
Abstract
A major determinant of cell fate is regulation of cell cycle. Tight regulation of this process is lost during the course of development and progression of various tumors. The ubiquitin-proteasome system (UPS) constitutes a universal protein degradation pathway, essential for the consistent recycling of a plethora of proteins with distinct structural and functional roles within the cell, including cell cycle regulation. High grade tumors, such as glioblastomas have an inherent potential of escaping cell cycle control mechanisms and are often refractory to conventional treatment. Here, we review the association of UPS with several UPS-targeted proteins and pathways involved in regulation of the cell cycle in malignant gliomas, and discuss the potential role of UPS inhibitors in reinstitution of cell cycle control.
Similar content being viewed by others


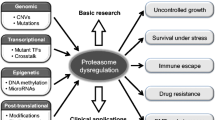
Introduction
Malignant gliomas constitute a spectrum of poorly differentiated primary brain tumors of astrocytic or oligodendroglial origin with a marked resistance to treatment, a high tendency of recurrence and a poor prognosis [1, 2].
Deregulation of cell cycle in most cancer cell types, including glioma is a critical mechanism of development, progression, and resistance to treatment [3]. Aberrant function of critical regulators of the cell cycle generally results in modification of growth, differentiation and apoptotic properties of the cell. Knowledge of the regulatory mechanisms that govern the function of cell cycle controllers is critical to enable design of new or use of already existing inhibitors with the aim of inducing a cell cycle related anticancer effect.
The ubiquitin-proteasome system (UPS) is long-known as a cellular tool for the marking and proteolytic degradation of proteins involved in a wide variety of structural and functional roles inside the cell. The UPS includes the “ubiquitously” expressed 76-amino acid protein ubiquitin (Ub), the multisubunit protein organelle 26S proteasome, consisting of one 20S catalytic and two 19S regulatoty subunits, and finally, a 3-step enzymatic cascade of Ub-activating (E1), Ub-conjugating (E2) and Ub-ligase (E3) enzymes which attach ubiquitin to the target protein [4, 5].
In cancer, a great number of cellular proteins with various roles, including cell cycle control, either comprise direct targets of an aberrant degradation machinery or have a close structural or/and functional connection with abnormal ubiquitin- or ubiquitin like-ligases, deubiquitinating enzymes and UPS-regulated signaling factors and pathways [6–8]. In this context, the involvement of UPS in cell cycle regulation is critical.
Ubiquitination and proteasomal degradation of glioma cell cycle proteins
The earliest evidence indicative of an association between the stability of cell cycle proteins and UPS in gliomas concerns the cyclin-dependent kinase inhibitor p27, which was proved to be degraded in a proteasome-dependent manner [9, 10]. p27 is a negative regulator of cyclin D-Cdk4, cyclin E-Cdk2 and cyclin A-Cdk2, being involved in G1-S transition, and its expression was observed to decrease with advancing anaplasia of astrocytic tumors. This feature was correlated with and, at least partially, attributed to increased levels of degradation activity [9]. This finding is further supported by evidence of an inverse correlation between levels of p27 and Skp2 (S-phase kinase-associated protein 2), a member of the ubiquitin ligase F-box family of proteins that promotes G1-S transition through targeting of p27 for degradation. p27 decreased with anaplasia and almost disappeared in glioblastomas (GBMs), whereas Skp2 was absent or poorly expressed in well-differentiated astrocytomas and it was diffusely or focally expressed in most GBMs [11].
p21 is another cell cycle-related protein target of the ubiquitin-proteasome pathway in gliomas. Being an established Cdk2 inhibitor (and likely Cdk1 and Cdk3 as well), p21 was found to be dependent on the ubiquitin ligase APC/CCdc20 (Anaphase Promoting Complex/Cyclosome and its activator Cdc20) for its proteolytic degradation by the proteasome in prometaphase [12]. The suggested model involves a positive feedback loop where Cdk1, by phosphorylating certain subunits of APC/CCdc20, promotes the activity of APC/CCdc20, consequently triggering the degradation of p21, resulting in a further activation of Cdk1. The degradation of p21 via APC/CCdc20 contributes to the full activation of Cdk1 in early M phase and prevents mitotic slippage during activation of the spindle assembly checkpoint [12]. The APC/CCdc20 regulates the UPS-dependent degradation of several proteins that drive the cell cycle, including cyclins A and B, while other APC/CCdc20 substrates (geminin, survivin, polo and aurora kinases) or regulators (Emi1, RASSF1A) are overexpressed in high grade gliomas [13]. p21 is also a transcriptional target of p53 and a negative regulator of the proteasome-dependent stability of p53- and Rb [14, 15] whose intimate associations with the UPS are well-established and analyzed below.
