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Stirnemann, J., Vigan, M., Hamroun, D. et al. The French Gaucher’s disease registry: clinical characteristics, complications and treatment of 562 patients. Orphanet J Rare Dis 7, 77 (2012). https://doi.org/10.1186/1750-1172-7-77
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1750-1172-7-77