
Abstract
Proteasomes are multicatalytic protease complexes in the cell, involved in the non-lysosomal recycling of intra-cellular proteins. Proteasomes play a critical role in regulation of cell division in both normal as well as cancer cells. In cancer cells this homeostatic function is deregulated leading to the hyperactivation of the proteasomes. Proteasome inhibitors (PIs) are a class of compounds, which either reversibly or irreversibly block the activity of proteasomes and induce cancer cell death. Interference of PIs with the ubiquitin proteasome pathway (UPP) involved in protein turnover in the cell leads to the accumulation of proteins engaged in cell cycle progression, which ultimately put a halt to cancer cell division and induce apoptosis. Upregulation of many tumor suppressor proteins involved in cell cycle arrest are known to play a role in PI induced cell cycle arrest in a variety of cancer cells. Although many PIs target the proteasomes, not all of them are effective in cancer therapy. Some cancers develop resistance against proteasome inhibition by possibly activating compensatory signaling pathways. However, the details of the activation of these pathways and their contribution to resistance to PI therapy remain obscure. Delineation of these pathways may help in checking resistance against PIs and deducing effective combinational approaches for improved treatment strategies. This review will discuss some of the signaling pathways related to proteasome inhibition and cell division that may help explain the basis of resistance of some cancers to proteasome inhibitors and underline the need for usage of PIs in combination with traditional chemotherapy.
Similar content being viewed by others


Introduction
Cancer cells differ from the normal cells of the body in their ability to divide indefinitely and evade programmed cell death [1]. In normal cells, the cell cycle is controlled by a complex series of signaling pathways by which a cell grows, replicates its DNA and divides. This process also includes mechanisms which ensure that any errors therein are corrected, and if not, the cells commit suicide in a systematic cellular process known as programmed cell death or apoptosis. In cancer, as a result of genetic mutations, this regulatory process malfunctions, resulting in uncontrolled cell proliferation. Alteration in these mechanisms commonly affect the expression of cell cycle regulatory proteins, causing overexpression of cyclins and loss of expression of cyclin dependent kinase inhibitors (CKIs). Therefore CDK inhibitors and related transcription factors have long been viewed as potential targets for anticancer therapeutics. Despite years of research and attempts directed at inhibiting cell cycle kinases or cell cycle regulating transcription factors, most of these approaches have not been successfully translated to the clinic as cancer therapeutics [2]. Therefore, targeting the pathways which bring about timely degradation of these proteins i.e. Proteasome pathway, provided the rationale for targeted anticancer drug development.
Cells normally depend upon two important pathways for degradation of cellular proteins, i.e. 1. the aggresome/lysosome pathway for extracellular proteins and 2. the ubiquitin proteasome pathway ( UPP) for intracellular proteins [3]. Proteasomes, key complexes of the latter pathway, are multicatalytic protease complexes engaged in non-lysosomal recycling of intra-cellular proteins of short life span. These small protein degradation machines are present ubiquitously in both cytoplasm and nucleus of mammalian cells. These are constituted of a ATP-dependent 26S core complex consisting of 20S catalytic core capped by a 19S regulatory subunit at both ends [3]. They guide the proteolytic cleavage and recycling of proteins whose functions are needed to be checked on a timely and stage specific manner [4–7]. Target proteins of the UPP consist of a polyubiquitin chain covalently attached to their lysine residues. This ubiquitin tag is recognized by the 19S regulatory subunit of the proteasome and the tagged protein is degraded in a ATP dependent manner [8]. Apart, from their presence in healthy cells they are found to be highly expressed and active in cancer cells. In cancer cells, proteasomes are engaged in proteolysis of many tumor suppressor proteins related to cell division. Owing to their established role in cancer progression, several compounds with proteasome inhibition activity has been tested and recognized as potential anti-cancer drugs against hematological malignancies and some solid cancers [9–12]. Based on their mode of action, compounds having proteasome inhibitory activity have either reversible or irreversible mechanisms of action [13]. Proteasome inhibitors can be of two types depending upon the source of origin i.e. synthetic and natural. Bortezomib ( Valcade, PS-341), a dipeptide boronic acid, first of its class of compounds, has been well studied for its proteasome inhibitory activity [14].Bortezomib was discovered to be a synthetic proteasome inhibitor with reversible mechanism of action [15]. Long tested for its anti cancer properties in multiple myeloma, it was approved by FDA for treatment of patients with multiple myeloma [16, 17]. Bortezomib has also been shown to exert anti-cancer activity against many cancer types [18, 19]. More recently compounds with irreversible mode of action are considered to be the second generation of proteasome inhibitors and have shown promising results in clinical trials namely Carfilzomib (PR-171), Salinosporamide A (NP-0052) and MLN9708 [20, 21]. Moreover many such compounds are also being tested currently in pre-clinical studies on multiple cancer cell lines [9, 22–26] (Table 1). Inhibition of proteasomal activity targets the cancer cell in a multipronged manner including, inhibition of proliferation and induction of cell cycle arrest, induction autophagy and apoptosis. As uncontrolled cell division is the root cause of any cell acquiring malignancy, cell cycle and cell division are popular targets for limiting cancer cell proliferation. Most of the PIs studied till date arrest cancer cells in different phases of cell cycle thereby deregulating cancer cell division (Table 2). Some cancer cells often develop resistance to proteasome inhibition thereby limiting their use in cancer therapy. However the details of the related signaling pathways and molecular mechanisms activated in response to proteasome inhibition require detailed investigation. This review will briefly discuss some of these signaling pathways / mechanisms regulated by PIs and their possible role in influencing resistance to proteasome inhibition therapy.