The migration and invasion inhibitor protein (MIIP, also known as IIp45) was discovered as a negative regulator of cell migration and invasion in glioma and its expression was reduced or undetectable in tissue samples obtained from patients with GBM. At the cellular level, an important role was revealed for MIIP in the inhibition of gliomagenesis and attenuation of mitotic transition based on its association with APC/C activity. MIIP interacts directly with Cdc20, and this interaction of inhibits APC/C-mediated degradation of cyclin B1, thereby causing attenuation of mitotic transition and increased mitotic catastrophe [16]. Thus, loss of MIIP promotes cell cycle progression and mitotic activity during development and progression of gliomas.
Cdk1 (also named Cdc2) and cyclin B1 are also targets of the proteasome as evidenced from a study using geldanamycin, a drug interfering with the function of the chaperone protein Hsp90 (Heat shock protein 90), in glioma cells. Geldamycin promotes proteasome degradation of client proteins by inhibiting Hsp90 function [17]. In addition to Cdk1, proteasome-mediated degradation of kinase chk1 (check point kinase 1), a Cdk1 regulator, is also promoted by geldanamycin [18].
The cyclin A/Cdk2 complex was also demonstrated to be a proteasome substrate after being tethered to a constructed chimeric protein composed of an F-box protein (TrCP) fused to a cyclin A/Cdc2 inhibitor binding peptide. Moreover, this proteasome-mediated destruction resulted in massive tumor cell (including glioma) apoptosis both in vitro and in vivo. The explanation provided, was based on unopposed E2F1 (a cell cycle regulatory transcription factor) activity which is further enhanced in Rb-deregulated cells [19].
UPS is also involved in the regulation of cyclin D1 protein levels. This is effected through its phosphorylation by the IKKα (Inhibitor of kappa B kinase alpha), which in turn drives the protein from nucleus to the cytoplasm, targeting it for ubiquitination and subsequent proteolytic degradation by the proteasome [20]. Of note is that this translocation preceding proteolytic degradation is favoured by acidic environment [21]. On the other hand, UPS is also “present” in cyclin D1 upregulation via the UPS-regulated transcription factor NF-κB, activation of which contributes to cell cycle progression and prevents differentiating GBM-initiating cells (GICs) from acquiring a mature postmitotic phenotype [22].
The E2F1 protein, a cellular mediator involved in the transition from G1 to S phase was found to be degraded by the proteasome in response to Δ9-tetrahydrocannabinol treatment of human GBM cells, thus blocking cell cycle progression and supporting a role for cannabinoids as anticancer agents [23]. The first report implicating the UPS in E2F1 regulation had revealed a degradation target sequence in a carboxyl-terminal region of E2F1 as well as a stabilizing role for Rb tumor suppressor protein in this process [24].
A relatively new cell cycle regulatory protein in malignant astrocytoma, Pescadillo, is subjected to post-translational modification by SUMO-1 (small ubiquitin-like modifier, a member of the ubiquitin protein family). Pescadillo protein expression is upregulated after loss of p53, contributing to cell cycle progression and proliferation. This SUMO-1 modification may be important for protein targeting and protein-protein interactions, although it is probably not involved in a proteolytic control of the protein-conjugate, as derived from data obtained from yeast and human cells, in which SUMO-1 conjugates remained unaffected by proteasome inhibition [25].
There are also several implications for an indirect role of UPS in cell cycle, mediated through the regulation of stability of the oncoprotein c-Myc, which is a known activator of cell cycle acceleration. Interestingly, the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) has emerged as a novel modulator of this ubiquitin-mediated c-Myc proteolysis via Akt /glycogen synthase kinase 3 (GSK3) pro-survival pathway in human glioma cells [26]. The importance of c-Myc downregulation in relation with cell cycle responses in astrocytoma cell lines involves a G1 arrest together with inhibition of DNA synthesis and delay in S phase transition. These effects emanate from loss of cyclin E and cyclin A proteins accompanied by inhibition of the cyclin D1/Cdk4 complex activity (as evaluated by presence of its active unphosphorylated target substrate, Rb) via p21 and p57 assembly [27]. Notably, Rb, which plays a key role in regulation of cell cycle, primarily via interaction with the E2F family of transcription factors in assembly of active repressor complexes to negatively regulate expression of E2F-dependent genes important for cell cycle progression, is itself a proteasome target protein. This regulation of Rb protein levels is governed by an Mdm2-mediated promotion of Rb-20S proteasome subunit interaction leading to a proteasome-dependent ubiquitin-independent degradation of Rb [28].
