
Abstract
Background
Pyoderma gangrenosum (PG) is a rare inflammatory skin disorder characterised by painful and rapidly progressing skin ulceration. PG can be extremely difficult to treat and patients often require systemic immunosuppression. Recurrent lesions of PG are common, but the relative rarity of this condition means that there is a lack of published evidence regarding its treatment. A systematic review published in 2005 found no randomised controlled trials (RCTs) relating to the treatment of PG. Since this time, one small RCT has been published comparing infliximab to placebo, but none of the commonly used systemic treatments for PG have been formally assessed. The UK Dermatology Clinical Trials Network’s STOP GAP Trial has been designed to address this lack of trial evidence.
Methods
The objective is to assess whether oral ciclosporin is more effective than oral prednisolone for the treatment of PG. The trial design is a two-arm, observer-blind, parallel-group, randomised controlled trial comparing ciclosporin (4 mg/kg/day) to prednisolone (0.75 mg/kg/day). A total of 140 participants are to be recruited over a period of 4 years, from up to 50 hospitals in the UK and Eire. Primary outcome of velocity of healing at 6 weeks is assessed blinded to treatment allocation (using digital images of the ulcers). Secondary outcomes include: (i) time to healing; (ii) global assessment of improvement; (iii) PG inflammation assessment scale score; (iv) self-reported pain; (v) health-related quality of life; (vi) time to recurrence; (vii) treatment failures; (viii) adverse reactions to study medications; and (ix) cost effectiveness/utility. Patients with a clinical diagnosis of PG (excluding granulomatous PG); measurable ulceration (that is, not pustular PG); and patients aged over 18 years old who are able to give informed consent are included in the trial. Randomisation is by computer generated code using permuted blocks of randomly varying size, stratified by lesion size, and presence or absence of underlying systemic disease (for example, rheumatoid arthritis).
Patients who require topical therapy are asked to enter a parallel observational study (case series). If topical therapy fails and systemic therapy is required, participants are then considered for inclusion in the randomised trial.
Trial registration
Current controlled trials: ISRCTN35898459. Eudract No.2008-008291-14.
Similar content being viewed by others


Background
Pyoderma gangrenosum (PG) is a rare inflammatory dermatosis characterised by painful and rapidly progressive skin ulceration [1, 2]. It most commonly affects the lower limbs, but can affect any other site including the peristomal area. It can be seen in association with a number of conditions including inflammatory bowel disease, rheumatoid arthritis, haematological disorders and malignancies [3–5]. It is a disease that causes considerable morbidity to patients, who often have a poor quality of life and high need for healthcare resources. Many patients require hospitalization for initial control, and regular changes of dressings by community care teams.
PG can be extremely difficult to treat and recurrent lesions are common [5]. There is a lack of published evidence regarding its treatment, largely because large-scale RCTs are difficult to conduct in a rare condition such as this. A systematic review in 2005 recommended the use of prednisolone, ciclosporin or high-dose intravenous steroids for large lesions; or potent topical steroids, tacrolimus or intralesional steroid injection for small lesions [6]. Other reviews have suggested a similar stepwise approach [7]. However, these recommendations were based on case series alone as no RCTs of these most commonly used treatments were identified and clinical guidelines are currently lacking. There has been one randomised controlled trial demonstrating superiority of infliximab over placebo [8], but such a potent drug would rarely be used as first line treatment. Many of the commonly used treatments are associated with unpleasant and potentially serious side effects, making their formal evaluation a matter of urgency. These treatments are currently being used for patients with PG without rigorous testing, or understanding of their relative efficacy, cost and side-effect profile.
In order to begin to address this lack of evidence, the first randomised controlled trial of systemic treatments for pyoderma gangrenosum is being carried out. The ‘Study of Treatments fOr Pyoderma GAngrenosum Patients’ (STOP GAP) will compare the two most commonly used systemic treatments: prednisolone and ciclosporin. The trial was granted ethics approval by the Northern and Yorkshire research ethics committee (MREC reference: 09/H0903/5), and all participants gave written informed consent. This paper is based on a summary of the current protocol (version 4.0, 30 August 2011. Further details are available on the STOP GAP Trial website at http://www.stopgaptrial.co.uk) [9].
