
Abstract
Amelogenesis imperfecta (AI) is a genetically heterogeneous group of diseases that result in defective development of tooth enamel. Mutations in several enamel proteins and proteinases have been associated with AI. The object of this study was to evaluate evidence of etiology for the six major candidate gene loci in two Brazilian families with AI. Genomic DNA was obtained from family members and all exons and exon-intron boundaries of the ENAM, AMBN, AMELX, MMP20, KLK4 and Amelotin gene were amplified and sequenced. Each family was also evaluated for linkage to chromosome regions known to contain genes important in enamel development. The present study indicates that the AI in these two families is not caused by any of the known loci for AI or any of the major candidate genes proposed in the literature. These findings indicate extensive genetic heterogeneity for non-syndromic AI.
Similar content being viewed by others

Background
Amelogenesis imperfecta (AI) is a group of inherited defects of dental enamel formation that show both clinical and genetic heterogeneity [1]. In its mildest form, AI causes discoloration, while in the most severe presentation the enamel is hypocalcified causing it to be abraded from the teeth shortly after their emergence into the mouth [2]. Both the primary and permanent dentitions may be affected. Enamel findings in AI are highly variable, ranging from deficient enamel formation to defects in the mineral and protein content [3]. Four main types of AI have been described: hypoplastic, hypocalcified, hypomaturation and hypomaturation-hypoplastic with taurodontism [4].
The AI phenotypes vary widely depending on the specific gene involved, the location and type of mutation, and the corresponding putative change at the protein level [5]. Different inheritance patterns such as X-linked, autosomal dominant and autosomal recessive types have been reported and 14 subtypes of AI are recognized [4].
The distribution of AI types is known to vary in different populations [3], suggesting allele frequency differences between ethnic groups [6]. The combined prevalence of all forms of AI has been reported as 1:14000 in the U.S. [7], 1:8000 in Israel [6] and 1:4000 in Sweden [8]. The autosomal dominant form of AI is most prevalent in the United States and Europe, while autosomal recessive AI is most prevalent in the Middle East [6, 7]. Different mutations in genes that encode principal matrix proteins and proteinases of enamel have been associated with the different phenotypes of AI.
The main structural proteins in forming enamel are amelogenin, ameloblastin, and enamelin. These proteins are proteolytically cleaved following their secretion. Some of the cleavage products accumulate in the enamel layer, while others are either degraded or reabsorbed by ameloblasts [9]. Different proteinases such as matrix metalloproteinase-20 and kallikrein-4, regulate the enamel matrix protein processing that ultimately defines the structure and composition of enamel [10].
Amelogenin, the protein product of the AMELX Xp22.3-p22.1 and AMELY Yp11 genes, is considered to be critical for normal enamel thickness and structure [11]. Amelogenin is the most abundant protein in developing enamel, accounting for more than 90% of total enamel protein [12], while ameloblastin and enamelin account for about 5% and 2% of total protein, respectively [9]. Amelogenin is thought to form a scaffold for enamel crystallites and to control their growth [11], but its exact functions are not fully known [13]. At least 14 mutations have been described in the X-chromosome amelogenin gene and are associated with hypoplastic and/or hypomineralization AI [12–19]. However, no cases of mutation in the Y-chromosome amelogenin gene have been reported [13], due to the fact that, the amino acid sequence of the X and Y chromosome amelogenin genes are not the same and only the X copy is critical for normal enamel development.
The chromosome 4q13 region contains at least 3 genes important in enamel development: enamelin, ameloblastin, and amelotin. Enamelin gene mutations have been identified in autosomal dominant AI [1, 5, 20, 21]. Recently it was reported that transgenic mice overexpressing ameloblastin develop AI [22]. In ameloblastin null mutant mice, ameloblasts regain some early phenotypes of undifferentiated dental epithelial cells, and the abnormalities occur when the cells detach indicating that ameloblastin is an adhesion molecule key for enamel formation [23].
Recently a novel gene coding for an ameloblast-specific protein, amelotin, was mapped close to the amelobastin and enamelin genes. It was hypothesed that amelotin is involved primarily in the maturation of enamel and thus the formation of its unique biomechanical characteristics during tooth development [24, 25].