Proteasome inhibitors and cancer cell division
Proteasomes are critical for maintenance of cancer cell division
Cell division in cells is the interplay of several cell cycle regulatory proteins with restricted life span. Major proteins related to cell cycle process are cyclins, cyclin dependent kinases (CDKs), CDK inhibitors (CKIs) and some transcription factors. Among these cyclins/CDK complexes remains to be the central players in ensuring rapid cell division by continuously reshuffling their core partners for subsequent cell cycle phase transitions. Each complex type is assigned a role in a particular cell cycle phase and every time the replaced partner is degraded to mark the onset of the next phase. Cyclin proteins are found to be highly upregulated in cases of aberrant cell division in cancer cells. Particularly cyclins D and E are frequently upregulated in different cancer types [27, 28]. This upregulation of cyclins is further supported by the down regulation of another class of CDK regulatory proteins, the CKIs, which bind, inactivate and degrade the cyclin/CDK complex. Rapid proteasomal degradation of CKIs in subsequent cell cycle phases contribute to the uncontrolled cell division in cancer cells. Recent studies have established the involvement of ubiquitin proteasome pathway (UPP) in degradation and recycling of the above mentioned class of proteins, crucial for cancer onset and progression [29]. The proteasomal degradation of proteins is well complemented by the ubiquitin conjugating enzymes E1, E2 and E3 ligases, which add multiple ubiquitin molecules to the proteasome substrate proteins. Mdm2, Skp 1/Cul 1/F-box protein (SCF), Anaphase promoting complex (APC), Ubiquitin-conjugating enzyme E2C (UBE2C) are among the busiest ligases involved in cell cycle regulation. Most of these enzymes target the proteolysis of tumor suppressor proteins like p53, p21 and p27. The expression and activity of some of these enzymes has also been reported to be altered by PIs in many cancer cells [29]. Proteasomes are also involved in the regulation of cell division in response to DNA damage response (DDR) signals mediating F-box protein triggered degradation of cyclin D1 protein and G1 cell cycle arrest [30].
PIs disrupt cancer cell division
Proteasome inhibitors inhibit the activity of the proteasome complex and in turn attenuate its protein degradation activity. Therefore, PIs lead to the accumulation of those proteins which are marked for ubiquitin-proteasome mediated degradation in cancer cells. These proteins includes CKIs, various transcription factors, DNA repair enzymes, kinases and cyclin dependent kinase inhibitors (CKIs). Generally, the prime function of CKIs lies in degrading cyclin/CDK complexes and disrupting cell division. CKIs, p21 and p27 are known to be suppressed in several cancer types and are attributed to cancer progression [31, 32]. Bortezomib, along with other PIs has been reported to significantly increase the expression of p21 and p27 proteins in many cancers thereby causing cell cycle arrest [33–35]. On the other side proteasome inhibition also cause accumulation of the tumor suppressor p53 which is a crucial component of cell cycle regulation. PIs abrogate the degradation of p53 in cancer cell reactivating its function in G1/S and G2/M arrest [29]. Another key component of PI mediated growth arrest and apoptosis is inhibition of NFkB signaling and its downstream target proteins mainly cyclin D, responsible for G1/S transition and commitment to DNA synthesis [36]. Accumulation of non degraded proteins increase unfolded protein response (UPR) leading to induction of endoplasmic reticulum stress which again causes cell cycle arrest to release cells from this cellular stress. PIs induced ROS generation in many cancers is responsible for DNA damage mediated cell cycle arrest wherein cells rigorously perform DNA repair mechanisms [36]. p53 along with some other kinases either help in this process through DNA repair enzymes or alternatively induce apoptosis in case of serious genotoxic stress [37]. PIs have also been reported to modulate the activity of MAPK pathways of cell proliferation [36].Relationship between these signaling pathways and proteasome inhibitors in cancer cells are discussed in the subsequent portions of this review.
PI induced signaling pathways
NFkB signaling pathway: a favorite choice
NFkB is one of the major transcription factors critical for inflammatory cytokine expression in many cancer cells. NFkB signaling in cancer cells is controlled by the ubiquitin proteasome pathway which directs the ubiquitination and proteolysis of its inhibitory partner, IĸB. Once freed from its inhibitor, NFkB gets localized in the nucleus where it allows transcription of many genes driving cancer cell proliferation [38]. NFkB pathway is constitutively activated in cancers and play a crucial role in conferring resistance to radiation and chemotherapeutic agents in cancers [39, 40].However, among many proteins activated by NFkB, Cyclin D plays a crucial role in carcinogenesis. Cyclin D binds to CDK 4/6 in the initial phase of G1 to S phase transition where it performs two major functions, hypophosphorylation of retinoblastoma protein (Rb) [41] and protection of cyclin E/ CDK2 complex from inhibitory effect of p21 and p27 [27] (Figure 1).

Regulation of NFkB, E2F/Rb and p53/p21 pathways of cell cycle arrest by PIs. Proteasome inhibitors block the degradation of IĸB, an inhibitor of NFĸB via proteasome. This causes the accumulation of NF-ĸB in cytosol and represses its transcriptional activity which further inhibits CyclinD1 expression. Decreased Cyclin D1 level terminates its binding to CDK4, which in turn fails to hypo-phosphorylate the Rb protein. Once hypo-phosphorylation of Rb protein is ceased its hyper-phosphorylation by Cyclin E/ CDK2 complex is abrogated and its dissociation from E2F and proteasomal degradation is also inhibited. Undissociated E2F fails to express CyclinA genes and genes involved in DNA synthesis and S phase progression. On the other hand, proteasome inhibitors also inhibit mdm2 mediated degradation of p53 protein leading to the reactivation of its transcriptional activity. p53 reactivation increases p21 expression, a CKI, which inhibit cyclin E/ CDK2 complex ceasing G1 phase progression. However p21 also is known to inhibit cyclin B/ CDK1 complex causing G2/M arrest. Proteins like APC and cdc25 family members are also key players in the sG2/M phase of cell cycle but their modulation by PIs still remains unclear.
MAPK pathway: modulating cellular division
Mitogen activated protein kinases (MAPKs) family proteins are involved in many processes of tumor development and progression [39]. MAPK are associated with cell survival and apoptosis in stressful cellular environments [39]. Among the family members, ERK1/2 (p44/42) is associated with cell survival and tumor progression where as the other two members, p38 MAPK and c-Jun N-Terminal kinases (JNK) are linked to either cell survival or apoptosis [39]. Though not much has been explored regarding how PIs modulate these pathways but there are evidences which clearly indicated crosstalk between them [39]. According to a previous report addition of PIs decreased the level of phosphorylated ERK1/2 in breast cancer cell lines through activation of MKP-1, a phosphatase, generally degraded by the proteasome pathway [39]. Ras / MEK/ERK pathway regulates the expression of Cyclin D1 by modulating AP-1 and ETS transcription factor, thereby playing a critical role in G1 progression [42–44]. Bortezomib has also been documented to abrogate the activation of p44/42 (ERK1/2) protein along with PI3K/Akt/mTOR and inhibition of the nuclear export of HIF-1α, however its relation to cell cycle arrest remains elusive [45]. Inactivation of ERK by PIs may lead to cell cycle arrest in cancer cells. Secondly, p38 MAP kinases signaling also modulate mammalian cell cycle by antagonizing the effects of ERK mediated Cyclin D expression and thus regulating the G1/ S cell cycle arrest. MG-132 treatment significantly increased the phosphorylation of p38 MAPK in Calu-6 cells and its inhibition partially inhibited MG-132 induced cell death [46]. Lastly, c-Jun-N-terminal Kinase, the third component of the MAPK pathway, has been reported to be activated in MG-132 induced apoptosis [47]. Bortezomib treatment activated JNK in glioblastma multiforme (GBM) cells causing G2/M arrest with subsequent upregulation of p21 and p27 proteins and simultaneous decrease in cyclin B, CDK2, CDK4 and E2F4 protein levels [48]. More recently Bortezomib has also been reported to activate JNK in head and neck cancer cells [49]. JNK1 binds and phosphorylates E2F and limit its ability to bind to DNA, this may induce G1 arrest subsequent to CDK mediated phosphorylation of pRb [50]. On the other hand its activation leads to cell proliferation by increasing the expression of cyclin D1 and repression of p53 and p21 proteins [51, 52]. However correlation of JNK to the cell cycle arrest is still a matter of investigation. Involvement of MAPK pathways in cell cycle and PI induced cell death pave the way for identification of many of the novel targets which are supposed to provide a mechanistic link between these two processes (Figure 2).