Methods
Design
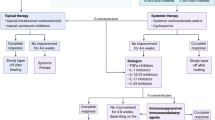
STOP GAP is a multicentre, parallel group, observer-blind, randomised controlled trial (Figure 1). This is a superiority trial, with prednisolone as the control intervention. The decision to power the STOP GAP trial on the basis of superiority was made as (i) ciclosporin is considerably more expensive than prednisolone (which means that it would need to be considerably better than prednisolone to warrant a change in clinical practice); (ii) case series and clinical experience give the impression that ciclosporin may gain control more quickly, and have a side-effect profile that is more suitable for long-term therapy; and (iii) pragmatically, for a rare condition such as PG, this was felt to be the best approach as an equivalence trial would require a much larger trial that could only be delivered internationally, which was beyond the scope of our funding.

Flow chart of trial recruitment.
The study aims to recruit 140 patients with PG over a 4-year period. Participants are randomised using a ratio of 1:1 to receive either oral prednisolone (0.75 mg/kg/day) or oral ciclosporin (4 mg/kg/day). Patients who initially require topical therapy are asked to enter a parallel observational study, the purpose of which is to provide an estimate of treatment response to topical therapy in a well defined prospective case series, and also to enhance recruitment by allowing those who fail on topical therapy to enter the main study (Figure 1).
Follow-up is continued until the lesion has healed, or for a maximum of 6 months (whichever is sooner).
Objectives
The primary objective for this trial is to assess the speed of response to treatment for the compared treatments.
Secondary objectives are to assess overall treatment response (especially time to healing), safety, and cost effectiveness of the compared treatments.
Participants
Patients aged over 18 years with a clinical diagnosis of PG (as diagnosed by the recruiting dermatologist) can be enrolled into the study. There must be measurable ulceration (that is, not pustular PG) for the patient to be included. In keeping with the pragmatic nature of the study, a positive skin biopsy is not required for diagnosis since histology is rarely diagnostic, but should be performed as part of normal care in cases of diagnostic uncertainty in order to exclude alternative diagnoses. For cases where there is doubt over the clinical diagnosis, an expert panel is available to provide assistance in confirming or refuting the diagnosis.
Patients are not eligible for the study if they: (i) have granulomatous PG (which is very rare, and may respond differently to treatment); (ii) have received oral prednisolone, oral ciclosporin or intravenous immunoglobulins within 1 month prior to randomisation; or (iii) are already participating in another clinical trial. In addition patients are not eligible for the RCT arm of the study if they (i) are pregnant, lactating or at risk of pregnancy; (ii) have known hypersensitivity to either of the study drugs; (iii) have a biopsy result that is consistent with a diagnosis other than PG; (iv) have clinically significant renal impairment that would result in the investigator not normally treating with either study drug; (v) have any pretreatment investigations, the results of which would prompt the investigator not to use either study drug; (vi) have any malignant of premalignant disease where treatments might interfere with ongoing therapy or might cause harm; (vii) have any concurrent medical condition which would preclude treatment with either of the study medications; (viii) administration of a live vaccine within 2 weeks prior to randomisation; or (ix) are currently taking Rosuvastatin (Crestor) for hypercholesterolaemia, since this is contraindicated in patients taking ciclosporin.
Interventions
Participants are randomised to receive either oral prednisolone 0.75 mg/kg/day or ciclosporin (Neoral) 4 mg/kg/day (in two divided doses), up to a ceiling dose of 75 mg/day prednisolone and 400 mg/day ciclosporin. The dose of study drug can be adjusted (up or down) by the investigator according to response after the week 2 visit. If possible the dose of study drug should not be altered for the first 2 weeks of the study.
The use of a double dummy design was not felt to be appropriate for this study as the side effect profile, monitoring requirements and dosing regimen for the compared drugs is very different, making such a design both costly and possibly ineffective. As a result the trial team opted to ensure blinded outcome assessment of the primary outcome (using digital images) and secure allocation concealment. Participants are prescribed study medication from standard pharmacy supplies at each recruiting hospital.
Participants of the RCT should not use any topical therapy (for example, corticosteroids or calcineurin inhibitors) after randomisation. If topical therapy has been prescribed while the participant is waiting to enter the study it should be stopped when the participant enters the RCT. All other concomitant medications should continue as per normal care. The use of live vaccines is not permitted during the intervention phase of the trial and clinicians will be advised not to start Rosuvastatin (Crestor) on any patients receiving ciclosporin as it is contraindicated. Participants are asked to assess how well they adhered to study drug at week 6 and at 6 months (or time of healing).
