
Abstract
A female patient, with normal familial history, developed at the age of 30 months an episode of diarrhoea, vomiting and lethargy which resolved spontaneously. At the age of 3 years, the patient re-iterated vomiting, was sub-febrile and hypoglycemic, fell into coma, developed seizures and sequels involving right hemi-body. Urinary excretion of hexanoylglycine and suberylglycine was low during this metabolic decompensation. A study of pre- and post-prandial blood glucose and ketones over a period of 24 hours showed a normal glycaemic cycle but a failure to form ketones after 12 hours fasting, suggesting a mitochondrial β-oxidation defect. Total blood carnitine was lowered with unesterified carnitine being half of the lowest control value. A diagnosis of mild MCAD deficiency (MCADD) was based on rates of 1-14C-octanoate and 9, 10-3H-myristate oxidation and of octanoyl-CoA dehydrogenase being reduced to 25% of control values. Other mitochondrial fatty acid oxidation proteins were functionally normal. De novo acylcarnitine synthesis in whole blood samples incubated with deuterated palmitate was also typical of MCADD. Genetic studies showed that the patient was compound heterozygous with a sequence variation in both of the two ACADM alleles; one had the common c.985A>G mutation and the other had a novel c.145C>G mutation. This is the first report for the ACADM gene c.145C>G mutation: it is located in exon 3 and causes a replacement of glutamine to glutamate at position 24 of the mature protein (Q24E). Associated with heterozygosity for c.985A>G mutation, this mutation is responsible for a mild MCADD phenotype along with a clinical story corroborating the emerging literature view that patients with genotypes representing mild MCADD (high residual enzyme activity and low urinary levels of glycine conjugates), similar to some of the mild MCADDs detected by MS/MS newborn screening, may be at risk for disease presentation.
Similar content being viewed by others

Introduction
Mitochondrial fatty acid oxidation defects (FAOD) are rare inherited diseases with clinical presentation ranging from severe outcome to asymptomatic status. Diagnosis of FAOD requires specific investigations including biochemical and genetic studies. The most common FAOD is represented by a deficiency in the activity of the medium-chain acyl-CoA dehydrogenase (MCAD, EC 3.1.2.20) protein, which is coded by the ACADM gene (chromosome 1p31) [1–4]. First reported in 1976 [5] and biochemically characterized in 1982 [6], MCAD deficiency (MCADD) has since been extensively described. Point mutations in the ACADM gene were reported early [7, 8] and many mutations have been documented subsequently, in part as a result of the large development of screening strategies [9–18]. As a general rule, these mutations lead to either truncated/absent protein synthesis, or replacement of one amino acid by another with a consequent drop in the activity of the mutant protein. The most common ACADM mutation is c.985A>G, which causes the replacement of a lysine by a glutamate at position 329 of the precursor protein (position 304 of the mature protein) [3, 17], being for this reason also referred to as the K329E (or K304E) protein variant. Eighty percent of symptomatic MCADD patients of European origin are homozygous for this mutation, and a further 18% of symptomatic MCADD are associated with the c.985A>G mutation in one allele and a rare mutation in the other allele [1, 3, 19, 20]. Mutations other than c.985A>G may predominate in non-European populations [21, 22].
MCADD is inherited as an autosomal recessive trait [2, 3, 23, 24]. Like other genetic β-oxidation defects, MCADD may cause in early childhood life-threatening events like hypoketotic/hypoglycaemic comas occurring during the course of catabolic states which disrupt the body energetic production/expense balance (infectious episodes, for instance) [2, 3, 23, 24]. In fact, MCADD may present phenotypically as either a classical (severe) or a mild form. In mild MCADD, the residual MCAD activity is intermediate between that observed in classical MCADD (usually less than a few percent) and their heterozygous carriers (around 50%) [25], the classical form of the disease resulting mainly from homozygosity for the c.985A>G mutation as mentioned above. The residual activity of the ACADM gene product and hence its impact on cell metabolism and patient clinical presentation depends on a series of factors including protein misfolding, MCAD tetramer stability, and thermosensitivity of the mutant protein [3, 26–28] along with contribution from the overlapping activities of other mitochondrial acyl-CoA dehydrogenases and efficacy of L-carnitine-based detoxification. In turn, a toxic "gain-of-function" pathogenesis has been also proposed for some MCAD variants [3].
Febrile episodes may negatively impact the disease outcome, especially when the mutant protein is thermosensitive, which is the case for the most common K304E protein variant [28]. The patient has then to face concomitantly not only the increased energy demand but also the sudden lowering of the fatty acid oxidation-derived energy production secondary to a drop in residual MCAD activity. This might explain why one quarter of the patients with classical MCADD do not survive their first life-threatening episode and why this disease is present in the etiology list for sudden infant death syndrome [29–31].