MAPK pathways and Proteasome inhibitors: Proteasome inhibitors modulate the members of MAPK pathway and control cell cycle of cancer cells. PIs inhibit the activation of ERK 1/2 protein, which is found to increase the expression of cyclin D1. PIs activate p38 MAPK which antagonizes the effect of ERK 1/2 and decrease cyclin D1. PIs also activate JNK which is known to inhibit the E2F activity.
p53/21 pathway: blocking the doorstep of cell cycle entry
Transcription factor p53 lies in very close association with both proteasome and cell cycle pathways [53]. It has been the most extensively studied protein of its class in relation to PIs. In cancer cells, p53 protein is marked for proteasomal degradation by mdm2 protein, a E3 type of ubiquitin ligase [53–55]. Mdm2 ligase is found to be highly expressed in cancers, though the underlying mechanism is still unclear, possibly to keep p53 protein away from tumor formation processes [56]. Proteasomal inhibition allows accumulation of p53 and its nuclear export in cancer cells and thereby increase the expression of its transcriptional target gene p21. p21, a potent CDK inhibitor binds and inactivate cyclin E and the CDK2 complex. This complex is essential for late G1 phase of cell cycle mediating entry in the S phase of cell cycle (Figure 1). However activation of p53 and p21 proteins leads to G0/G1 cell cycle arrest but probably there are several other mediators of this pathway regulating PI induced G0/G1 arrest.
Rb/E2F pathway: regulating DNA synthesis
Rapid proteasomal degradation of the retinoblastoma protein is evident in several cancers. Many oncoproteins are engaged in triggering ubiquitin proteasome degradation of Rb including viral oncoprotein E7 of human pappiloma virus type 16, Epstein-Barr virus nuclear antigen 3C (EBNA3C) and gankyrin [57–62]. Rb act as a tumor suppressor protein playing crucial role in cell cycle regulation, DNA replication, DNA damage repair and many other cellular processes. The preliminary role of Rb in cell cycle regulation is binding and stabilization of E2F family of proteins causing their transcriptional repression. Transcriptionally repressed E2F becomes ineffective in activating genes required for DNA synthesis and S phase progression [63]. Administration of PIs mediates Rb protein escape from proteasomal degradation. Non-degraded Rb still remains bounded to E2F which indirectly inhibits E2F mediated expression of S phase progression genes thereby arresting cancer cells in G0/G1 or S phase (Figure 1). According to a recent study , Bortezomib induced cell cycle arrest and apoptosis in BCR/Abl expressing, imatinib resistant and sensitive CML cells. The molecular mechanism involved inhibition of both NFĸB and phosphorylation of Rb eventually leading to caspase dependent apoptosis [64]. However, more elaborate mechanism of cell cycle regulation by Rb/E2F pathway is still not understood with respect to the proteasome inhibitors.
DNA damage check point pathway: enforcing G2/M phase arrest
Generally DNA damage checkpoint is imposed by formation of double stranded breaks to the intact DNA by any DNA damaging agent or generation of intracellular ROS [65–68]. DNA damage and break in DNA replication process most often cause G2/M or S phase cell cycle arrest [69–72]. Several reports have established that PIs induced G2/M arrest in cancer cells by activation of p53 and p21 proteins [73, 74] (Figure 3). A recent study has shown that PSI-341 treatment lead to p53 dependent G2/M arrest upon co-treatment with DNA damaging agent SN-38 [39]. PSI-341 also reduced the expression of two p53 regulated proteins 14-3-3sigma and survivin both of which regulate G2/M progression and apoptosis [75]. p53 dependent G2/M arrest after generation of ROS has also been reported in isothiocyanate treated multiple myeloma cells [34]. MG-132 has been reported to cause generation of ROS and depletion of the antioxidant GSH in Calu-6 lung cancer cells and cervical cancer leading to S and G2/M phase arrest respectively [46, 76]. Other signaling proteins viz, ATM, ATR, Chk1, Chk2 kinases and cdc25 family of phosphatases also lie close to the pathway DDR and promote cell cycle arrest both in p53 dependent and independent manner. Recycling of these proteins is performed by the UPP [77, 78]. Though their role in PI induced cell cycle arrest is a matter of further research.

Proteasome inhibitor induced ROS generation and cell cycle arrest. Proteasome inhibitors increase the endogenous level of reactive oxygen species which causes DNA damage by inducing double strand breaks in intact DNA. PIs treatment leads to accumulation of p53 which senses this DNA damage. p53 reactivation upregulates p21 expression, a CDK inhibitor. p21 binds and inactivates Cyclin B and CDK2 and causes G2/M cell cycle arrest in response to PIs.