Participants in the observational arm of the study receive whichever topical therapy is prescribed as per normal care at the recruiting hospital (this is most likely to be clobetasol propionate (Dermovate) or tacrolimus (Protopic 0.1% or 0.03%) as there is reasonable evidence to support the use of both of these treatments [10, 11].
Randomisation and blinding (masking)
The randomisation schedule has been created by the Nottingham Clinical Trials Unit data manager using the ralloc Strata (College Station, TX, USA) add-on. Participants are randomised to treatment allocation using a computer-generated pseudorandom list using permuted blocks of randomly varying size between 2 and 6. Randomisation is stratified by lesion size (≥20 cm2 vs lesions <20 cm2) and presence or absence of underlying systemic disease. The randomisation schedule will be concealed until interventions are all assigned and recruitment, data collection, data cleaning and analysis are complete.
This is an observer-blind study. The primary outcome (velocity of healing) is assessed on the basis of digital images of the target lesion in order to protect against detection bias. The digital images will be assessed by two assessors who are blinded to treatment allocation, using patient number as the identifier. The images will also be used to assess global improvement in disease and the PG inflammation scale (if possible, depending on the quality of the images).
Both clinicians and participants know their treatment allocation and so no special measures are required to allow for breaking of treatment codes. However, treatment allocation will not be revealed to the recruiting physician until participants’ details and key stratification variables have been irrevocably entered by the physician onto the web-based randomisation site.
Primary outcome
The primary outcome is velocity of healing at 6 weeks. This will be captured for a single target lesion per patient and measured using digital photography and Verge Videometry VEV MD (Vista Medical, Winnipeg, Canada) computerised planimetry [12]. If multiple lesions are present, the target lesion should be the lesion that is most able to be photographed on a single plane (that is, not around the curvature of a limb). Digital images will be taken at baseline, 6 weeks and when the ulcer has healed (maximum 6 months). In addition, maximum longitudinal length and maximum perpendicular length will be measured in order to provide some measure of improvement in case of difficulties with the digital images, or if the lesion extends around the curvature of a limb. This will be converted to approximate area by the formula: length × width × 0.785, which approximates to an ellipse for the purpose of randomisation and analysis.
Velocity of healing at 6 weeks was chosen as the primary outcome as it is unlikely to be compromised by the single blind nature of the study, and does not require lengthy follow-up, thus minimising the possibility of missing data. Previous work in patients with venous leg ulcers suggests that velocity of healing is a good surrogate for subsequent healing [13].
Secondary outcomes
Secondary outcomes include the following: (1) time to healing assessed by participants (estimated to the nearest week) based on the time at which sterile dressings are no longer required for the wound. Healing is confirmed by the clinician, and documented using digital photography at the first opportunity. Time to healing is important clinically as it will be used to confirm the primary outcome as a useful surrogate measure. It also gives an indication of duration of treatment, which therefore provides information on cumulative drug toxicity. (2) PG specific global assessment of efficacy (derived from a study by Foss et al.[14]) as assessed by the clinician and the patient at 2 weeks, 6 weeks and at 6 months (or healed). This will also be assessed on the digital images by an independent assessor. (3) Inflammation assessment scale. This is a combination scale including erythema, border elevation and exudate (based on a scale reported by Foss et al.[14]). The inflammation assessment scale is recorded by the clinician and the participant at baseline, 2 weeks, 6 weeks and when the ulcer has healed (maximum 6 months). This will also be assessed on the digital images by an independent assessor. (4) Self-reported pain. Each day for the first 6 weeks of the trial participants report the severity of the pain in a study diary (none, mild, moderate, severe or extreme), and whether or not painkillers were taken. (5) Health-related quality of life assessed at baseline, 6 weeks and 6 months (or healed), using validated questionnaires: Dermatology Life Quality Index [15] and the EQ-5day [16]. (6) Time to recurrence assessed at the end of the trial. A recurrence is defined as the occurrence of a further episode of PG (at any site) that appears after the target lesion has been confirmed as being healed by a physician (or nurse). Self-reported healing that has not been confirmed by a medical professional, and which subsequently recurs, will not be classed as a recurrence and handled as a continuation of the initial episode. (7) Number of treatment failures. Treatment failures are defined as being participants who withdraw (or are withdrawn) from their randomized treatment because of treatment intolerance or worsening of the PG, or those who continue to have any unhealed ulcers after 6 months of follow-up. (8) Adverse reactions to study medications. Adverse events that are classed as possibly, probably or definitely relating to the study medication. (9) Cost effectiveness of the compared treatments.