Like patients with classical MCADD, patients with mild MCADD may develop metabolic decompensation in their early childhood. They may have to cope throughout life with neurological sequels or other irreversible lesions caused by early childhood metabolic decompensation. These sequels do not necessarily reflect the MCADD genotype but rather are the consequences of the life threatening episode before medical intervention. Recent work, however, suggests that the link between susceptibility to develop metabolic decompensation and MCADD patient genotype is not straightforward and that this susceptibility is largely dependent on environmental triggers such as starvation stress, infection and/or fever [3, 18]. We report here on a patient with mild MCADD having undergone a severe metabolic decompensation and subsequent clinical sequels. Genetic studies revealed a previously unreported mutation (c.145C<G) in one of the ACADM alleles combined with the common c.985A>G mutation in the other allele. Biochemical investigations of the patient are reported along with clinical data from the same time and an assessment of the impact of the new mutant protein variant is discussed.
Materials and methods
Solvents, reagents and internal calibrators for in vitro biological explorations
Chemical reagents and solvents were HPLC grade and were obtained from Merck, acetonitrile from JT Baker, 1-butanol and methanol from Prolabo, and stable isotopes including [16-2H3, 15-2H2]-palmitate (substrate), [16-2H3]-pamitoylcarnitine, [8-2H3]-octanoylcarnitine and [3-2H3]-propionylcarnitine (internal standards) from CDN-Isotopes Inc. L-carnitine and other chemicals were from Sigma (St Louis, MO).
Determinations of body fluid organic acids, acylglycines, carnitine (total, unesterified and esterified forms) and acylcarnitines
Trimethylsilyl derivatives of organic acids were quantified using capillary gas chromatography (GC) coupled to mass spectrometry (MS), and total and unesterified carnitine levels were determined as previously described [32, 33]. Individual acylcarnitines in body fluids were analyzed by fast atom bombardment (FAB)-MS or MS-MS of a blood spot on filter paper (Guthrie card) [33–35].
Preparation of lymphocytes and culture of fibroblasts
Mononuclear cells were isolated from 10 mL of blood, using a Ficoll-Paque procedure. The pellet was suspended in Dulbecco's phosphate buffered saline with 0.9 mM Ca2+ and 0.5 mM Mg2+, pH 7.35 (PBS), in order to adjust total cell proteins of the cell suspension to 1 ± 0.4 mg/ml for enzymatic determinations.
Skin fibroblasts were cultured as previously described [36, 37].
Fatty acid oxidation rate studies on lymphocytes and fibroblasts
Tritiated water release experiments [38] were performed in triplicate, in 24-well microtiter plates. [9, 10(n)-3H] palmitic and myristic acids bound to albumin (0.5 mg/ml) were used as substrates, at a final concentration of 100 μmol/l in PBS.
The capacity to oxidize 14C lapelled substrates was examined essentially as described previously [36, 37].
Measurement of individual enzymes of fatty acid oxidation
Mitochondrial acyl-CoA dehydrogenases were assayed by the ETF reduction assay [38]. Other fatty acid oxidation proteins were assayed by well standardized procedures [39, 40].
Genetic studies
Preparation of DNA samples of blood origin and PCR, purification and sequence analysis of exons of the ACADM gene were performed as described previously [12].
Procedure relative to de novo synthesis of acylcarnitines by whole blood samples
De novo synthesis of acylcarnitines by whole blood samples incubated with deuterated palmitate was monitored essentially as described in Dessein et al. [34] with the ESI tandem mass spectrometry performed with either a Perkin Elmer mass or a Micromass Quattro PremierTM mass spectrometer.
Case description
First clinical symptoms and biological data
The patient was born normally as a result of a twin pregnancy, her twin brother being clinically healthy. Her eldest brother has also been healthy. She had a normal gestation and familial history.
At the age of 30 months, she was hospitalized for diarrhea without fever, vomiting and lethargy. Electroencephalogram and brain scanner examinations were normal. Glycaemia was normal as well as aminoacidogram. Discrete biological liver cytolysis was noticed with GOT and GPT increased at 45 and 90 IU/l, respectively (upper control values at 30 IU/l) along with echographical signs of mild hepatomegaly. Viral and bacterial serology was negative. This episode spontaneously resolved in a couple of days, and the patient recovered rapidly to a healthy status and normal life activities.
At the age of 3 years, she re-iterated an episode of vomiting which was sub-febrile (38.5°C) and followed by a coma, requiring a new hospitalization. On this occasion, marked hypoglycaemia (0.17 g/L) was documented and the patient presented with tonic-clonic seizures on the right side which persisted for 3 days and was treated by combined phenobarbital and phenytoin. The recovery of this status epilepticus was progressive. The child developed, however, sequels including a deficit of the right hemi-body, delay in the language acquisition, and electroencephalographic abnormalities in the rear area of the left hemisphere. The biological analyses revealed a liver cytolysis (increased blood levels of GPT [96 IU/l] and GOT [134 IU/l]) without signs of hepatic failure. A study of pre- and post-prandial blood glucose and ketones over a period of 24 hours showed normal glycaemias but a failure to form ketones after 12 hours fasting. This prompted a study of mitochondrial fatty acid oxidation via metabolite analyses in body fluids samples and biochemical measurements in cultured skin fibroblasts and lymphocytes. The diagnostic biological/biochemical investigations were completed by genetic studies and functional assessment of de novo acylcarnitine synthesis by whole blood samples.