Mutations in the predominant enamel proteinases [9] have also been associated with AI. MMP20 is secreted into the enamel matrix in the secretory and transition developmental stages [10, 26, 27]. This enzyme accounts for most of the proteolytic activity of the enamel matrix and is thought to be responsible for the processing of the amelogenin protein causing the tyrosine-rich amelogenin peptide (TRAP) to form [28, 29]. Kallikrein-4 is thought to be the major enzyme responsible for the degradation of enamel proteins during the maturation stage, and has been shown to cleave amelogenin [30]. The human MMP20 and KLK4 genes map to chromosome 11 and 19, respectively [31]. Two different mutations in MMP20 gene and one in KLK4 gene confirm that mutations in theses genes have been associated with autosomal-recessive forms of AI [32, 33].
The purpose of this study was to evaluate evidence for a genetic etiology for the six major candidate gene loci (ENAM, AMBN, AMELX, MMP20, KLK4, Amelotin) in two Brazilian families segregating AI. All exons and intron-exon junctions of these genes were sequenced, and polymorphic DNA loci spanning candidate genes in seven chromosomal regions were genotyped to evaluate support for linkage. Results of these studies provide further evidence for genetic heterogeneity of AI.
Materials and methods
Family and phenotype analyses
This study was carried out with the approval of the FOP/UNICAMP Ethics Committee (protocol 127/03) and informed consent was obtained from all subjects. Two families segregating AI were identified. All available family members were examined clinically and in some cases radiographically. Oral examinations included visual examination in a dental clinic using artificial light and dental mirror evaluations of teeth and supporting tissues. Affected and unaffected individuals were also evaluated clinically for the presence of skin, hair, fingernail and osseous abnormalities know to be associated with systemic or syndromic conditions that can be associated with enamel defects. No history of nutritional disturbances was reported by the affected members of the two families.
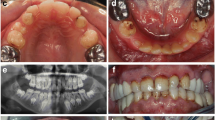
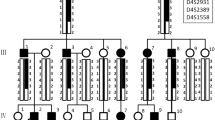
Affected status of family 1 was established clinically by the presence of a generalized yellow-brown discoloration of primary and permanent dentitions. The deficiency in the enamel mineral content was evidenced by a lack of radiographic enamel opacity and a pathological loss of enamel through wear and fracturing. The clinical phenotype and family history suggested an autosomal recessive hypocalcified AI (Fig 1).

Clinical phenotype and pedigree of Family 1. Family 1: A phenotype demonstrating generalized yellow-brown discoloration of the dentition (A1 patient III-2, A2 patient III-5); B X-ray showing lack of enamel opacity and a pathological loss of enamel (B1 patient III-2, B2 patient III-5); C pedigree of Family 1.
The enamel of affected members of family 2 was thin with rough and pitted surface (hypoplastic AI, Family 2). Both primary and permanent dentitions were affected. The clinical phenotype and family history did not allow determining the pattern of gene inheritance (Fig 2).

Clinical phenotype and pedigree of Family 2. Family 2: A phenotype of patient III-4 demonstrating points of yellow-brown discoloration of the dentition, and areas with thin enamel. (A1 dentition, A2 detail); B radiographic patient III-4; C pedigree of Family 2 suggested X-link AI.
Blood was obtained by venepuncture (Vacutainer system) and DNA extracted using Kit Puregene (Gentra Systems) for genotyping and sequence analysis.
Genotyping studies
Members of each family were evaluated for linkage to chromosomal regions known to contain genes important in enamel development at previously described [24, 32–38]. Table 1 shows studied markers for linkage to chromosome regions known to contain genes important in enamel development. The PCR reactions were performed using 20 ng of genomic DNA in a final volume of 7.5 μl, as reported previously [39]. All electrophoretic evaluations of the marker gene allele sizes were performed on an ABI 3100XL automated DNA sequencer using POP-7, 37 cm capillary and an internal size standard (ROX GS 400 standard (Applied Biosystems, Foster City, CA, USA)). Allele calling was done using the genescan software (Applied Biosystems, Foster City, CA, USA).