For participants in the observational study, only efficacy outcomes are being collected (no safety or cost-effectiveness outcomes will be reported).
Sample size
A total of 140 patients (randomised 1:1 to prednisolone or ciclosporin) will give the study at least 80% power at a 5% level of significance using a two-sided two-sample t test to detect a difference in means of 0.5 SDs of the primary outcome of velocity of healing at 6 weeks. The velocity of healing at 6 weeks is defined as the percentage change in surface area (measured by planimetry using digital photographs) over baseline of the target lesion. This sample size allows for an approximate 10% loss to follow-up at 6 weeks. These calculations were performed using Nquery 6.0.2 on Windows XP (Microsoft, Redmond, WA, USA).
Statistical analysis
The primary analysis will be according to the intention to treat principle, and will adjust for known prognostic baseline covariates. There are no formal planned interim analyses, but progress reports on all data issues are presented to a Data Monitoring Committee (DMC).
The primary outcome is to be assessed at 6 weeks, which reduces the likelihood of having missing data for this outcome. If digital images are not available for any participants, then the physical length and width measurements recorded by the clinician will be used in place of the computer generated planimetry measurements. If neither a digital image, nor physical measurements are available at 6 weeks, multiple imputation will be used using the assumption that the data are missing at random.
All primary outcome data will be double data entered and 10% of other data will be entered twice and checked for errors.
Categorical variables will be summarised with the number and proportion of participants falling in each category. Continuous variables will be summarised using the number available, number missing, mean and standard deviation (SD), or median, 25th and 75th quartiles, and minimum and maximum values as appropriate.
Differences between treatment groups for the primary and secondary outcomes will be assessed using Student’s t test. Linear regression models, adjusting for baseline covariates, will be used to compare the treatment groups at 6 weeks for the primary outcome, and any other continuous secondary outcomes that fit the assumptions. Cox regression models, adjusting for baseline covariates, will be used to compare the treatment groups in the analysis of the time to healing of the target lesion and the time to recurrence. Proportional odds models, adjusting for baseline covariates, will be used to analyse the categorical secondary outcomes, including global assessment of improvement and the inflammation assessment scale. The self-reported pain and the EQ-5D score will be summarised by the area under the curve (AUC), using Generalized linear models with the appropriate distribution. Patients are required to have the data recorded at the initial timepoint, and at the 6 week timepoint (for the 6 week analyses) or the final visit (for the final visit analyses), so only patients with at least the first observation and the last observation for the self-reported pain data, or those with both timepoints for the EQ-5D data, will be analysed in the first instance. Sensitivity analyses will be carried out for the self-reported pain outcome using the ‘last value carried forward’ method for patients who have missing interim data.
Cost effectiveness
Costs of the compared treatments will be assessed from the perspective of the health service, using resources such as inpatient stays, outpatient attendances, primary care appointments (at surgery), home visits by GP, district nurse visits and PG-related treatment costs. Cost effectiveness will be presented as the cost per healed ulcer at 6 months.
Health related quality of life will be estimated from responses to the Dermatology Life Quality Index. Quality-adjusted life years will be assessed from responses to the EQ-5D questionnaire, and the incremental cost per quality-adjusted life years gained will be presented for the compared treatments. Health state utilities will be calculated using UK population tariffs. Estimates of resource use (from medical records and patient diaries) will be combined with unit costs, to derive total costs. Unit costs will be based on study specific estimates and data from standard sources. Point estimates of mean incremental costs, QALYs, cost per QALY and cost per resolved episode at 6 months will be reported.
Study organisation and funding
The study is being coordinated through the UK Dermatology Clinical Trials Network [17] and the Nottingham Clinical Trials Unit at the University of Nottingham. It is funded by the National Institute for Health Research (NIHR) as part of an NIHR Programme Grant (RP-PG-0407-10177), and sponsored by the Nottingham University Hospitals NHS Trust. Further research nurse support is provided through the NIHR Clinical Research Networks [18].