Diagnostic biological and biochemical explorations
- Metabolite analyses
The results of metabolite analyses are given in Table 1. Urinary organic acid profiles obtained during the comatose metabolic crisis showed increased medium-chain dicarboxylic acids but neither hexanoylglycine nor suberylglycine were detected by routine GC-MS analysis. 3-Hydroxybutyrate was increased in the urine indicating some ability to form ketone bodies. The patient presented with hypocarnitinemia affecting both total and unesterified carnitine levels which were reduced to half of the control values. This hypocarnitinemia was treated by oral L-carnitine supplementation (100 mg/kg/day) maintained throughout life (with a dose limit of 1.5 g/day). Several other metabolite analyses in blood and urine were performed when the patient was under L-carnitine therapy. Under these conditions, blood total and unesterified carnitine levels were normalized, and the esterified to unesterified carnitine ratio was at the upper control limit. Blood acylcarnitine analysis showed an increase in octanoylcarnitine with a C8/C10 acylcarnitine ratio higher than 5 and evocative of a defect in octanoate oxidation. In the patient urine, C8 acylcarnitines represented more than half of the total acylcarnitine content in contrast to the normal urine samples in which, even under carnitine load, the major acylcarnitine is acetylcarnitine [33].
- Biochemical determinations
Fatty acid oxidation rates measured on intact lymphocytes (Table 2) were within control range for butyrate and palmitate labeled at carbon 1, whereas octanoate oxidation was deficient. Using either myristate or palmitate labeled by tritium substitution of carbons 9 and 10 of the fatty acid indicated residual fatty acid oxidation activities of 11 and 24% of controls, respectively.
Biochemical determinations were also performed on patient fibroblast preparations (Table 3). Overall fibroblast fatty acid oxidation rates investigated with either [9, 10-3H]-myristic acid or [9, 10-3H]-palmitic acid confirmed a fatty acid oxidation defect. Other measurements of mitochondrial oxidation performed on intact fibroblasts indicated that the fatty acid oxidation defect involved medium-chain fatty acid (octanoate) β-oxidation. This location of the defect was confirmed, using either octanoyl-CoA or phenylpropionyl-CoA as a substrate, by the reduction of MCAD activity along with normal VLCAD and SCAD activities. Reduced oxidation of phenylpropionate by intact fibroblasts (10% of control values) further confirmed deficient MCAD activity (data not shown).
Residual MCAD activity studied by the various procedures was between 10 to 28% of control values and overall fatty acid oxidation rates were lowered to similar extents. The activities of other enzymes involved in mitochondrial β-oxidation, including enoyl-CoA hydratases, β-hydroxyacyl-CoA dehydrogenases and β-ketoacyl-CoA thiolases as well as the ketolysis enzyme succinyl-CoA ketoacid transferase, were in normal range values. The proteins involved in cellular utilization of carnitine and its esters, including carnitine palmitoyltransferase types I and II and the plasma membrane carnitine transporter(s) also exhibited normal activities in fibroblast preparations (Table 2).
Genetic studies
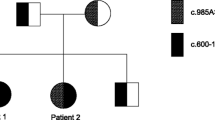
PCR amplification of all exons of the human ACADM gene, including part of the flanking intron sequences, was carried out as previously described [12] using intron-located primers under standard conditions. The PCR products were subjected to bi-directional cycle sequencing using DNA dye terminator sequencing kits (BigDye, Perkin-Elmer). Patient DNA samples were found to be heterozygous for the common c.985A>G mutation in exon 11. Another heterozygous mutation, consisting of C replacement by G at position 145 of the cDNA sequence (c.145C>G) in exon 3 was identified. Sequence analysis of exons 3 and 11 was then performed on DNA samples from parents and siblings. A pedigree tree for the two ACADM mutations is shown in Figure 1.

Pedigree diagram showing haplotypes for the ACADM allele mutations in the patient, siblings and parents.
De novo syntheses of acylcarnitines from deuterated palmitate
De novo syntheses of acylcarnitines from deuterated palmitate were studied in whole blood samples from the patient and healthy controls. Relative median [D5]-acylcarnitine production rates generated from incubation of whole blood samples with [16-2H3, 15-2H2]-[D5]-palmitate along with C8 on C4 acylcarnitine production rate ratio are illustrated in Figure 2 for the patient and a healthy control. Compared with the profile in this healthy control and with median reference values originating from 59 distinct healthy control subjects, the data from our patient with mild MCADD appeared to be pathological. Both the profile of deuterated acylcarnitine produced from stable-labeled palmitate and the ratio between rates of production of C8- and C4-acylcarnitines were abnormal, aligning on profiles in three patients with classical severe MCADD (Figure 2, bottom panels). This datum is clearly distinguishable from profiles in a carrier adult who was heterozygous for the c.985A>G MCAD gene mutation (Figure 2, top panels). The classical MCADD patient 1 was among those reported in previous work [34]; patients 2 and 3 being sister and daughter of the classical MCADD patient 1 and of the adult carrier, respectively.