Mutation analysis
PCRs were carried out in a Perkin-Elmer GeneAmp 2400 thermal cycler and total volume of 50 μl, containing 500 ng genomic DNA, 10 mM Tris-HCl (pH 8,3), 50 mM KCl, 1.5 mM MgCl2, 1 μM of each primer, 200 mM each dNTPs, and 1 units Taq DNA polymerase (Amersham Pharmacia Biotech AB, Uppsala, Sweden). PCR was performed by an initial denaturation at 95°C for 5 min, followed by 35 cycles of 1 min at 95°C, annealing for 1 min at temperature listed in Table 2, extension at 72°C for 1 min, and a final extension at 72°C for 7 min. The primer sequences and PCR conditions are shown in Table 2.
The PCR products were electrophoresed through 1% agarose gels and the amplicons extracted using GFX™ PCR DNA and Gel Band Purification Kit (Amersham Pharmacia Biotech). Extracted amplicons were sequenced using do Big Dye Terminator Kit (Perkin Elmer) and an ABI Prism 377 DNA Sequencer™.
Results and Discussion
Examinations of all affected and unaffected members from both families studied indicated 4 of the 17 family members evaluated were affected (2 members affected in each family). Affected individuals showed no signs of syndromic conditions or systemic illnesses associated with defective enamel development. None of the unaffected family members had generalized enamel defects clinically and showed no evidence of radiographic enamel defects, taurodontism or dental abnormalities. There was variability in the severity of expression of the AI phenotype in family 2. Individual III-4 of family 2 showed more severe pitting than his mother (individual II-6). This difference in severity between males and females may be indicative of X-linked AI form. The presence of only one male and one female affected, however, did not allow confirming this pattern of inheritance. Additionally sequencing of amelogenin X gene did not reveal any mutations in this gene that could be associated with enamel phenotype. Radiographically, enamel was very thin but in some areas it was possible to note that enamel displayed a radiodensity similar to that of normal enamel (Fig. 2).
Affected individuals of family 1 reported variable dental hypersensitivity ranging from mild dental discomfort with thermal or chemical stimulation to normal dental sensitivity. Radiographically the teeth displayed enamel that had a radiodensity similar to that of dentin (Fig. 1).
A number of genes involved in enamel formation have been identified, and based on their expression and function, several of these genes have been proposed as candidates for AI. This study all available family members were genotyped for multiple short tandem repeat polymorphism (STRP) type markers spanning each AI candidate gene locus. Haplotyped genotype results did not show support for linkage to any of the chromosomal regions tested, clearly rejecting the linkage hypothesis throughout all six candidate regions.
The exons and intron/exon junctions of the AMELX, ENAM, AMBN, MMP20, KLK4 and Amelotin genes were sequenced and no gene mutations were identified in any individuals. A novel polymorphism was identified in the amelotin gene next exon 5 this gene. This SNP is characterized by a change of A to G in base 7125 (NCBI35:4:71564458:71579819:1). However, this SNP does not change the amino acid coded for by the triplet codon sequence and, therefore, does not appear to be associated with AI in the studied families. Figure 3 shows the position of this polymorphism.

A single nucleotide polymorphism in amelotin gene: change of A to G in base 7125 (NCBI35:4:71564458:71579819:1).
While we did not find exon mutations, it is possible that others types of mutations may be involved, such as promoter or intron mutations or deletions that encompass whole exons. However, results of the genotyping analyses do not support genetic linkage to the interval, suggesting that theses regions are not involved with AI in the studied families.
Others failed to show association between mutation in known genes involved in enamel formation and AI [40]. It has been known for some time that defects in known and suspected candidate genes can not explain all AI cases. Kim et al. (2006) [41] showed that the current list of AI candidate genes was insufficient to identify the causative gene defect in most families studied, suggesting that unknown genes/proteins that are critical for dental enamel formation. Our results indicate that additional locus coding for genes involved in ameloblast cytodifferentiation and function remain unidentified. Recently, Mendoza et al. (2006) [42] have mapped a new locus for autosomal dominant amelogenesis imperfecta on the long arm of chromosome 8 at 8q24.3.