Mutations of the gene PARK2, which encodes an E3 ubiquitin ligase, parkin, is common not only in early-onset familial Parkinson’s disease but also in cancer, as they occur in the same domains, and sometimes at the same residues. The significance of PARK2 mutations in various cancers, including GBM, lies in the fact that the subsequent decrease in PARK2’s E3 ligase activity compromises its ability to ubiquitinate cyclin E and thereby results in mitotic instability [29, 30]. Parkin expression is dramatically reduced in glioma cells. Restoration of parkin expression promotes G1 phase cell cycle arrest and attenuates the proliferation rate of glioma cells in vitro and in vivo. Notably, parkin-expressing glioma cells have reduced levels of cyclin D1, but not cyclin E, and a selective downregulation of Akt serine-473 phosphorylation and VEGF receptor levels. Reversely, parkin-null mouse models exhibit increased levels of cyclin D1, VEGF receptor, and Akt phosphorylation, and divide significantly faster when compared with wild-type cells, with suppression of these changes following parkin reintroduction. At the clinical level, a prognostic role of the parkin pathway has emerged from studying the survival outcome of patients with glioma according to parkin status. Parkin expression in GBM patients is associated with lower grade and improved survival [31].
Regulator of Cullins-1 (ROC1) or Ring Box Protein-1 (RBX1) is a RING component of SCF (Skp-1, cullins, F-box proteins) E3 ubiquitin ligases implicated in cell growth and cell cycle progression of GBM. The latter was shown to be attenuated after ROC1 silencing by siRNA which caused growth inhibition, induction of senescence, apoptosis and G2/M arrest in U87 GBM cells. Senescence induction was coupled with DNA damage in p53/p21- and p16/Rb-independent manners. Apoptosis was associated with accumulation of Puma (p53 upregulated modulator of apoptosis) and reduction of Bcl-2, Mcl-1, (Myeloid cell leukemia sequence 1) and survivin, and G2/M arrest was associated with accumulation of 14-3-3sigma and elimination of cyclin B1 and Cdc2 [32].
Last but not least, the induction of G0/G1 or G2/M cell cycle arrest and/or apoptosis, whenever indicated as a response to various stressors, could not be ensured without the critical involvement of p53, whose levels and activation within the ARF-Mdm2-p53 axis are under tight UPS-dependent regulation. Phosphorylation in response to genotoxic stimuli, acetylation and deubiquitination enhance p53 stabilization and activation via attenuated interaction with Mdm2 and inhibition or reversal of ubiquitination. Phosphorylation of the E3-ligase Mdm2 may either suspend nuclear export of p53 or promote nuclear translocation of Mdm2 with adverse consequences on p53 stability, depending on the inducing kinase (ATM signaling stabilizes p53, Akt signalling destabilizes it). ARF can induce the accumulation of p53 by repressing Mdm2, and all three members of the axis (ARF, Mdm2, p53) exhibit non-overlapping activities and participate in autoregulatory feedback loops involving p53-Mdm2 and p53-ARF doublets [33]. The p53 pathway plays a crucial role in the development of secondary GBMs, thought to be derived to the malignant progression to grade II or III astrocytomas as p53 mutations are rather common (65 %) in this subtype [34, 35]. Amplification of Mdm2 is rare but amplification and overexpression of MdmX, which is a p53-binding protein with close homology to Mdm2, has been found in certain malignant gliomas that have wild-type p53 [36]. Further, the alternative splicing of Mdm2 has been correlated with stabilized wild-type p53 in certain human GBM cells [37]. Newer studies have ascribed a role for merlin, a neurofibromatosis 2 (NF2)-related tumor suppressor in promotion of p53 stability and activity by inducing Mdm2 degradation in glioma cells [38]. p53 protection from Mdm2-mediated degradation is also conferred by PTEN via inhibition of PI3K/Akt signaling that promotes Mdm2 nuclear translocation. Furthermore, activated p53 induces PTEN gene expression, providing evidence for a positive feedback loop, amplifying sensitization of glioma cells to chemotherapy that relies on p53 activity [39]. The process of post-translational p53 stabilization further involves reversible protein phosphorylation by a kinase/phosphatase pair. In general, phosphorylation of p53 blocks the p53-Mdm2 interaction and thus stabilizes p53 in stressed cells. A newly identified complex GAS41-PP2Cβ is specifically required for dephosphorylation of serine 366 on p53 and this is highly relevant in human glioma given that GAS41 is frequently amplified in this type of malignancy [40]. Thus, p53 regulation by UPS in malignant gliomas is a promising target, in terms of proteasome inhibition, or ubiquitination-resistant p53 protein transduction therapy [41]. A stable mutant p53 in which all lysines were replaced by arginines making it ubiquitination-resistant was found to be transcriptionally active and to inhibit glioma cell proliferation. If transduction efficacy could be ensured in vivo, a stable ubiquitination-resistant p53 could become a therapeutically useful intervention in malignant gliomas.