De novo individual acylcarnitine synthesis by whole blood samples from the mild MCADD patient, a healthy control and a adult carrier (top panels), and from patients with classical MCADD (bottom panels). De novo acylcarnitine synthesis rates generated from deuterated palmitate in the presence of added L-carnitine by whole blood samples and derived C8/C4 acylcarnitine production rates have been determined in patient (compound heterozygous for c.985A>G and c.145C>G ACADM mutations) and a adult healthy volunteer and is compared to the same exploration performed in a adult carrier for the c.985A>G ACADM mutation (see top panels). The series of bottom panels report results of these measurements in individual patients with the classical form of MCADD and with proven homozygocy for the c.985A>G ACADM gene mutation. Formations of individual [D5]-acylcarnitines in whole blood samples are relative median rates expressed as percentages of median control values (framed profiles). The ratio between the rates of production of the major C8-acylcarnitine and the C4-acylcarnitine is expressed as relative median units, i.e. as the number of fold (and not the number of percents of) median control values. Values of C8 acylcarnitine production rates in disrupted histograms are given at the top of these histograms, and values for C8/C4 acylcarnitine ratios are given below each ratio histogram. The 5th to 95th percentile range for relative median production rate values is given in back position of each framed series of acylcarnitine production rates. The 5th to 95th percentile range for relative median C8/C4 acylcarnitine ratios is represented by the small green rectangle present at the basis of each C8/C4 ratio histogram and was exceeded by all MCADD patient values but neither carrier nor control values.
Discussion
Our patient undoubtedly suffered from a deficiency of MCAD, presenting with a mild phenotype of the disease as ascertained by relatively high residual activity from lymphocytes and fibroblasts and by a low urinary excretion of hexanoylglycine and suberylgycine during metabolic decompensation. At the time of this urine analysis, acylglycines were determined by GC/MS (with a detection limit of 2 mmol/mol creatine) and could not be discriminated from control values. Better analytical detections of urinary acylglycines are obtained by GC/MS-MS or GC-MS stable-isotope dilution analysis (SID) of methyl esters, with detection limits less than 0.7-1 mmol/mol creatinine, allowing for upper limit of controls a clear cut-off value close to 1.1 mmol/mol [25, 41].
Biochemically, our patient resembled the four mild MCADD patients described by Zschocke et al. [25] who had a residual MCAD activity (10-20%) of the same order of magnitude as in our patient. In fact, these four cases and our patient all show intermediate (between controls and severe MCADD patients) values for both enzyme activities and acylcarnitine levels and ratios. The residual enzyme activity observed in classical MCADD, caused by homozygosity for c.985A>G, is usually below 1%. Two of the four patients previously described by Zschocke et al. [25] were homozygous for ACADM gene mutations (c.734C>T or c.799G>A [exon 9]) distinct from the c.985A>G mutation; the other two patients were compound heterozygous for the c.985A>G mutation in one allele and the c.199T>C (exon 3) mutation in the other ACADM allele. Genotypically, our patient resembled the latter two patients, with the c.985A>G mutation (exon 11) in one allele and a mutation in exon 3 (namely, c.145C>G mutation) in the other allele. This mutation has never been observed in controls. To our knowledge, this is the first report for the c.145 C>G mutation in the ACADM gene.
As a general rule, amino acid changes introduced in a mature protein affect its conformation and this may impact its final stability and function/activity. This has been well documented for the MCAD mutant protein (K304E) resulting from the c.985A>G mutation. The MCAD protein requires assembly of the monomers in a tetramer, which represents the active functional form of MCAD. In the case of monomers resulting from the wild-type ACADM gene, the MCAD tetramer is stable. In the case of K304E monomers, the tetramer dissociates abnormally rapidly [26], giving rise to a reduced steady-state concentration of the tetrameric form. Another contributing factor is impairment of intramitochondrial processes including altered monomer folding or management [27]. Several studies indicate that protein misfolding is the key event in the loss of MCAD activity from many protein variants, and that overexpressing chaperonins may rescue monomers from aggregation and enhance residual MCAD activity (see [3] and references therein). As a result of protein misfolding, accelerated temperature-driven unfolding may affect MCAD protein variants. This abnormal MCAD loss of function and thermosensitivity first documented for the K304E variant, has been recently extended to a number of MCAD variants [18, 42], questioning the view that some of the missense mutations detected by newborn screening are less life-threatening than those clinically proven to be disease causing. In this respect, the relatively high residual activity found in our mild MCADD patient has not prevented the occurrence of a severe metabolic decompensation which might have been fatal in the absence of adequate medical management. A patient with the R28C mutant protein who had a mutation (c.157C>T), also less severe than the c.985A>G mutation, died unexpectedly in early childhood [43].