Endoplasmic reticulum stress pathway inducing autophagy mediating cell death
PIs increase the load of misfolded and ubiquitinated proteins in cancer cells which elicit endoplasmic reticulum stress. ER stress induced non-functional protein overload leads to formation of autophagosome to digest these intracellular proteins by a process known as macroautophagy [49]. Macroautophgy induction is thought to overcome the protein load and restore normal cell division [79, 80]. Autophagy is the process of degradation of intracellular proteins next to ubiquitin proteasome pathway and possess dual role in PI induced cancer cell death or survival. A recent study demonstrated the involvement of eIF2α-dependent pathway of autophagy induction in prostate cancer cells upon PI treatment. This autophagy induction induced a cytoprotective effect in prostate cancer cells which was alleviated when cells were co-treated with autophagy inhibitors [81]. The exact mechanism of autophagy induction followed by PI treatment still remains obscure and needs detailed investigation. Bortezomib treatment induces autophagy in MCF-7 cells as a compensatory mechanism to escape cell death which can be restored upon co-treatment with 3-MA, an autophagy inhibitor [82]. MG-132 induced G2/M arrest and apoptosis was also enhanced upon abolition of autophagy in gastric cancer cells [83]. Similarly, celastrol, a natural PI, resulted in the induction of autophagy and G2/M arrest in HeLa cells. Interestingly it also caused paraptosis, formation of cytoplasmic vacuoles and apoptosis in multiple cancer cell types [84]. On the contrary, MG-132 induced cell death was partially attenuated on treatment with autophagy inhibitor, 3-MA, which ensures the importance of autophagy in PI, triggered cell death. However, induction of autophagy is supposed to be primarily a defense mechanism to bypass the incidence of cell death after PI treatment, still it remains to be a crucial event in PI induced growth arrest of cancer cells (Figure 4). These studies clearly indicate that the intricate mechanisms of association of ER stress, autophagy and resistance to PI therapy needs further investigation.

Association of ER stress pathway and autophagy in Proteasome inhibitor mediated cell cycle arrest. Proteasome inhibitor treatment arrests the normal functioning of the proteasome and increases the load of ubiquitinated and misfolded proteins in cancer cells. This accumulation generates unfolded protein response (UPR) and consequently ER stress. ER stress generation enforces cells to be arrested in cell cycle phases and induce autophagy in order to compensate and reduce the non-functional protein load. At this stage autophagy can resolve the normal cell state by digesting the non-functional proteins and promote cancer cell survival or can trigger apoptosis leading to cancer cell death. Autophagy inhibitors 3-MA and Bafilomycin A can modulate the effect of autophagy induced cell cycle arrest or apoptosis in association with PIs.
Conclusions and future perspectives
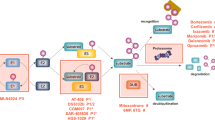
Development of chemoresistance against PIs and its inefficacy against many solid cancers necessitates a thorough assessment of the treatment strategies involving PIs. Though not much has been explored in this context but certain reports suggest that induction of autophagy post PI treatment is a major cause of resistance to PI therapy [85–87]. Therefore exploring synergistic drug combinations with PIs for effectively combating chemoresistance and limitations of targeted therapy could be a logical therapeutic strategy. As discussed above proteasome inhibitor induced cell cycle arrest is attributed to modulation of one or more pathways. Therefore, the combination of PIs along with the modulators of one of these signaling pathways, may possibly be explored for their therapeutic potential in PI resistant cancer types. Already some reports have indicated the potential of PIs in combination with different class of compounds (Figure 5). Combination of Bortezomib and autophagy inhibitor, Bafilomycin A1 increases the cytotoxic effects against multiple myeloma cells. Similarly, combination of PIs and ER stress inhibitor, salubrinal potentiates toxicity in therapy resistant multiple myeloma cells [88, 89]. Recent studies also suggest the synergy between HDAC inhibitors (HDACi) and PIs against multiple myeloma cell lines and have shown promising results in clinical trials [89]. PIs in combination with HDAC inhibitors also induce cell cycle arrest via NFkB and ROS pathways [90]. These findings provide new insights for combination therapy of HDAC inhibitors and PIs [91, 92]. Bortezomib has also been tested in combination with Nelfinavir, an HIV protease inhibitor and the results have indicated induction of ER stress, cell cycle arrest in cervical cancer cells [93]. Several other combinations with PIs worked out on different cancer cells given encouraging results for combination therapy [94–100] is summarized in Additional file 1: Table S1. However, there is an ever increasing need for such synergistic combinations with drugs from the synthetic and natural source for effectively targeting cancer cells with PI resistance.

Refinement in PIs therapy via synergistic approaches. PIs can be used in combination with different class of compounds for effective targeting of PI resistant cancer cells and overcoming chemoresistance. PIs can be combined with ER-stress modulators which can together cause cell cycle arrest via unfolded protein response and sometimes autophagy. Autophagy inhibitors have also shown to cause cell cycle arrest in combination with PIs thereby surpassing chemoresistance induced by PIs in alone. One more class of compounds, HDAC inhibitors have also shown promising results in combination with PIs. This combination is reported to decrease NFkB activation and induces ROS leading to cell cycle arrest in cancer cells.
Authors’ information
The research group headed by DPM is dedicated to study of molecular mechanisms of cell death in cancer, neuronal and male germ cells. We are currently screening for proteasome inhibitors using commercially available drug libraries and natural compound libraries synthesized at the CSIR- Central Drug Research Institute, Lucknow, India.
Abbreviations
- PI:
-
Proteasome inhibitor
- CDK:
-
Cyclin dependent kinase
- CKI:
-
Cyclin dependent kinase inhibitor
- UPP:
-
Ubiquitin proteasome pathway
- UPR:
-
Unfolded protein response
- ER stress:
-
Endoplasmic reticulum stress
- 3-MA:
-
3-Methyladenine
- NF-ĸB:
-
Nuclear factor kappa B
- SCF:
-
Skp 1/Cul 1/F-box protein
- APC:
-
Anaphase promoting complex
- UBE2C:
-
Ubiquitin-conjugating enzyme E2C
- ROS:
-
Reactive oxygen species.
- JNK:
-
c-jun-N-terminal kinase
- MAPK:
-
Mitogen activated protein kinase
- DDR:
-
DNA damage response
- HDACi:
-
Histone deacetylase inhibitors.
References
Hanahan D, Weinberg RA: Hallmarks of cancer: the next generation. Cell 144: 646–74.
Pentimalli F, Giordano A: Promises and drawbacks of targeting cell cycle kinases in cancer. Discov Med 2009, 8: 177–80.
Adams J: The proteasome: a suitable antineoplastic target. Nat Rev Cancer 2004, 4: 349–60. 10.1038/nrc1361
Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, Elliott PJ: Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res 1999, 59: 2615–22.
Rivett AJ: Proteasomes: multicatalytic proteinase complexes. Biochem J 1993,291(Pt 1):1–10.