Most important cell cycle regulators that are intimately associated with the UPS, either members of the basal cell cycle machinery (cyclins, Cdks, Cdk inhibitors) or cell cycle-related transription factors are depicted in Figures 1 and 2, respectively.

UPS-dependent regulation of cyclins, Cdks and Cdk inhibitors in GBM.

UPS-dependent regulation of cell cycle-associated transcription factors in GBM.
The role of proteasome inhibition in glioma cell cycle
The regulation of turnover of many cell cycle effectors by the UPS in gliomas has led to the consideration of proteasome inhibition as a potential modulator of cell cycle abnormalities in these tumors. Overall, an illustrative picture of the changes in cell cycle of GBM cell lines, induced by a proteasome inhibitor, includes a G2/M cell cycle arrest with a concomitant decreased percentage of cells in S phase. This is associated with an increased expression of p21, p27, cyclin D1 and decreased levels of Cdk2, Cdk4 and E2F4 (member of the E2F family of transcription factors) [42]. As data from basic and preclinical research continue to accumulate, existing information on the role of UPS in glioma cell cycle regulation is steadily confirmed by the use of novel proteasome inhibitors, such as SC68896 and celastrol [43, 44]. Accumulation of G2/M phase-related proteins p21 and p27 was the most prominent cell cycle-related event after treatment with SC68896. This was accompanied by caspase cleavage, and sensitization of glioma cells to TRAIL and CD95L via Upregulation of cell surface expression of the death receptors DR4 and DR5 [43]. Upregulation of p21, p27 and cyclin B1 together with downregulation of Cdk2 were observed with celastrol. Cell cycle arrest was followed by proapoptotic Bax and caspase-3 stabilization as well as reduced levels of antiapoptotic Bcl-2 and XIAP proteins [44]. In addition, a new role has been revealed for the gamma-secretase inhibitor LLNIe as it was shown to cause G2/M arrest and subsequent apoptosis in cells with GBM tumor-initiating cells (TICs) phenotype by inducing proteasome inhibition and proteolytic stress [45]. High concentrations of ritonavir, an anti-retroviral medication recently found to be a proteasome inhibitor, induced a cell cycle arrest in the G1 phase followed by apoptosis of GL15 glioma cells [46].
Conclusions
This review was intended to offer an insight to the role of the UPS in regulation of ubiquitination and stability of several cell cycle proteins rather than cover all aspects of cell cycle events being under the control of UPS in malignant gliomas, as well as of the complexity and extent of this control. The use of proteasome inhibition as an anticancer treatment is possibly meaningful in terms of cell cycle control, and combinations with other agents with established activity against aberrant cell cycle progression or under investigation might enable a more efficient strategy for rapidly proliferating tumors such as malignant gliomas.