Our mild MCADD patient combines MCAD K304E and Q24E variant monomers. Conformational characteristics of the MCAD K304E variant have been repeatedly described in the literature. The amino acid change is located in the MCAD C terminal α domain which is involved in helix-helix interactions taking place in tetramer assembly (see [18] and references therein). As a result, tetramer assembly is compromised: unemployed monomers aggregate and are subjected to degradation. Experimental prevention of monomer aggregation by overexpressed chaperonins has been shown to result in increased tetramer assembly along with higher residual activity (see [3] and references therein).
The Q24E variant has not been previously described. The amino acid change here occurs in the MCAD N-terminal α domain. Some of the protein variants with mutations in this protein region may present with normal or close to normal enzyme activity [3, 18]. Mutations of this portion of the MCAD protein are also shown to induce moderate alterations in tetramer assembly and to disrupt thermal stability. This is the case for the Y42 H mutant protein for which temperature-driven loss of activity is greater than normal though lower than that of common K304E mutant protein [42]. De novo synthesis of acylcarnitines monitored on whole blood samples from our patient was consistent with MCADD. The major acylcarnitine produced from deuterated palmitate was octanoylcarnitine, other medium-chain (hexanoyl- and decanoyl-) carnitine esters being also generated in excess. The C8 to C4 acylcarnitine production rate ratio was 54-fold that of controls. As a whole, profile and ratio of acyl-carnitine production rates in our mild MCADD patient resemble those found in patients with classical MCADD, indicating clearly functional repercussion of the enzyme defect on the disposal of acyl-CoA. The carnitine added to incubation media appeared to mobilize acyl-CoAs unmetabolised by β-oxidation and, by comparing acylcarnitines production rates to control values, revealed the level(s) at which β-oxidation was defective. The acylcarnitine production rates and increased C8/C4 acylcarnitine de novo synthesis ratio of our mild MCADD patient are clearly different from those of the adult carrier of the c.985A>G mutation (figure 2).
The last but not least issue concerns the carnitine status of our patient at the time of metabolic decompensation. The patient was then not receiving L-carnitine therapy and her systemic carnitine levels were below normal (Table 1). This chronic hypocarnitinaemia was concomitant with a low urinary excretion of hexanoylglycine and suberylglycine. Interestingly, no elevation of urinary suberylglycine was reported in the urine from one patient with hypocarnitinaemia in the description of four MCADD asymptomatic siblings (compound heterozygotes for the common c.985A>G and the c.842G>C ACADM gene mutations) provided by Albers and coworkers [44]. Zschocke and coworkers on the other hand discussed elevated acylcarnitine and undetectable 5-hydroxyhexanoic acid and suberylglycine in their four mild MCADD patients [25]. Patients with genotypes representing mild MCADD (high residual enzyme activity and low urinary levels of glycine conjugates) should get the same advice as patients with classical MCADD regarding risks associated with catabolic states and carnitine deficiency.
Consent
Written informed consent was obtained by mother of the patient for publication of this case report.
References
Gregersen N, Bross P, Andresen BS: Genetic defects in fatty acid beta-oxidation and acyl-CoA dehydrogenases. Molecular pathogenesis and genotype-phenotype relationships. Eur J Biochem. 2004, 271: 470-482. 10.1046/j.1432-1033.2003.03949.x.
Roe CR, Ding JH: Mitochondrial fatty acid oxidation disorders. The metabolic and molecular bases of inherited disease. Edited by: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B. McGraw-Hill, New York, 2005, 8: 2297-2326.
Gregersen N, Andresen BS, Pedersen CB, Olsen RK, Corydon TJ, Bross P: Mitochondrial fatty acid oxidation defects--remaining challenges. J Inherit Metab Dis. 2008, 31: 643-657. 10.1007/s10545-008-0990-y.
Kompare M, Rizzo WB: Mitochondrial fatty-acid oxidation disorders. Semin Pediatr Neurol. 2008, 15: 140-149. 10.1016/j.spen.2008.05.008.
Gregersen N, Lauritzen R, Rasmussen K: Suberylglycine excretion in the urine from a patient with dicarboxylic aciduria. Clin Chim Acta. 1976, 70: 417-425. 10.1016/0009-8981(76)90355-7.
Kølvraa S, Gregersen N, Christensen E, Hobolth N: In vitro fibroblast studies in a patient with C6-C10-dicarboxylic aciduria: evidence for a defect in general acyl-CoA dehydrogenase. Clin Chim Acta. 1982, 126: 53-67. 10.1016/0009-8981(82)90361-8.