Goldberg AL: Functions of the proteasome: the lysis at the end of the tunnel. Science 1995, 268: 522–3. 10.1126/science.7725095
Hershko A: Roles of ubiquitin-mediated proteolysis in cell cycle control. Curr Opin Cell Biol 1997, 9: 788–99. 10.1016/S0955-0674(97)80079-8
Orlowski RZ, Stinchcombe TE, Mitchell BS, Shea TC, Baldwin AS, Stahl S, Adams J, Esseltine DL, Elliott PJ, Pien CS, Guerciolini R, Anderson JK, Depcik-Smith ND, Bhagat R, Lehman MJ, Novick SC, O'Connor OA, Soignet SL: Phase I trial of the proteasome inhibitor PS-341 in patients with refractory hematologic malignancies. J Clin Oncol 2002, 20: 4420–7. 10.1200/JCO.2002.01.133
Nam S, Smith DM, Dou QP: Ester bond-containing tea polyphenols potently inhibit proteasome activity in vitro and in vivo. J Biol Chem 2001, 276: 13322–30. 10.1074/jbc.M004209200
Wu WK, Cho CH, Lee CW, Wu K, Fan D, Yu J, Sung JJ: Proteasome inhibition: a new therapeutic strategy to cancer treatment. Cancer Lett 2010, 293: 15–22. 10.1016/j.canlet.2009.12.002
Mitsiades CS, McMillin D, Kotoula V, Poulaki V, McMullan C, Negri J, Fanourakis G, Tseleni-Balafouta S, Ain KB, Mitsiades N: Antitumor effects of the proteasome inhibitor bortezomib in medullary and anaplastic thyroid carcinoma cells in vitro. J Clin Endocrinol Metab 2006, 91: 4013–21. 10.1210/jc.2005-2472
Frezza M, Schmitt S, Dou QP: Targeting the ubiquitin-proteasome pathway: an emerging concept in cancer therapy. Curr Top Med Chem 2011, 11: 2888–905. 10.2174/156802611798281311
Bogyo M, Wang EW: Proteasome inhibitors: complex tools for a complex enzyme. Curr Top Microbiol Immunol 2002, 268: 185–208. 10.1007/978-3-642-59414-4_8
Adams J, Behnke M, Chen S, Cruickshank AA, Dick LR, Grenier L, Klunder JM, Ma YT, Plamondon L, Stein RL: Potent and selective inhibitors of the proteasome: dipeptidyl boronic acids. Bioorg Med Chem Lett 1998, 8: 333–8. 10.1016/S0960-894X(98)00029-8
LeBlanc R, Catley LP, Hideshima T, Lentzsch S, Mitsiades CS, Mitsiades N, Neuberg D, Goloubeva O, Pien CS, Adams J, Gupta D, Richardson PG, Munshi NC, Anderson KC: Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res 2002, 62: 4996–5000.
Kane RC, Bross PF, Farrell AT, Pazdur R: Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003, 8: 508–13. 10.1634/theoncologist.8-6-508
Kane RC, Farrell AT, Sridhara R, Pazdur R: United States Food and Drug Administration approval summary: bortezomib for the treatment of progressive multiple myeloma after one prior therapy. Clin Cancer Res 2006, 12: 2955–60. 10.1158/1078-0432.CCR-06-0170
Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Dunner K Jr, Huang P, Abbruzzese JL, McConkey DJ: Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res 2005, 65: 11658–66. 10.1158/0008-5472.CAN-05-2370
Wunderlich A, Arndt T, Fischer M, Roth S, Ramaswamy A, Greene BH, Brendel C, Hinterseher U, Bartsch DK, Hoffmann S: Targeting the proteasome as a promising therapeutic strategy in thyroid cancer. J Surg Oncol 2012, 105: 357–64. 10.1002/jso.22113
Chari A, Mazumder A, Jagannath S: Proteasome inhibition and its therapeutic potential in multiple myeloma. Biologics 2010, 4: 273–87.
Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN, Jiang J, Laidig GJ, Lewis ER, Parlati F, Shenk KD, Smyth MS, Sun CM, Vallone MK, Woo TM, Molineaux CJ, Bennett MK: Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res 2007, 67: 6383–91. 10.1158/0008-5472.CAN-06-4086
Piva R, Ruggeri B, Williams M, Costa G, Tamagno I, Ferrero D, Giai V, Coscia M, Peola S, Massaia M, Pezzoni G, Allievi C, Pescalli N, Cassin M, di Giovine S, Nicoli P, de Feudis P, Strepponi I, Roato I, Ferracini R, Bussolati B, Camussi G, Jones-Bolin S, Hunter K, Zhao H, Neri A, Palumbo A, Berkers C, Ovaa H, Bernareggi A, Inghirami G: CEP-18770: a novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib. Blood 2008, 111: 2765–75. 10.1182/blood-2007-07-100651
Yang H, Zhou P, Huang H, Chen D, Ma N, Cui QC, Shen S, Dong W, Zhang X, Lian W, Wang X, Dou QP, Liu J: Shikonin exerts antitumor activity via proteasome inhibition and cell death induction in vitro and in vivo. Int J Cancer 2009, 124: 2450–9. 10.1002/ijc.24195
Milacic V, Banerjee S, Landis-Piwowar KR, Sarkar FH, Majumdar AP, Dou QP: Curcumin inhibits the proteasome activity in human colon cancer cells in vitro and in vivo. Cancer Res 2008, 68: 7283–92. 10.1158/0008-5472.CAN-07-6246
Yang H, Landis-Piwowar KR, Chen D, Milacic V, Dou QP: Natural compounds with proteasome inhibitory activity for cancer prevention and treatment. Curr Protein Pept Sci 2008, 9: 227–39. 10.2174/138920308784533998
Dai Y, Desano J, Tang W, Meng X, Meng Y, Burstein E, Lawrence TS, Xu L: Natural proteasome inhibitor celastrol suppresses androgen-independent prostate cancer progression by modulating apoptotic proteins and NF-kappaB. PLoS One 2010, 5: e14153. 10.1371/journal.pone.0014153
Diehl JA, Ponugoti B: Ubiquitin-dependent proteolysis in G1/S phase control and its relationship with tumor susceptibility. Genes Cancer 2010, 1: 717–724. 10.1177/1947601910382902
Masamha CP, Benbrook DM: Cyclin D1 degradation is sufficient to induce G1 cell cycle arrest despite constitutive expression of cyclin E2 in ovarian cancer cells. Cancer Res 2009, 69: 6565–72. 10.1158/0008-5472.CAN-09-0913
Bavi P, Uddin S, Ahmed M, Jehan Z, Bu R, Abubaker J, Sultana M, Al-Sanea N, Abduljabbar A, Ashari LH, Alhomoud S, Al-Dayel F, Prabhakaran S, Hussain AR, Al-Kuraya KS: Bortezomib stabilizes mitotic cyclins and prevents cell cycle progression via inhibition of UBE2C in colorectal carcinoma. Am J Pathol 2011, 178: 2109–20. 10.1016/j.ajpath.2011.01.034
Santra MK, Wajapeyee N, Green MR: F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature 2009, 459: 722–5. 10.1038/nature08011
Abbas T, Dutta A: p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 2009, 9: 400–14. 10.1038/nrc2657
Chu IM, Hengst L, Slingerland JM: The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer 2008, 8: 253–67. 10.1038/nrc2347
Sterz J, Jakob C, Kuckelkorn U, Heider U, Mieth M, Kleeberg L, Kaiser M, Kloetzel PM, Sezer O, von Metzler I: BSc2118 is a novel proteasome inhibitor with activity against multiple myeloma. Eur J Haematol 2010, 85: 99–107.