In this study, exclusion of six candidate genes suggests that this common AI type is caused by alteration of a gene that is either not known or not considered to be a major contributor to enamel formation. Continued mutational analysis of families with AI will allow a comprehensive standardized nomenclature system to be developed for this group of disorders that will include molecular delineation as well as a mode of inheritance and phenotype.
Conclusion
The present study indicates that the autosomal recessive hypocalcified and a hypoplastic form of AI in two distinct families are not caused by mutations in any of the known loci for amelogenesis imperfecta. This suggests that many additional genes potentially contribute to the etiology of AI.
References
Rajpar MH, Harley K, Laing C, Davies RM, Dixon MJ: Mutation of the gene encoding the enamel-specific protein, enamelin, causes autosomal-dominant amelogenesis imperfecta. Hum Mol Genet. 2001, 10 (16): 1673-1677. 10.1093/hmg/10.16.1673.
Wright JT, Deaton TG, Hall KI, Yamauchi M: The mineral and protein content of enamel in amelogenesis imperfecta. Connect Tissue Res. 1995, 32 (1–4): 247-252.
Nusier M, Yassin O, Hart TC, Samimi A, Wright JT: Phenotypic diversity and revision of the nomenclature for autosomal recessive amelogenesis imperfecta. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004, 97 (2): 220-230.
Witkop CJ: Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited:problems in classification. J Oral Pathol. 1988, 17: 547-553. 10.1111/j.1600-0714.1988.tb01332.x.
Hart PS, Michalec MD, Seow WK, Hart TC, Wright JT: Identification of the enamelin (g.8344delG) mutation in a new kindred and presentation of a standardized ENAM nomenclature. Arch Oral Biol. 2003, 48 (8): 589-596. 10.1016/S0003-9969(03)00114-6.
Chosack A, Eidelman E, Wisotski I, Cohen T: Amelogenesis imperfecta among Israeli Jews and the description of a new type of local hypoplastic autosomal recessive amelogenesis imperfecta. Oral Surg Oral Med Oral Pathol. 1979, 47 (2): 148-156. 10.1016/0030-4220(79)90170-1.
Witkop C, Sauk JJ: Heritable defects of enamel. Oral Facial Genetics. 1976, St Louis: CV Mosby Company, 1: 151-1226.
Sundell S, Koch G: Hereditary amelogenesis imperfecta. I. Epidemiology and clinical classification in a Swedish child population. Swed Dent J. 1985, 9 (4): 157-169.
Simmer JP, Hu JC: Expression, structure, and function of enamel proteinases. Connect Tissue Res. 2002, 43 (2–3): 441-449.
Bartlett JD, Simmer JP, Xue J, Margolis HC, Moreno EC: Molecular cloning and mRNA tissue distribution of a novel matrix metalloproteinase isolated from porcine enamel organ. Gene. 1996, 183 (1–2): 123-128. 10.1016/S0378-1119(96)00525-2.
Fincham AG, Lau EC, Simmer J, Zeichner-David M: Amelogenin biochemistry-form and function. Amsterdam: Elsevier Science. 1992, 1: 187-201.
Fincham AG, Moradian-Oldak J, Simmer JP: The structural biology of the developing dental enamel matrix. J Struct Biol. 1999, 126 (3): 270-299. 10.1006/jsbi.1999.4130.
Hart PS, Aldred MJ, Crawford PJ, Wright NJ, Hart TC, Wright JT: Amelogenesis imperfecta phenotype-genotype correlations with two amelogenin gene mutations. Arch Oral Biol. 2002, 47 (4): 261-265. 10.1016/S0003-9969(02)00003-1.
Aldred MJ, Crawford PJ, Roberts E, Thomas NS: Identification of a nonsense mutation in the amelogenin gene (AMELX) in a family with X-linked amelogenesis imperfecta (AIH1). Hum Genet. 1992, 90 (4): 413-416. 10.1007/BF00220469.
Lench NJ, Winter GB: Characterisation of molecular defects in X-linked amelogenesis imperfecta (AIH1). Hum Mutat. 1995, 5 (3): 251-259. 10.1002/humu.1380050310.
Kindelan SA, Brook AH, Gangemi L, Lench N, Wong FS, Fearne J, Jackson Z, Foster G, Stringer BM: Detection of a novel mutation in X-linked amelogenesis imperfecta. J Dent Res. 2000, 79 (12): 1978-1982.