The trial is being overseen by a Trial Steering Committee, which includes an independent chair and three other independent members (one of whom is a patient with PG). The DMC meets annually (or more frequently as required), and oversee all ethical and safety issues in accordance with current regulations and MRC guidelines for data monitoring committees. All members are independent of the study team, although the Trial Manager and some other members of the Trial Management Group (TMG) attend the open sessions in order to inform the DMC of trial progress. This committee meet at least once a year.
Discussion
The STOP GAP trial is a unique study that could not be achieved without the collaborative efforts of large numbers of participating dermatologists, nurses, NHS Trusts and research networks. Recruitment is taking place at approximately 50 UK (and Ireland) secondary care dermatology departments, all of whom are contributing just 1 or 2 participants per year. Investigators are contributing on a voluntary basis because they believe that this is an important clinical question that will help to inform their clinical practice. Such a recruitment model means that it is not possible to provide dedicated STOP GAP research nurses at individual sites, and that the recruiting dermatologists are sometimes isolated. However, the development of the National Institute for Health Research (NIHR) Clinical Research infrastructure in the England (and CRC Cymru in Wales) means that support is now available to deliver collaborative trials of this kind, and many nurses are available within participating NHS Trusts to assist with patient recruitment and follow-up.
As a model for conducting trials of rare skin conditions, the STOP GAP trial is methodologically interesting in that efforts have been made to include all PG patients who are willing to take part, by inclusion in either the RCT or the observational study. This means that the trial will not only deliver the largest RCT ever to have been conducted in PG patients, but also the largest prospective case series of patients treated with topical therapy.
The use of a parallel observational study alongside the RCT also has advantages for recruitment as it means that patients with mild disease, who are initially considered to be ineligible for the main RCT, remain in contact with the trial team. If topical treatment fails and systemic therapy is indicated, these patients can then be considered for inclusion in the RCT. This approach is particularly useful for rare conditions, where few patients are seen at recruiting centres and the evidence- base for treatment is poor (making a prospective case series more valuable).
Trial status
Recruitment into this trial started in May 2009 and is taking place in approximately 50 secondary care hospitals in the UK and Ireland. It is anticipated that recruitment will be completed by December 2012 at the latest.
References
Craig F, Ormerod A: Pyoderma gangrenosum: pathogenesis, diagnosis, classification and treatment. J Clin Dermatol. 2011, 1: 1-9.
Powell FC, Daniel Su WP, Perry HO: Pyoderma gangrenosum: classification and management. J Am Acad Dermatol. 1996, 34: 395-409. 10.1016/S0190-9622(96)90428-4.
Bennett ML, Jackson JM, Jorizzo JL, Fleischer AB, White WL, Callen J: Pyoderma gangrenosum: a comparison of typical and atypical forms with an emphasis on time to remission. Case review of 86 patients from 2 institutions. Medicine (Baltimore). 2000, 79: 37-46. 10.1097/00005792-200001000-00004.
Powell FC, Schroeter AL, Su WP, Perry HO: Pyoderma gangrenosum: a review of 86 patients. Q J Med. 1985, 55: 173-186.
von den Driesch P: Pyoderma gangrenosum: a report of 44 cases with follow-up. Br J Dermatol. 1997, 167: 1000-1005.
Reichrath J, Bens G, Bonowitz A: Treatment recommendations for pyoderma gangrenosum: an evidence-based review of the literature based on more than 350 patients. J Am Acad Dermatol. 2005, 53: 273-283. 10.1016/j.jaad.2004.10.006.
Prajapati V, Man J, Brassard A: Pyoderma gangrenosum: common pitfalls in management and a stepwise, evidence-based, therapeutic approach. J Cutan Med Surg. 2009, 13: S2-S11.
Brooklyn TN, Dunnill MG, Shetty A, Bowden JJ, Williams JD, Griffiths CE, Forbes A, Greenwood R, Probert CS: Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006, 55: 505-509. 10.1136/gut.2005.074815.
STOP GAP Trial: STOP GAP Trial website. [http://www.stopgaptrial.co.uk]
Lyon CC, Stapleton M, Smith AJ, Mendelsohn S, Beck MH, Griffiths CE: Topical tacrolimus in the management of peristomal pyoderma gangrenosum. J Dermatolog Treat. 2001, 12: 13-17. 10.1080/095466301750163518.