Mi L, Gan N, Chung FL: Isothiocyanates inhibit proteasome activity and proliferation of multiple myeloma cells. Carcinogenesis 2011, 32: 216–23. 10.1093/carcin/bgq242
Huang H, Liu N, Zhao K, Zhu C, Lu X, Li S, Lian W, Zhou P, Dong X, Zhao C, Guo H, Zhang C, Yang C, Wen G, Lu L, Li X, Guan L, Liu C, Wang X, Dou QP, Liu J: Sanggenon C decreases tumor cell viability associated with proteasome inhibition. Front Biosci (Elite Ed) 2011, 3: 1315–25.
Joyce D, Albanese C, Steer J, Fu M, Bouzahzah B, Pestell RG: NF-kappaB and cell-cycle regulation: the cyclin connection. Cytokine Growth Factor Rev 2001, 12: 73–90. 10.1016/S1359-6101(00)00018-6
Lane DP: Cancer. p53, guardian of the genome. Nature 1992, 358: 15–6. 10.1038/358015a0
Palombella VJ, Rando OJ, Goldberg AL, Maniatis T: The ubiquitin-proteasome pathway is required for processing the NFkB1 precursor protein and the activation of NFkB. Cell 1994, 78: 773–85. 10.1016/S0092-8674(94)90482-0
Orlowski RZ, Small GW, Shi YY: Evidence that inhibition of p44/42 mitogen-activated protein kinase signaling is a factor in proteasome inhibitor-mediated apoptosis. J Biol Chem 2002, 277: 27864–71. 10.1074/jbc.M201519200
Elliott PJ, Ross JS: The proteasome: a new target for novel drug therapies. Am J Clin Pathol 2001, 116: 637–46. 10.1309/44HW-5YCJ-FLLP-3R56
Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC: Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999, 98: 859–69. 10.1016/S0092-8674(00)81519-6
Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A, Pestell RG: Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem 1995, 270: 23589–97. 10.1074/jbc.270.40.23589
Lavoie JN, L'Allemain G, Brunet A, Muller R, Pouyssegur J: Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem 1996, 271: 20608–16. 10.1074/jbc.271.34.20608
Liu JJ, Chao JR, Jiang MC, Ng SY, Yen JJ, Yang-Yen HF: Ras transformation results in an elevated level of cyclin D1 and acceleration of G1 progression in NIH 3 T3 cells. Mol Cell Biol 1995, 15: 3654–63.
Befani CD, Vlachostergios PJ, Hatzidaki E, Patrikidou A, Bonanou S, Simos G, Papandreou CN, Liakos P: Bortezomib represses HIF-1alpha protein expression and nuclear accumulation by inhibiting both PI3K/Akt/TOR and MAPK pathways in prostate cancer cells. J Mol Med (Berl). 2012, 90: 45–54. 10.1007/s00109-011-0805-8
Han YH, Park WH: Treatment with p38 inhibitor partially prevents Calu-6 lung cancer cell death by a proteasome inhibitor, MG132. Cancer Genet Cytogenet 2010, 199: 81–8. 10.1016/j.cancergencyto.2010.02.004
Meriin AB, Gabai VL, Yaglom J, Shifrin VI, Sherman MY: Proteasome inhibitors activate stress kinases and induce Hsp72. Diverse effects on apoptosis, J Biol Chem. 1998, 273: 6373–9.
Yin D, Zhou H, Kumagai T, Liu G, Ong JM, Black KL, Koeffler HP: Proteasome inhibitor PS-341 causes cell growth arrest and apoptosis in human glioblastoma multiforme (GBM). Oncogene 2005, 24: 344–54. 10.1038/sj.onc.1208225
Li C, Johnson DE: Bortezomib induces autophagy in head and neck squamous cell carcinoma cells via JNK activation. Cancer Lett 2012, 314: 102–7. 10.1016/j.canlet.2011.09.020
Wang S, Nath N, Minden A, Chellappan S: Regulation of Rb and E2F by signal transduction cascades: divergent effects of JNK1 and p38 kinases. EMBO J 1999, 18: 1559–70. 10.1093/emboj/18.6.1559
Wisdom R, Johnson RS, Moore C: c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J 1999, 18: 188–97. 10.1093/emboj/18.1.188
Schreiber M, Kolbus A, Piu F, Szabowski A, Mohle-Steinlein U, Tian J, Karin M, Angel P, Wagner EF: Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev 1999, 13: 607–19. 10.1101/gad.13.5.607
Momand J, Zambetti GP, Olson DC, George D, Levine AJ: The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69: 1237–45. 10.1016/0092-8674(92)90644-R
Haupt Y, Maya R, Kazaz A, Oren M: Mdm2 promotes the rapid degradation of p53. Nature 1997, 387: 296–9. 10.1038/387296a0
Honda R, Tanaka H, Yasuda H: Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett 1997, 420: 25–7. 10.1016/S0014-5793(97)01480-4
Marine JC, Lozano G: Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ 2010, 17: 93–102. 10.1038/cdd.2009.68
Berezutskaya E, Bagchi S: The human papillomavirus E7 oncoprotein functionally interacts with the S4 subunit of the 26 S proteasome. J Biol Chem 1997, 272: 30135–40. 10.1074/jbc.272.48.30135
Berezutskaya E, Yu B, Morozov A, Raychaudhuri P, Bagchi S: Differential regulation of the pocket domains of the retinoblastoma family proteins by the HPV16 E7 oncoprotein. Cell Growth Differ 1997, 8: 1277–86.
Boyer SN, Wazer DE, Band V: E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res 1996, 56: 4620–4.