References
Preusser M, Haberler C, Hainfellner JA: Malignant glioma: neuropathology and neurobiology. Wien Med Wochenschr 2006, 156: 332–337. 10.1007/s10354-006-0304-7
Wen PY, Kesari S: Malignant gliomas in adults. N Engl J Med 2008, 359: 492–507. 10.1056/NEJMra0708126
Hanahan D, Weinberg RA: Hallmarks of cancer: the next generation. Cell 2011, 144: 646–674. 10.1016/j.cell.2011.02.013
Ciechanover A, Orian A, Schwartz AL: Ubiquitin-mediated proteolysis: biological regulation via destruction. Bioessays 2000, 22: 442–451. 10.1002/(SICI)1521-1878(200005)22:5<442::AID-BIES6>3.0.CO;2-Q
Adams J: The proteasome: structure, function, and role in the cell. Cancer Treat Rev 2003,29(Suppl 1):3–9.
Mani A, Gelmann EP: The ubiquitin-proteasome pathway and its role in cancer. J Clin Oncol 2005, 23: 4776–4789. 10.1200/JCO.2005.05.081
Burger AM, Seth AK: The ubiquitin-mediated protein degradation pathway in cancer: therapeutic implications. Eur J Cancer 2004, 40: 2217–2229. 10.1016/j.ejca.2004.07.006
Hoeller D, Hecker CM, Dikic I: Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat Rev Cancer 2006, 6: 776–788. 10.1038/nrc1994
Piva R, Cancelli I, Cavalla P, Bortolotto S, Dominguez J, Draetta GF, Schiffer D: Proteasome-dependent degradation of p27/kip1 in gliomas. J Neuropathol Exp Neurol 1999, 58: 691–696. 10.1097/00005072-199907000-00002
Pamarthy D, Tan M, Wu M, Chen J, Yang D, Wang S, Zhang H, Sun Y: p27 degradation by an ellipticinium series of compound via ubiquitin-proteasome pathway. Cancer Biol Ther 2007, 6: 360–366. 10.4161/cbt.6.3.3703
Schiffer D, Cavalla P, Fiano V, Ghimenti C, Piva R: Inverse relationship between p27/Kip.1 and the F-box protein Skp2 in human astrocytic gliomas by immunohistochemistry and Western blot. Neurosci Lett 2002, 328: 125–128. 10.1016/S0304-3940(02)00483-4
Amador V, Ge S, Santamaría PG, Guardavaccaro D, Pagano M: APC/C(Cdc20) controls the ubiquitin-mediated degradation of p21 in prometaphase. Mol Cell 2007, 27: 462–473. 10.1016/j.molcel.2007.06.013
Lehman NL, Tibshirani R, Hsu JY, Natkunam Y, Harris BT, West RB, Masek MA, Montgomery K, van de Rijn M, Jackson PK: Oncogenic regulators and substrates of the anaphase promoting complex/cyclosome are frequently overexpressed in malignant tumors. Am J Pathol 2007, 170: 1793–1805. 10.2353/ajpath.2007.060767
Broude EV, Demidenko ZN, Vivo C, Swift ME, Davis BM, Blagosklonny MV, Roninson IB: p21 (CDKN1A) is a negative regulator of p53 stability. Cell Cycle 2007, 6: 1468–1471.
Broude EV, Swift ME, Vivo C, Chang BD, Davis BM, Kalurupalle S, Blagosklonny MV, Roninson IB: p21(Waf1/Cip1/Sdi1) mediates retinoblastoma protein degradation. Oncogene 2007, 26: 6954–6958. 10.1038/sj.onc.1210516
Ji P, Smith SM, Wang Y, Jiang R, Song SW, Li B, Sawaya R, Bruner JM, Kuang J, Yu H, Fuller GN, Zhang W: Inhibition of gliomagenesis and attenuation of mitotic transition by MIIP. Oncogene 2010, 29: 3501–3508. 10.1038/onc.2010.114
Nomura N, Nomura M, Newcomb EW, Zagzag D: Geldanamycin induces G2 arrest in U87MG glioblastoma cells through downregulation of Cdc2 and cyclin B1. Biochem Pharmacol 2007, 73: 1528–1536. 10.1016/j.bcp.2007.01.022
Nomura M, Nomura N, Yamashita J: Geldanamycin-induced degradation of Chk1 is mediated by proteasome. Biochem Biophys Res Commun 2005, 335: 900–905. 10.1016/j.bbrc.2005.07.160
Chen W, Lee J, Cho SY, Fine HA: Proteasome-mediated destruction of the cyclin a/cyclin-dependent kinase 2 complex suppresses tumor cell growth in vitro and in vivo. Cancer Res 2004, 64: 3949–3957. 10.1158/0008-5472.CAN-03-3906
Kwak YT, Li R, Becerra CR, Tripathy D, Frenkel EP, Verma UN: IkappaB kinase alpha regulates subcellular distribution and turnover of cyclin D1 by phosphorylation. J Biol Chem 2005, 280: 33945–33952. 10.1074/jbc.M506206200