Nybaek H, Olsen AG, Karlsmark T, Jemec GB: Topical therapy for peristomal pyoderma gangrenosum. J Cutan Med Surg: Inc Med Surg Dermatol. 2004, 8: 220-223.
Wunderlich RP, Peters EJ, Armstrong DG, Lavery LA: Reliability of digital videomatery and acetate tracing in measuring the surface area of cutaneous wounds. Diabetes Res Clin Pract. 2000, 49: 87-92. 10.1016/S0168-8227(00)00145-5.
Kantor J, Margolis DJ: A multi-centre study of percentage change in venous leg ulcer area as a prognostic index of healing at 24 weeks. Br J Dermatol. 2000, 142: 960-964. 10.1046/j.1365-2133.2000.03478.x.
Foss CE, Clark AR, Inabinet R, Camacho F, Jorizzo JL: An open-label pilot study of alefacept for the treatment of pyoderma gangrenosum. J Euro Acad Dermatol. 2008, 22: 943-949. 10.1111/j.1468-3083.2008.02680.x.
Finlay AY, Khan GK: Dermatology Life Quality Index (DLQI): a simple practical measure for routine clinical use. Clin Exp Dermatol. 1994, 19: 210-216. 10.1111/j.1365-2230.1994.tb01167.x.
Brookes R: EuroQol - the current state of play. Health Policy. 1996, 37: 53-72. 10.1016/0168-8510(96)00822-6.
UK Dermatology Clinical Trials Network: UK Dermatology Clinical Trials Network's website. [http://www.ukdctn.org]
NIHR Clinical Research Network: NIHR Clinical Research Network website. [http://www.crncc.nihr.ac.uk]
Nottingham Clinical Trials Unit: Nottingham Clinical Trials Unit website. [http://ctu.nottingham.ac.uk]
Acknowledgements
This trial would not have been possible without the enthusiasm and commitment of clinicians, nurses and patients throughout the UK and Eire. Full details of recruiting centres are listed below under contributors. The trial has been developed and continues to be supported by the UK Dermatology Clinical Trials Network [17]; a collaborative group of clinicians, nurses, researchers and patients with an interest in the treatment and prevention of skin disease. Additional nursing support is being been provided by the NIHR Comprehensive Local Research Networks [18]. The trial is being managed through the Nottingham Clinical Trials Unit [19] and sponsored by Nottingham University Hospitals NHS Trust.
Trial management group
FFC, KST, EJM, HCW, JN, JMM, ADO, plus Nicola Greenlaw (statistician, University of Glasgow), Diane Whitham (Deputy Director, Nottingham CTU), Daniel Simpkins (Senior Data Manager, Nottingham CTU), Katharine Foster (STOP GAP Trial Manager, maternity leave cover), Julie Barnes (STOP GAP administrator), Sally Kucyj (Trial Administrator).
Trial steering committee
Independent Chair: Frank Powell (Mater Private Hosp, Dublin), Sarah Meredith, (MRC CTU), Daniel Wallach (Hôpital Tarnier, Paris), Paul Mussell (patient representative, York).
Data monitoring committee
Chair: Julia Schofield (University of Hertfordshire), Angela Crook (MRC CTU), Alison McDonald (CHaRT, Aberdeen), Ian Ford (for preparation of unblinded reports to the DMC, Robertson Centre for Biostatistics).