Ikeda Y, Hale DE, Keese SM, Coates PM, Tanaka K: Biosynthesis of variant medium chain acyl-CoA dehydrogenase in cultured fibroblasts from patients with medium chain acyl-CoA dehydrogenase deficiency. Pediatr Res. 1986, 20: 843-847. 10.1203/00006450-198609000-00007.
Kelly DP, Kim JJ, Billadello JJ, Hainline BE, Chu TW, Strauss AW: Nucleotide sequence of medium-chain acyl-CoA dehydrogenase mRNA and its expression in enzyme-deficient human tissue. Proc Natl Acad Sci USA. 1987, 84: 4068-4072. 10.1073/pnas.84.12.4068.
Yokota I, Coates PM, Hale DE, Rinaldo P, Tanaka K: Molecular survey of a prevalent mutation, 985A-to-G transition, and identification of five infrequent mutations in the medium chain Acyl-CoA dehydrogenase (MCAD) gene in 55 patients with MCAD deficiency. Am J Hum Genet. 1991, 49: 1280-1291.
Coates PM, Tanaka K: Molecular basis of mitochondrial fatty acid oxidation defects. J Lipid Res. 1992, 33: 1099-1110.
Tanaka K, Yokota I, Coates PM, Strauss AW, Kelly DP, Zhang Z, Gregersen N, Andresen BS, Matsubara Y, Curtis D, Chen YT: Mutations in the medium chain acyl-CoA dehydrogenase (MCAD) gene. Hum Mutat. 1992, 1: 271-279. 10.1002/humu.1380010402.
Andresen BS, Bross P, Udvari S, Kirk J, Gray G, Kmoch S, Chamoles N, Knudsen I, Winter V, Wilcken B, Yokota I, Hart K, Packman S, Harpey JP, Saudubray JM, Hale DE, Bolund L, Kølvraa S, Gregersen N: The molecular basis of medium-chain acyl-CoA dehydrogenase (MCAD) deficiency in compound heterozygous patients: is there correlation between genotype and phenotype?. Hum Mol Genet. 1997, 6: 695-707. 10.1093/hmg/6.5.695.
Küchler B, Abdel-Ghany AG, Bross P, Nandy A, Rasched I, Ghisla S: Biochemical characterization of a variant human medium-chain acyl-CoA dehydrogenase with a disease -associated mutation localized in the active site. Biochem J. 1999, 337: 225-230. 10.1042/0264-6021:3370225.
Andresen BS, Dobrowolski SF, O'Reilly L, Muenzer J, McCandless SE, Frazier DM, Udvari S, Bross P, Knudsen I, Banas R, Chace DH, Engel P, Naylor EW, Gregersen N: Medium-chain acyl-CoA dehydrogenase (MCAD) mutations identified by MS/MS-based prospective screening of newborns differ from those observed in patients with clinical symptoms: identification and characterization of a new, prevalent mutation that results in mild MCAD deficiency. Am J Hum Genet. 2001, 68: 1408-1418. 10.1086/320602.
Gregersen N, Andresen BS, Corydon MJ, Corydon TJ, Olsen RK, Bolund L, Bross P: Mutation analysis in mitochondrial fatty acid oxidation defects: Exemplified by acyl-CoA dehydrogenase deficiencies, with special focus on genotype-phenotype relationship. Hum Mutat. 2001, 18: 169-189. 10.1002/humu.1174.
Korman SH, Gutman A, Brooks R, Sinnathamby T, Gregersen N, Andresen BS: Homozygosity for a severe novel medium-chain acyl-CoA dehydrogenase (MCAD) mutation IVS3-1G>C that leads to introduction of a premature termination codon by complete missplicing of the MCAD mRNA and is associated with phenotypic diversity ranging from sudden neonatal death to asymptomatic status. Mol Genet Metab. 2004, 82: 121-129. 10.1016/j.ymgme.2004.03.002.
Rhead WJ: Newborn screening for medium-chain acyl-CoA dehydrogenase deficiency: a global perspective. J Inherit Metab Dis. 2006, 29: 370-377. 10.1007/s10545-006-0292-1.
Maier EM, Gersting SW, Kemter KF, Jank JM, Reindl M, Messing DD, Truger MS, Sommerhoff CP, Muntau AC: Protein misfolding is the molecular mechanism underlying MCADD identified in newborn screening. Hum Mol Genet. 2009, 18: 1612-1623. 10.1093/hmg/ddp079.
Tanaka K, Gregersen N, Ribes A, Kim J, Kølvraa S, Winter V, Eiberg H, Martinez G, Deufel T, Leifert B, Santer R, François B, Pronicka E, László A, Kmoch S, Kremensky I, Kalaydjicva L, Ozalp I, Ito MA: A survey of the newborn populations in Belgium, Germany, Poland, Czech Republic, Hungary, Bulgaria, Spain, Turkey, and Japan for the G985 variant allele with haplotype analysis at the medium chain Acyl-CoA dehydrogenase gene locus: clinical and evolutionary consideration. Pediatr Res. 1997, 41: 201-209. 10.1203/00006450-199702000-00008.