Schnier JB, Nishi K, Harley WR, Gorin FA: An acidic environment changes cyclin D1 localization and alters colony forming ability in gliomas. J Neurooncol 2008, 89: 19–26. 10.1007/s11060-008-9591-8
Nogueira L, Ruiz-Ontañon P, Vazquez-Barquero A, Lafarga M, Berciano MT, Aldaz B, Grande L, Casafont I, Segura V, Robles EF, Suarez D, Garcia LF, Martinez-Climent JA, Fernandez-Luna JL: Blockade of the NFκB pathway drives differentiating glioblastoma-initiating cells into senescence both in vitro and in vivo. Oncogene 2011, 30: 3537–3548. 10.1038/onc.2011.74
Galanti G, Fisher T, Kventsel I, Shoham J, Gallily R, Mechoulam R, Lavie G, Amariglio N, Rechavi G, Toren A: Delta 9-tetrahydrocannabinol inhibits cell cycle progression by downregulation of E2F1 in human glioblastoma multiforme cells. Acta Oncol 2008, 47: 1062–1070. 10.1080/02841860701678787
Campanero MR, Flemington EK: Regulation of E2F through ubiquitin-proteasome-dependent degradation: stabilization by the pRB tumor suppressor protein. Proc Natl Acad Sci U S A 1997, 94: 2221–2226. 10.1073/pnas.94.6.2221
Kinoshita Y, Jarell AD, Flaman JM, Foltz G, Schuster J, Sopher BL, Irvin DK, Kanning K, Kornblum HI, Nelson PS, Hieter P, Morrison RS: Pescadillo, a novel cell cycle regulatory protein abnormally expressed in malignant cells. J Biol Chem 2001, 276: 6656–6665. 10.1074/jbc.M008536200
An J, Yang DY, Xu QZ, Zhang SM, Huo YY, Shang ZF, Wang Y, Wu DC, Zhou PK: DNA-dependent protein kinase catalytic subunit modulates the stability of c-Myc oncoprotein. Mol Cancer 2008, 7: 32. 10.1186/1476-4598-7-32
Amendola D, De Salvo M, Marchese R, Verga Falzacappa C, Stigliano A, Carico E, Brunetti E, Moscarini M, Bucci B: Myc down-regulation affects cyclin D1/cdk4 activity and induces apoptosis via Smac/Diablo pathway in an astrocytoma cell line. Cell Prolif 2009, 42: 94–109. 10.1111/j.1365-2184.2008.00576.x
Ying H, Xiao ZX: Targeting retinoblastoma protein for degradation by proteasomes. Cell Cycle 2006, 5: 506–508. 10.4161/cc.5.5.2515
Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, Janakiraman M, Schultz N, Hanrahan AJ, Pao W, Ladanyi M, Sander C, Heguy A, Holland EC, Paty PB, Mischel PS, Liau L, Cloughesy TF, Mellinghoff IK, Solit DB, Chan TA: Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet 2010, 42: 77–82. 10.1038/ng.491
Veeriah S, Morris L, Solit D, Chan TA: The familial Parkinson disease gene PARK2 is a multisite tumor suppressor on chromosome 6q25.2–27 that regulates cyclin E. Cell Cycle 2010, 9: 1451–1452. 10.4161/cc.9.8.11583
Yeo CW, Ng FS, Chai C, Tan JM, Koh GR, Chong YK, Koh LW, Foong CS, Sandanaraj E, Holbrook JD, Ang BT, Takahashi R, Tang C, Lim KL: Parkin pathway activation mitigates glioma cell proliferation and predicts patient survival. Cancer Res 2012, 72: 2543–53. 10.1158/0008-5472.CAN-11-3060
Jia L, Soengas MS, Sun Y: ROC1/RBX1 E3 ubiquitin ligase silencing suppresses tumor cell growth via sequential induction of G2-M arrest, apoptosis, and senescence. Cancer Res 2009, 69: 4974–4982. 10.1158/0008-5472.CAN-08-4671
Yang Y, Li CC, Weissman AM: Regulating the p53 system through ubiquitination. Oncogene 2004, 23: 2096–2106. 10.1038/sj.onc.1207411
Steinbach JP, Weller M: Apoptosis in gliomas: molecular mechanisms and therapeutic implications. J Neurooncol 2004, 70: 245–254. 10.1007/s11060-004-2753-4
Ohgaki H, Kleihues P: Genetic pathways to primary and secondary glioblastoma. Am J Pathol 2007, 170: 1445–1453. 10.2353/ajpath.2007.070011
Riemenschneider MJ, Büschges R, Wolter M, Reifenberger J, Boström J, Kraus JA, Schlegel U, Reifenberger G: Amplification and overexpression of the MDM4 (MDMX) gene from 1q32 in a subset of malignant gliomas without TP53 mutation or MDM2 amplification. Cancer Res 1999, 59: 6091–6096.