Ravassipour DB, Hart PS, Hart TC, Ritter AV, Yamauchi M, Gibson C, Wright JT: Unique enamel phenotype associated with amelogenin gene (AMELX) codon 41 point mutation. J Dent Res. 2000, 79 (7): 1476-1481.
Aldred MJ, Hall RK, Kilpatrick N, Bankier A, Savarirayan R, Lamande SR, Lench NJ, Crawford PJ: Molecular analysis for genetic counselling in amelogenesis imperfecta. Oral Dis. 2002, 8 (5): 249-253. 10.1034/j.1601-0825.2002.02835.x.
Greene SR, Yuan ZA, Wright JT, Amjad H, Abrams WR, Buchanan JA, Trachtenberg DI, Gibson CW: A new frameshift mutation encoding a truncated amelogenin leads to X-linked amelogenesis imperfecta. Arch Oral Biol. 2002, 47 (3): 211-217. 10.1016/S0003-9969(01)00111-X.
Mardh CK, Backman B, Simmons D, Golovleva I, Gu TT, Holmgren G, MacDougall M, Forsman-Semb K: Human ameloblastin gene: genomic organization and mutation analysis in amelogenesis imperfecta patients. Eur J Oral Sci. 2001, 109 (1): 8-13. 10.1034/j.1600-0722.2001.00979.x.
Kida M, Ariga T, Shirakawa T, Oguchi H, Sakiyama Y: Autosomal-dominant hypoplastic form of amelogenesis imperfecta caused by an enamelin gene mutation at the exon-intron boundary. J Dent Res. 2002, 81 (11): 738-742.
Paine ML, Wang HJ, Luo W, Krebsbach PH, Snead ML: A transgenic animal model resembling amelogenesis imperfecta related to ameloblastin overexpression. J Biol Chem. 2003, 278 (21): 19447-1952. 10.1074/jbc.M300445200.
Fukumoto S, Kiba T, Hall B, Iehara N, Nakamura T, Longenecker G: Ameloblastin is a cell adhesion molecule required for maintaining the differentiation state of ameloblasts. J Cell Biol. 2004, 167 (5): 973-983. 10.1083/jcb.200409077.
Iwasaki K, Bajenova E, Somogyi-Ganss E, Miller M, Nguyen V, Nourkeyhani H, Gao Y, Wendel M, Ganss B: Amelotin--a Novel Secreted, Ameloblast-specific Protein. J Dent Res. 2005, 84 (12): 1127-1132.
Moffatt P, Smith CE, St-Arnaud R, Simmons D, Wright JT, Nanci A: Cloning of rat amelotin and localization of the protein to the basal lamina of maturation stage ameloblasts and junctional epithelium. Biochem J. 2006, 399 (1): 37-46.
Fukae M, Tanabe T, Uchida T, Lee SK, Ryu OH, Murakami C, Wakida K, Simmer JP, Yamada Y, Bartlett JD: Enamelysin (matrix metalloproteinase-20): localization in the developing tooth and effects of pH and calcium on amelogenin hydrolysis. J Dent Res. 1998, 77 (8): 1580-1588.
Bartlett JD, Simmer JP: Proteinases in developing dental enamel. Crit Rev Oral Biol Med. 1999, 10 (4): 425-441.
Ryu OH, Fincham AG, Hu CC, Zhang C, Qian Q, Bartlett JD, Simmer JP: Characterization of recombinant pig enamelysin activity and cleavage of recombinant pig and mouse amelogenins. J Dent Res. 1999, 78 (3): 743-750.
Palosaari H, Pennington CJ, Larmas M, Edwards DR, Tjaderhane L, Salo T: Expression profile of matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs in mature human odontoblasts and pulp tissue. Eur J Oral Sci. 2003, 111 (2): 117-127. 10.1034/j.1600-0722.2003.00026.x.
Ryu O, Hu JC, Yamakoshi Y, Villemain JL, Cao X, Zhang C, Bartlett JD, Simmer JP: Porcine kallikrein-4 activation, glycosylation, activity, and expression in prokaryotic and eukaryotic hosts. Eur J Oral Sci. 2002, 110 (5): 358-365. 10.1034/j.1600-0722.2002.21349.x.