Wang J, Sampath A, Raychaudhuri P, Bagchi S: Both Rb and E7 are regulated by the ubiquitin proteasome pathway in HPV-containing cervical tumor cells. Oncogene 2001, 20: 4740–9. 10.1038/sj.onc.1204655
Knight JS, Sharma N, Robertson ES: Epstein-Barr virus latent antigen 3C can mediate the degradation of the retinoblastoma protein through an SCF cellular ubiquitin ligase. Proc Natl Acad Sci U S A 2005, 102: 18562–6. 10.1073/pnas.0503886102
Higashitsuji H, Itoh K, Nagao T, Dawson S, Nonoguchi K, Kido T, Mayer RJ, Arii S, Fujita J: Reduced stability of retinoblastoma protein by gankyrin, an oncogenic ankyrin-repeat protein overexpressed in hepatomas. Nat Med 2000, 6: 96–9. 10.1038/71600
Ying H, Xiao ZX: Targeting retinoblastoma protein for degradation by proteasomes. Cell Cycle 2006, 5: 506–8. 10.4161/cc.5.5.2515
Albero MP, Vaquer JM, Andreu EJ, Villanueva JJ, Franch L, Ivorra C, Poch E, Agirre X, Prosper F, Perez-Roger I: Bortezomib decreases Rb phosphorylation and induces caspase-dependent apoptosis in Imatinib-sensitive and -resistant Bcr-Abl1-expressing cells. Oncogene 2010, 29: 3276–86. 10.1038/onc.2010.81
Cadet J, Delatour T, Douki T, Gasparutto D, Pouget JP, Ravanat JL, Sauvaigo S: Hydroxyl radicals and DNA base damage. Mutat Res 1999, 424: 9–21. 10.1016/S0027-5107(99)00004-4
Beckman KB, Ames BN: Oxidative decay of DNA. J Biol Chem 1997, 272: 19633–6. 10.1074/jbc.272.32.19633
Pan J, She M, Xu ZX, Sun L, Yeung SC: Farnesyltransferase inhibitors induce DNA damage via reactive oxygen species in human cancer cells. Cancer Res 2005, 65: 3671–81. 10.1158/0008-5472.CAN-04-2744
Wiseman H, Halliwell B: Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J 1996,313(Pt 1):17–29.
Moon DO, Kim MO, Choi YH, Hyun JW, Chang WY, Kim GY: Butein induces G(2)/M phase arrest and apoptosis in human hepatoma cancer cells through ROS generation. Cancer Lett 2010, 288: 204–13. 10.1016/j.canlet.2009.07.002
Vilenchik MM, Knudson AG: Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc Natl Acad Sci U S A 2003, 100: 12871–6. 10.1073/pnas.2135498100
Petruccelli LA, Dupere-Richer D, Pettersson F, Retrouvey H, Skoulikas S, Miller WH Jr: Vorinostat induces reactive oxygen species and DNA damage in acute myeloid leukemia cells. PLoS One 2011, 6: e20987. 10.1371/journal.pone.0020987
He L, Nan MH, Oh HC, Kim YH, Jang JH, Erikson RL, Ahn JS, Kim BY: Asperlin induces G/M arrest through ROS generation and ATM pathway in human cervical carcinoma cells. Biochem Biophys Res Commun 2011, 409: 489–93. 10.1016/j.bbrc.2011.05.032
Wang Y, Rishi AK, Puliyappadamba VT, Sharma S, Yang H, Tarca A, Dou QP, Lonardo F, Ruckdeschel JC, Pass HI, Wali A: Targeted proteasome inhibition by Velcade induces apoptosis in human mesothelioma and breast cancer cell lines. Cancer Chemother Pharmacol 2011, 66: 455–66.
Russo A, Bronte G, Fulfaro F, Cicero G, Adamo V, Gebbia N, Rizzo S: Bortezomib: a new pro-apoptotic agent in cancer treatment. Curr Cancer Drug Targets 2010, 10: 55–67. 10.2174/156800910790980250
Vaziri SA, Hill J, Chikamori K, Grabowski DR, Takigawa N, Chawla-Sarkar M, Rybicki LR, Gudkov AV, Mekhail T, Bukowski RM, Ganapathi MK, Ganapathi R: Sensitization of DNA damage-induced apoptosis by the proteasome inhibitor PS-341 is p53 dependent and involves target proteins 14–3-3sigma and survivin. Mol Cancer Ther 2005, 4: 1880–90. 10.1158/1535-7163.MCT-05-0222
Han YH, Moon HJ, You BR, Park WH: The effect of MG132, a proteasome inhibitor on HeLa cells in relation to cell growth, reactive oxygen species and GSH. Oncol Rep 2009, 22: 215–21.
Eymin B, Claverie P, Salon C, Brambilla C, Brambilla E, Gazzeri S: p14ARF triggers G2 arrest through ERK-mediated Cdc25C phosphorylation, ubiquitination and proteasomal degradation. Cell Cycle 2006, 5: 759–65. 10.4161/cc.5.7.2625
Bassermann F, Pagano M: Dissecting the role of ubiquitylation in the DNA damage response checkpoint in G2. Cell Death Differ 2011, 17: 78–85.
Yang W, Monroe J, Zhang Y, George D, Bremer E, Li H: Proteasome inhibition induces both pro- and anti-cell death pathways in prostate cancer cells. Cancer Lett 2006, 243: 217–27. 10.1016/j.canlet.2005.11.033
Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM: Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 2007, 171: 513–24. 10.2353/ajpath.2007.070188
Zhu K, Dunner K Jr, McConkey DJ: Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 2010, 29: 451–62. 10.1038/onc.2009.343
Yao F, Wang G, Wei W, Tu Y, Tong H, Sun S: An autophagy inhibitor enhances the inhibition of cell proliferation. Mol Med Report. 2012, 5: 84–8.