Recruiting centers
Aberdeen Royal Infirmary, NHS Grampian: Dr Anthony Ormerod (PI), Dr Fiona Craig, Linda Lawson; Aneurin Bevan Health Board: Professor Alex Anstey (PI), Catherine Watkins, Sarah Mitchell, Dr Richard Goodwin, Cilla Benge; Barts & The London NHS Trust: Dr Frances Lawlor (PI); Basildon & Thurrock University Hospitals NHS Foundation Trust: Dr Malgorzata Skibinska (PI), Dr N Ariffin, Janice Armitt, Nhlanhla Mguni, Maxwell Masuku, Kerry Goodsell, Linda Johnson; Betsi Cadwaladr University Health Board: Dr Diane Williamson (PI), Dr Richard Williams, Dr Ewa Turczanska, Dr Alison Devine, Angela Steen, Val Loftus, Corrina Marsden; Cardiff & Vale University Health Board: Dr John Ingram (PI), Dr Girish Patel, Dr Mabs Chowdhury, Dr Richard Motley, Anne Thomas, Dr Colin Long, Dr Andrew Morris, Prof Vincent Piguet, Dr Manju Kalavala, Dr Ru Katugampla; Bradford Teaching Hospitals NHS Foundation Trust: Dr Andrew Wright (PI), Jenny Ott; Brighton & Sussex University Hospitals NHS Trust: Dr Paul Farrant (PI), Mary Flowerdew, Wendy Harman, Dr Lindsay Atkinson, Dr Jessie Felton, Dr Claudia deGiovanni; Cambridge University Hospitals NHS Foundation Trust: Dr Jane Sterling (PI), Dr Moumita Chattopadhyay, Dr Galia Ben-Zvi, Dr Shaheen Haque-Hussain; Chesterfield Royal Hospital NHS Foundation Trust: Dr Francisca Ezughah (PI), Dr Graham Colver, Amanda Whileman, Amanda Gascoigne; City Hospitals Sunderland NHS Foundation Trust: Dr Catherine Blasdale (PI), Dr Stephanie Lateo, Dr Neil Rajan, Anne Thomson, Sivakumar Natarajan; Cork University Hospitals: Dr Johnny Bourke (PI), Dr Maeve McAleer; Countess of Chester Hospital NHS Foundation Trust: Dr Asad Salim (PI), Helen Doyle; County Durham & Darlington NHS Foundation Trust: Dr Shyamal Wahie (PI), Dr Therese Sripathy, Dr Maneesha Vatve, Dr Vrinda Bajaj, Anne Thomson, Dr Keith Freeman, Dr Mary Carr; Craigavon Area Hospital Southern Health & Social Care Trust: Dr Rosemary Black (PI), Dr David Eedy; Derby Hospitals NHS Foundation Trust: Dr Adam Ferguson (PI), Katherine Riches; East Kent Hospitals University NHS Foundation Trust: Dr Emilia Duarte-Williamson (PI), Dr Claire Fuller, Anthea Potter, Laura Brockway, Dr Susannah Baron, Dr Ashley Cooper, Susan Thompson; East Sussex Hospitals NHS Trust: Dr Mahmud Ali (PI), Emma Edmunds; Frimley Park Hospital NHS Foundation Trust: Dr Fiona Antony (PI), Stephanie Atkinson, Lisa Moore, Jean Herzke; Great Western Hospitals NHS Foundation Trust: Dr Sam Gibbs (PI), Dr Deirdre Buckley, Dr Lindsay Whittam, Dr Hartmut Hempel, Dr Mohamed Alrawi, Sue Toft, Jayne Gingell, Jacqueline Arnold; Guys' & St Thomas' NHS Foundation Trust: Dr Catherine Smith (PI), Gemma Minifie, Naomi Hare, Kate Thornberry, Dr Shika Gupta, Dr Sinead Langan; Harrogate & District NHS Foundation Trust: Dr Alison Layton (PI), Angela Wray, Dr Benjamin Walker, Gayle Law, Elizabeth Marshall; Hull & East Yorkshire Hospitals NHS Trust: Dr Shernaz Walton (PI), Katherine Ashton, Angela Oswald, Dr Deborah Graham, Peter Jones, Dr Vanessa Smith; Hywel Dda Health Board: Dr Debbie Shipley (PI), Claire Duggan, Sarah Jones, Carol Thomas, Sally-Ann Rolls, Dr Emma Veysey; James Paget University Hospitals NHS Foundation Trust: Dr Robert Graham (PI), Dr Ingrid Salvary, Susan Simmons; Newcastle Upon Tyne Hospitals NHS Foundation Trust: Dr Simon Meggitt (PI); NHS Lanarkshire Monklands Hospital: Dr Christopher Evans (PI), Suzanne Clements, Gayle Moreland, Margaret Nisbet; Norfolk & Norwich University Hospitals NHS Foundation Trust: Dr Nick Levell (PI), Dr Kevin Lee, Dr Pariyawan Rakvit, Dr George Millington, Karen Banks-Dunnell, Dr Natasha Chetty, Dr Clive Grattan, Dr Syed