DuPont BR, Hu CC, Reveles X, Simmer JP: Assignment of serine protease 17 (PRSS17) to human chromosome bands 19q13.3-->q13.4 by in situ hybridization. Cytogenet Cell Genet. 1999, 86 (3–4): 212-213. 10.1159/000015340.
Ozdemir D, Hart PS, Ryu OH, Choi SJ, Ozdemir-Karatas M, Firatli E, Piesco N, Hart TC: MMP20 active-site mutation in hypomaturation amelogenesis imperfecta. J Dent Res. 2005, 84 (11): 1031-1035.
Hart PS, Hart TC, Michalec MD, Ryu OH, Simmons D, Hong S, Wright JT: Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J Med Genet. 2004, 41 (7): 545-549. 10.1136/jmg.2003.017657.
Collier PM, Sauk JJ, Rosenbloom SJ, Yuan ZA, Gibson CW: An amelogenin gene defect associated with human X-linked amelogenesis imperfecta. Arch Oral Biol. 1997, 42 (3): 235-242. 10.1016/S0003-9969(96)00099-4.
MacDougall M, DuPont BR, Simmons D, Reus B, Krebsbach P, Karrman C, Holmgren G, Leach RJ, Forsman K: Ameloblastin gene (AMBN) maps within the critical region for autosomal dominant amelogenesis imperfecta at chromosome 4q21. Genomics. 1997, 41 (1): 115-118. 10.1006/geno.1997.4643.
Deutsch D, Palmon A, Dafni L, Mao Z, Leytin V, Young M, Fisher LW: Tuftelin--aspects of protein and gene structure. Eur J Oral Sci. 1998, 106 (Suppl 1): 315-323.
Vieira H, Gregory-Evans K, Lim N, Brookes JL, Brueton LA, Gregory-Evans CY: First genomic localization of oculo-oto-dental syndrome with linkage to chromosome 20q13.1. Invest Ophthalmol Vis Sci. 2002, 43 (8): 2540-2545.
Lukusa T, Fryns JP: Syndrome of facial, oral, and digital anomalies due to 7q21.2-->q22.1 duplication. Am J Med Genet. 1998, 80 (5): 454-458. 10.1002/(SICI)1096-8628(19981228)80:5<454::AID-AJMG4>3.0.CO;2-O.
Zhang GW, Kotiw M, Daggard G: A RAPD-PCR genotyping assay which correlates with serotypes of group B streptococci. Lett Appl Microbiol. 2002, 35 (3): 247-250. 10.1046/j.1472-765X.2002.01177.x.
Hart PS, Wright JT, Savage M, Kang G, Bensen JT, Gorry MC, Hart TC: Exclusion of candidate genes in two families with autosomal dominant hypocalcified amelogenesis imperfecta. Eur J Oral Sci. 2003, 111 (4): 326-331. 10.1034/j.1600-0722.2003.00046.x.
Kim JW, Simmer JP, Lin BP, Seymen F, Bartlett JD, Hu JC: Mutational analysis of candidate genes in 24 amelogenesis imperfecta families. Eur J Oral Sci. 2006, 114 (1): 3-12. 10.1111/j.1600-0722.2006.00278.x.
Mendoza G, Pemberton TJ, Lee K, Scarel-Caminaga R, Mehrian-Shai R, Gonzalez-Quevedo C, Ninis V, Hartiala J, Allayee H, Snead ML, Leal SM, Line SR, Patel PI: A new locus for autosomal dominant amelogenesis imperfecta on chromosome 8q24.3. Hum Genet. 2007, 120 (5): 653-662. 10.1007/s00439-006-0246-6.
Acknowledgements
This study was supported by FAPES grant 03/09128-8 and CAPES grant BEX1914/05-7.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Santos, M.C., Hart, P.S., Ramaswami, M. et al. Exclusion of known gene for enamel development in two Brazilian families with amelogenesis imperfecta. Head Face Med 3, 8 (2007). https://doi.org/10.1186/1746-160X-3-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1746-160X-3-8