Wu WK, Cho CH, Lee CW, Wu YC, Yu L, Li ZJ, Wong CC, Li HT, Zhang L, Ren SX, Che CT, Wu K, Fan D, Yu J, Sung JJ: Macroautophagy and ERK phosphorylation counteract the antiproliferative effect of proteasome inhibitor in gastric cancer cells. Autophagy 2010, 6: 228–38. 10.4161/auto.6.2.11042
Wang WB, Feng LX, Yue QX, Wu WY, Guan SH, Jiang BH, Yang M, Liu X, Guo DA: Paraptosis accompanied by autophagy and apoptosis was induced by celastrol, a natural compound with influence on proteasome, ER stress and Hsp90. J Cell Physiol 2012, 227: 2196–2206. 10.1002/jcp.22956
Rzymski T, Milani M, Singleton DC, Harris AL: Role of ATF4 in regulation of autophagy and resistance to drugs and hypoxia. Cell Cycle 2009, 8: 3838–47. 10.4161/cc.8.23.10086
Markovina S, Callander NS, O'Connor SL, Kim J, Werndli JE, Raschko M, Leith CP, Kahl BS, Kim K, Miyamoto S: Bortezomib-resistant nuclear factor-kappaB activity in multiple myeloma cells. Mol Cancer Res 2008, 6: 1356–64. 10.1158/1541-7786.MCR-08-0108
Oerlemans R, Franke NE, Assaraf YG, Cloos J, van Zantwijk I, Berkers CR, Scheffer GL, Debipersad K, Vojtekova K, Lemos C, van der Heijden JW, Ylstra B, Peters GJ, Kaspers GL, Dijkmans BA, Scheper RJ, Jansen G: Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112: 2489–99. 10.1182/blood-2007-08-104950
Kawaguchi T, Miyazawa K, Moriya S, Ohtomo T, Che XF, Naito M, Itoh M, Tomoda A: Combined treatment with bortezomib plus bafilomycin A1 enhances the cytocidal effect and induces endoplasmic reticulum stress in U266 myeloma cells: crosstalk among proteasome, autophagy-lysosome and ER stress. Int J Oncol 2011, 38: 643–54.
Schewe DM, Aguirre-Ghiso JA: Inhibition of eIF2alpha dephosphorylation maximizes bortezomib efficiency and eliminates quiescent multiple myeloma cells surviving proteasome inhibitor therapy. Cancer Res 2009, 69: 1545–52. 10.1158/0008-5472.CAN-08-3858
Hideshima T, Richardson PG, Anderson KC: Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol Cancer Ther 2011, 10: 2034–42. 10.1158/1535-7163.MCT-11-0433
Carew JS, Giles FJ, Nawrocki ST: Histone deacetylase inhibitors: mechanisms of cell death and promise in combination cancer therapy. Cancer Lett 2008, 269: 7–17. 10.1016/j.canlet.2008.03.037
Bhalla S, Balasubramanian S, David K, Sirisawad M, Buggy J, Mauro L, Prachand S, Miller R, Gordon LI, Evens AM: PCI-24781 induces caspase and reactive oxygen species-dependent apoptosis through NF-kappaB mechanisms and is synergistic with bortezomib in lymphoma cells. Clin Cancer Res 2009, 15: 3354–65. 10.1158/1078-0432.CCR-08-2365
Bruning A, Vogel M, Mylonas I, Friese K, Burges A: Bortezomib targets the caspase-like proteasome activity in cervical cancer cells, triggering apoptosis that can be enhanced by nelfinavir. Curr Cancer Drug Targets 2011, 11: 799–809. 10.2174/156800911796798913
Baradari V, Hopfner M, Huether A, Schuppan D, Scherubl H: Histone deacetylase inhibitor MS-275 alone or combined with bortezomib or sorafenib exhibits strong antiproliferative action in human cholangiocarcinoma cells. World J Gastroenterol 2007, 13: 4458–66.
Paoluzzi L, Scotto L, Marchi E, Zain J, Seshan VE, O'Connor OA: Romidepsin and belinostat synergize the antineoplastic effect of bortezomib in mantle cell lymphoma. Clin Cancer Res 2010, 16: 554–65. 10.1158/1078-0432.CCR-09-1937
Iwata S, Saito T, Ito Y, Kamakura M, Gotoh K, Kawada J, Nishiyama Y, Kimura H: Antitumor activities of valproic acid on Epstein-Barr virus-associated T and natural killer lymphoma cells. Cancer Sci 2012, 103: 375–81. 10.1111/j.1349-7006.2011.02127.x
Jin L, Tabe Y, Kojima K, Zhou Y, Pittaluga S, Konopleva M, Miida T, Raffeld M: MDM2 antagonist Nutlin-3 enhances bortezomib-mediated mitochondrial apoptosis in TP53-mutated mantle cell lymphoma. Cancer Lett 2010, 299: 161–70. 10.1016/j.canlet.2010.08.015
Raje N, Hideshima T, Mukherjee S, Raab M, Vallet S, Chhetri S, Cirstea D, Pozzi S, Mitsiades C, Rooney M, Kiziltepe T, Podar K, Okawa Y, Ikeda H, Carrasco R, Richardson PG, Chauhan D, Munshi NC, Sharma S, Parikh H, Chabner B, Scadden D, Anderson KC: Preclinical activity of P276–00, a novel small-molecule cyclin-dependent kinase inhibitor in the therapy of multiple myeloma. Leukemia 2009, 23: 961–70. 10.1038/leu.2008.378
Menu E, Garcia J, Huang X, Di Liberto M, Toogood PL, Chen I, Vanderkerken K, Chen-Kiang S: A novel therapeutic combination using PD 0332991 and bortezomib: study in the 5T33MM myeloma model. Cancer Res 2008, 68: 5519–23. 10.1158/0008-5472.CAN-07-6404
Huang X, Di Liberto M, Jayabalan D, Liang J, Ely S, Bretz J, Shaffer AL 3rd, Louie T, Chen I, Randolph S, Hahn WC, Staudt LM, Niesvizky R, Moore MA, Chen-Kiang S: Prolonged early G1 arrest by selective CDK4/CDK6 inhibition sensitizes myeloma cells to cytotoxic killing through cell cycle-coupled loss of IRF4. Blood 2012, 120: 1095–106. 10.1182/blood-2012-03-415984
Acknowledgements
We wish to apologize to all colleagues whose work, because of lack of space, could not be cited. We thank all the members of the DP Mishra laboratory for helpful discussions. This work was supported by the Fast Track Grant grant from the Department of Science and Technology (GAP-00 56) to DP Mishra. Namrata Rastogi acknowledges the support by the senior research fellowship from the Council of Scientific and Industrial Research, New Delhi.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors declare that there is no competing interest.
Authors’ contribution
NR collected and reviewed the literature and wrote the manuscript. DPM corrected and revised the manuscript. Both authors read and approved the final manuscript.
Electronic supplementary material
13008_2012_173_MOESM1_ESM.doc
Additional file 1: Table S1. List of combinations of PIs with different class of compounds targeting cancer cell division. (DOC 32 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Rastogi, N., Mishra, D. Therapeutic targeting of cancer cell cycle using proteasome inhibitors. Cell Div 7, 26 (2012). https://doi.org/10.1186/1747-1028-7-26
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1747-1028-7-26