Shah, Donna Butcher; North Cumbria University Hospitals NHS Trust: Dr Marinela Nik (PI), Kathleen Gilbanks, Dr Neil Cox; Northern Devon Healthcare NHS Trust: Dr Karen Davies (PI), Nick Lawton; Nottingham University Hospitals NHS Trust: Dr John English (PI), Dr Ruth Murphy, Dr William Perkins, Professor Hywel Williams, Dr Sheelagh Littlewood, Dr Jan Bong, Dr Moona Malik, Dr Jonathan Batchelor, Dr Catriona Wootton, Sue Davies-Jones, Joanne Llewellyn, Dr Suzanne Cheng, Dr Maulina Sharma, Dr Janet Angus, Dr Sandeep Varma, Dr Stuart Cohen; Oxford University Hospitals NHS Trust: Dr Graham Ogg (PI), Dr Susan Burge, Dr Vanessa Venning, Dr Susan Cooper, Dr Tess McPherson, Lisa Matter, Gwendoline Reeves; Raigmore Hospital, NHS Highland: Dr James Vestey (PI), Paula Martin, Sue Ross, Charlotte Barr; Royal Berkshire NHS Foundation Trust: Dr Daron Seukeran (PI), Jennie King, Dr Janet Dua, Karen Wilmott; Royal Devon & Exeter NHS Foundation Trust: Dr Christopher Bower (PI), Robert James; South London Healthcare NHS Trust: Dr Anna Chapman (PI), Dr Natalie Miller, Dr Yana Estfan; Taunton & Somerset NHS Foundation Trust: Dr Rachel Wachsmuth (PI), Dr Victoria Lewis; Sandwell & West Birmingham Hospitals NHS Trust: Dr Shireen Velangi (PI), Dr Weronika Szczecinska, Tinomuda Shumba; Sherwood Forest Hospitals NHS Foundation Trust: Dr Jane Ravenscroft (PI), Dr John English, Dr Jan Bong, Dr Azaharry Yaakub, Dr Hong Trinh; South Devon Healthcare NHS Foundation Trust: Dr Alison Clegg (PI), Dr Jill Adams, Sarah Burns, Dr Tessa Frost; The Royal Liverpool & Broadgreen University Hospitals NHS Trust: Dr Hazel Bell (PI), Dr Richard Azurdia, Dr Maeve Walsh, Dr Caroline Angit, Dr Kok Ngan, Andrea Young, Julie Murgaza, Paula Taylor, Dr Hamish Hunter; University Hospitals Birmingham NHS Foundation Trust: Dr Agustin Martin-Clavijo (PI), Dr Renuga Raghavendran, Dr Lucy Evriviades, Dr Helen Lewis; University Hospitals Bristol NHS Foundation Trust: Dr Giles Dunnill (PI), Dr Adam Bray, Dr David De Berker; University Hospitals of Leicester NHS Trust: Dr Graham Johnston (PI), Dr John McKenna, Catherine Shelley, Dr Mohammad Ghazavi, Alison Hill; Weston Area Health NHS Trust: Dr Margaret Kirkup (PI), Glenn Saunders, Hugh Lloyd-Jones, Dawn Simmons, Donna Cotterill; Whipps Cross University Hospital NHS Trust: Dr Anthony Bewley (PI), Michael Galivo, Jane Watts, Dr Karen Gibbon, Dr Anshoo Sahota; Yeovil District Hospital NHS Foundation Trust: Dr Rachel Wachsmuth (PI), Michelle Davey; York Teaching Hospital NHS Foundation Trust: Dr Calum Lyon (PI), Jill Green, Dr Julia Stainforth.
Funding
This paper presents independent research commissioned by the National Institute for Health Research (NIHR) under its Programme Grants for Applied Research funding scheme (RP-PG-0407-10177). Pilot work for the STOP GAP trial was funded by the British Skin Foundation (grant reference: S317).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors contributed to the design and writing of the full STOP GAP protocol and are members of the Trial management Group. FC and KT wrote the first draft of this paper; all authors commented and amended drafts of the paper and approved the final version. All authors read and approved the final manuscipt.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Craig, F.F., Thomas, K.S., Mitchell, E.J. et al. UK Dermatology Clinical Trials Network’s STOP GAP trial (a multicentre trial of prednisolone versus ciclosporin for pyoderma gangrenosum): protocol for a randomised controlled trial. Trials 13, 51 (2012). https://doi.org/10.1186/1745-6215-13-51
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1745-6215-13-51