Waddell L, Wiley V, Carpenter K, Bennetts B, Angel L, Andresen BS, Wilcken B: Medium-chain acyl-CoA dehydrogenase deficiency: genotype-biochemical phenotype correlations. Mol Genet Metab. 2006, 87: 32-9. 10.1016/j.ymgme.2005.09.020.
Ensenauer R, Winters JL, Parton PA, Kronn DF, Kim JW, Matern D, Rinaldo P, Hahn SH: Genotypic differences of MCAD deficiency in the Asian population: novel genotype and clinical symptoms preceding newborn screening notification. Genet Med. 2005, 7: 339-343. 10.1097/01.GIM.0000164548.54482.9D.
Maier EM, Liebl B, Röschinger W, Nennstiel-Ratzel U, Fingerhut R, Olgemöller B, Busch U, Krone N, v Kries R, Roscher AA: Population spectrum of ACADM genotypes correlated to biochemical phenotypes in newborn screening for medium-chain acyl-CoA dehydrogenase deficiency. Hum Mutat. 2005, 25: 443-452. 10.1002/humu.20163.
Vianey-Liaud C, Divry P, Gregersen N, Mathieu M: The inborn errors of mitochondrial fatty acid oxidation. J Inherit Metab Dis. 1987, 10 (Suppl 1): 159-200. 10.1007/BF01812855.
Iafolla AK, Thompson RJ, Roe CR: Medium-chain acyl-coenzyme A dehydrogenase deficiency: clinical course in 120 affected children. J Pediatr. 1994, 124: 409-415. 10.1016/S0022-3476(94)70363-9.
Zschocke J, Schulze A, Lindner M, Fiesel S, Olgemöller K, Hoffmann GF, Penzien J, Ruiter JP, Wanders RJ, Mayatepek E: Molecular and functional characterisation of mild MCAD deficiency. Hum Genet. 2001, 108: 404-408. 10.1007/s004390100501.
Yokota I, Saijo T, Vockley J, Tanaka K: Impaired tetramer assembly of variant medium-chain acyl-coenzyme A dehydrogenase with a glutamate or aspartate substitution for lysine 304 causing instability of the protein. J Biol Chem. 1992, 267: 26004-26010.
Saijo T, Welch WJ, Tanaka K: Intramitochondrial folding and assembly of medium-chain acyl-CoA dehydrogenase (MCAD). Demonstration of impaired transfer of K304E-variant MCAD from its complex with hsp60 to the native tetramer. J Biol Chem. 1994, 269: 4401-4408.
Bross P, Jespersen C, Jensen TG, Andresen BS, Kristensen MJ, Winter V, Nandy A, Kräutle F, Ghisla S, Bolund L, Kim JJP, Gregersen N: Effects of two mutations detected in medium chain acyl-CoA dehydrogenase (MCAD)-deficient patients on folding, oligomer assembly, and stability of MCAD enzyme. J Biol Chem. 1995, 270: 10284-10290. 10.1074/jbc.270.17.10284.
Lundemose JB, Kølvraa S, Gregersen N, Christensen E, Gregersen M: Fatty acid oxidation disorders as primary cause of sudden and unexpected death in infants and young children: an investigation performed on cultured fibroblasts from 79 children who died aged between 0-4 years. Mol Pathol. 1997, 50: 212-217. 10.1136/mp.50.4.212.
Pollitt RJ, Leonard JV: Prospective surveillance study of medium chain acyl-CoA dehydrogenase deficiency in the UK. Arch Dis Child. 1998, 79: 116-119. 10.1136/adc.79.2.116.
Opdal SH, Rognum TO: The sudden infant death syndrome gene: does it exist?. Pediatrics. 2004, 114: e506-e512. 10.1542/peds.2004-0683.
Fontaine M, Porchet N, Largilliere C, Marrakchi S, Lhermitte M, Aubert JP, Degand P: Biochemical contribution to diagnosis and study of a new case of D-glyceric acidemia/aciduria. Clin Chem. 1989, 35: 2148-2151.
Fontaine M, Briand G, Vallée L, Ricart G, Degand P, Divry P, Vianey-Saban C, Vamecq J: Acylcarnitine removal in a patient with acyl-CoA beta-oxidation deficiency disorder: effect of L-carnitine therapy and starvation. Clin Chim Acta. 1996, 252: 109-122. 10.1016/0009-8981(96)06323-1.
Dessein AF, Fontaine M, Dobbelaere D, Mention-Mulliez K, Martin-Ponthieu A, Briand G, Vamecq J: Deuterated palmitate-driven acylcarnitine formation by whole-blood samples for a rapid diagnostic exploration of mitochondrial fatty acid oxidation disorders. Clin Chim Acta. 2009, 406: 23-26. 10.1016/j.cca.2009.04.026.