Kraus A, Neff F, Behn M, Schuermann M, Muenkel K, Schlegel J: Expression of alternatively spliced mdm2 transcripts correlates with stabilized wild-type p53 protein in human glioblastoma cells. Int J Cancer 1999, 80: 930–934. 10.1002/(SICI)1097-0215(19990315)80:6<930::AID-IJC20>3.0.CO;2-M
Kim H, Kwak NJ, Lee JY, Choi BH, Lim Y, Ko YJ, Kim YH, Huh PW, Lee KH, Rha HK, Wang YP: Merlin neutralizes the inhibitory effect of Mdm2 on p53. J Biol Chem 2004, 279: 7812–7818.
Mayo LD, Donner DB: The PTEN, Mdm2, p53 tumor suppressor-oncoprotein network. Trends Biochem Sci 2002, 27: 462–467. 10.1016/S0968-0004(02)02166-7
Park JH, Smith RJ, Shieh SY, Roeder RG: The GAS41-PP2Cbeta complex dephosphorylates p53 at serine 366 and regulates its stability. J Biol Chem 2011, 286: 10911–10917. 10.1074/jbc.C110.210211
Michiue H, Tomizawa K, Matsushita M, Tamiya T, Lu YF, Ichikawa T, Date I, Matsui H: Ubiquitination-resistant p53 protein transduction therapy facilitates anti-cancer effect on the growth of human malignant glioma cells. FEBS Lett 2005, 579: 3965–3969. 10.1016/j.febslet.2005.06.021
Yin D, Zhou H, Kumagai T, Liu G, Ong JM, Black KL, Koeffler HP: Proteasome inhibitor PS-341 causes cell growth arrest and apoptosis in human glioblastoma multiforme (GBM). Oncogene 2005, 24: 344–354. 10.1038/sj.onc.1208225
Roth P, Kissel M, Herrmann C, Eisele G, Leban J, Weller M, Schmidt F: SC68896, a novel small molecule proteasome inhibitor, exerts antiglioma activity in vitro and in vivo. Clin Cancer Res 2009, 15: 6609–6618. 10.1158/1078-0432.CCR-09-0548
Ge P, Ji X, Ding Y, Wang X, Fu S, Meng F, Jin X, Ling F, Luo Y: Celastrol causes apoptosis and cell cycle arrest in rat glioma cells. Neurol Res 2010, 32: 94–100. 10.1179/016164109X12518779082273
Monticone M, Biollo E, Fabiano A, Fabbi M, Daga A, Romeo F, Maffei M, Melotti A, Giaretti W, Corte G, Castagnola P: z-Leucinyl-leucinyl-norleucinal induces apoptosis of human glioblastoma tumor-initiating cells by proteasome inhibition and mitotic arrest response. Mol Cancer Res 2009, 7: 1822–1834. 10.1158/1541-7786.MCR-09-0225
Laurent N, de Boüard S, Guillamo JS, Christov C, Zini R, Jouault H, Andre P, Lotteau V, Peschanski M: Effects of the proteasome inhibitor ritonavir on glioma growth in vitro and in vivo. Mol Cancer Ther 2004, 3: 129–136.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
PJV performed literature review and drafted the manuscript. IAV and CNP revised and approved the final manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Vlachostergios, P.J., Voutsadakis, I.A. & Papandreou, C.N. The ubiquitin-proteasome system in glioma cell cycle control. Cell Div 7, 18 (2012). https://doi.org/10.1186/1747-1028-7-18
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1747-1028-7-18