Van Hove JL, Zhang W, Kahler SG, Roe CR, Chen YT, Terada N, Chace DH, Iafolla AK, Ding JH, Millington DS: Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency: diagnosis by acylcarnitine analysis in blood. Am J Hum Genet. 1993, 52: 958-966.
Vianey-Saban C, Mousson B, Bertrand C, Stamm D, Dumoulin R, Zabot MT, Divry P, Floret D, Mathieu M: Carnitine palmitoyl transferase I deficiency presenting as a Reye-like syndrome without hypoglycaemia. Eur J Pediatr. 1993, 152: 334-338. 10.1007/BF01956748.
Saudubray JM, Coudé FX, Demaugre F, Johnson C, Gibson KM, Nyhan WL: Oxidation of fatty acids in cultured fibroblasts: a model system for the detection and study of defects in oxidation. Pediatr Res. 1982, 16: 877-881. 10.1203/00006450-198210000-00015.
Brivet M, Slama A, Saudubray JM, Legrand A, Lemonnier A: Rapid diagnosis of long chain and medium chain fatty acid oxidation disorders using lymphocytes. Ann Clin Biochem. 1995, 32: 154-159.
Bertrand C, Dumoulin R, Divry P, Mathieu M, Vianey-Saban C: Purification of electron transfer flavoprotein from pig liver mitochondria and its application to the diagnosis of deficiencies of acyl-CoA dehydrogenases in human fibroblasts. Clin Chim Acta. 1992, 210: 75-91. 10.1016/0009-8981(92)90047-T.
IJlst L, Poggi F, Bonnefont JP, Munnich A, Brivet M, Rabier D, Saudubray J-M: Human trifunctional protein deficiency. Biochem Biophys Res Commun. 1992, 188: 1139-1145. 10.1016/0006-291X(92)91350-Y.
IJlst L, Mandel H, Oostheim W, Ruiter JPN, Gutman A, Wanders RJA: Molecular Basis of Hepatic Carnitine Palmitoyltransferase I Deficiency. J Clin Invest. 1998, 102: 527-531. 10.1172/JCI2927.
Downing M, Manning NJ, Dalton RN, Krywawych S, Oerton J: Detection of urinary hexanoylglycine in the diagnosis of MCAD deficiency from newborn screening. J Inherit Metab Dis. 2008, 31-550.
O'Reilly L, Bross P, Corydon TJ, Olpin SE, Hansen J, Kenney JM, McCandless SE, Frazier DM, Winter V, Gregersen N, Engel PC, Andresen BS: The Y42 H mutation in medium-chain acyl-CoA dehydrogenase, which is prevalent in babies identified by MS/MS-based newborn screening, is temperature sensitive. Eur J Biochem. 2004, 271: 4053-4063. 10.1111/j.1432-1033.2004.04343.x.
Andresen BS, Bross P, Jensen TG, Winter V, Knudsen I, Kølvraa S, Jensen UB, Bolund L, Duran M, Kim JJ, Curtis D, Divry P, Vianey-Saban C, Gregersen N: A rare disease-associated mutation in the medium-chain acyl-CoA dehydrogenase (MCAD) gene changes a conserved arginine, previously shown to be functionally essential in short-chain acyl-CoA dehydrogenase (SCAD). Am J Hum Genet. 1993, 53: 730-739.
Albers S, Levy HL, Irons M, Strauss AW, Marsden D: Compound heterozygosity in four asymptomatic siblings with medium-chain acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis. 2001, 24: 417-418. 10.1023/A:1010533408635.
Acknowledgements
We thank the patient, their parents and laboratory volunteer controls for participating in this study. The authors gratefully acknowledge J.C. Vienne, I. Kumorek, F. Hottevart, A. Kerkove, Dr. M. Kouach and Pr. N. Porchet for their help in either technical assistance or valuable comments. This work was supported by grants from the French Ministère de la Santé (PHRC 2003R/1903) and FMO (Fédération des Maladies Orphelines).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
AFD and MF contributed equally to this work. AFD, MF, AMP, GB and JV designed and performed measurements of de novo acylcarnitine syntheses by whole blood samples. BSA and NG contributed to genetic studies. MB, CVS and RJAW performed the biochemical and enzyme measurements. SNG, DD and KMM contributed to the clinical follow-up of studied patients. DR determined organic acids in body fluids and DSM performed acylcarnitine analysis on dried blood spots. Each author read the text and JV coordinated the writing of the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Dessein, AF., Fontaine, M., Andresen, B.S. et al. A novel mutation of the ACADM gene (c.145C>G) associated with the common c.985A>G mutation on the other ACADM allele causes mild MCAD deficiency: a case report. Orphanet J Rare Dis 5, 26 (2010). https://doi.org/10.1186/1750-1172-5-26
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1750-1172-5-26