
Abstract
Background
The cause of almost all cases of Parkinson’s disease (PD) remains unknown. Recent years have seen an explosion in the rate of discovery of genetic defects linked to PD. Different racial and geographical populations may have different distributions of genetic variants.
Methods
In the current study, we screened the following genetic variants, including some rare mutations and single nucleotide polymorphisms (SNPs), in a pedigree and cases-controls. To best of our knowledge, we first screened these variants known to be associated with neurodegeneration disease, E46K (rs104893875) in SNCA, A1442P in LRRK2, IVS9 in PARK2, A350V in SLC41A1, P268S (rs2066842), R702W (rs2066844), G908R (rs2066845), 1007fs (rs2066847) in NOD2 and G2385R (rs34778348) in LRRK2 from southern China population. Genotyping was performed by jointly using primers overlapping polymerase chain reaction (PCR) site-directed mutagenesis, restriction fragment length polymorphism (RFLP), and capillary electrophoresis (CE).
Results
We didn’t discover above 9 variants in the family members of the pedigree. Furthermore, of 237 patients with sporadic Parkinson’s disease and 190 controls, no heterozygosity or homozygosity were found from E46K, A1442P, A350V, R702W, G908R, or 1007fs but heterozygosity onto G2385R, IVS9, and P268S. No significant difference between cases and controls was found in both allele frequency (P = 0.572) and genotype frequency (P = 0.348) of IVS9. However, significant differences in genotype frequency (P = 0.009) of G2385R were consistent with prior observation. Eight patients with Parkinson’s disease (2 women and 6 men are over the age of 50 years at onset of PD) carried the P268S heterozygous variation in NOD2. There was no heterozygosity or homozygosity of P268S in the controls. Genotype frequency of P268S (P = 0.0450) had significant differences.
Conclusions
Our results suggested that the P268S variant in NOD2 might be a risk factor for susceptibility to sporadic Parkinson’s disease in Chinese populations. It also implied that the inflammatory response may play a role in PD.
Similar content being viewed by others


Background
Parkinson’s disease (PD) is a complex neurodegenerative disease caused by a variety of factors. The incidence in the population over the age of 65 is about 1.8% [1]. The major clinical features of PD include resting tremor, bradykinesia and rigidity [2]. The pathological features of PD include progressive loss of dopaminergic neurons from the substantia nigra pars compacta and the presence of intercellular Lewy bodies in surviving neurons [3]. The cause of PD is unclear, but it is generally considered to be associated with the impairment in degradation of ubiquitin proteasome system (UPS), mitochondrial dysfunction, and oxidative stress [4–6]. We hypothesize that genetic and environmental factors and aging together lead to the development of PD. In this study, we investigated five genes, including SNCA, LRRK2, PARK2, SLC41A1, and NOD2, all of which might be involved in the cytological mechanisms of neurodegeneration disease aiming to identify genetic variants associated with sporadic PD in Chinese patients and understand the genetic etiology of PD.
The first gene to be identified as associated with PD was SNCA (PARK1/4), which encodes α-synuclein. The major mutations in SNCA include A53T, A30P, and E46K [7–9]. Although these mutations account for less than 1% of the cases, patients carrying these mutations have obviously clinical phenotypes [8]. The A53T and A30P mutations have been found in Chinese patients with sporadic PD [10]. However, the association of E46K with sporadic PD has not been reported in Chinese patients.
LRRK2 (PARK8) encodes a large protein containing five functional domains, involved in a number of physiological functions, including substrate binding, protein phosphorylation and protein interactions [11]. The most common mutation G2019 of LRRK2 in European populations presents less than 0.1% in Asian individuals [12]. The G2385R was at a significantly higher frequency in Asian patients than in controls [13]. One of the purposes of this study is to re-validate this variant in southern China. In addition, Yue Huang found A1442P in pedigrees of the Australian patients and considered it pathogenic, because it is conserved across many species and a substitution of Ala with Pro can change the secondary structure of proteins [14]. However, A1442P has not been further confirmed from a large number of cases and other populations.
The most common cause of young-onset sporadic PD is autosomal recessive PARK2 mutation [15]. The E3 ubiquitin ligase, parkin, which is encoded by PARK2, can specifically degrade UPS through tagging ubiquitin on protein [16]. Exon deletion, insertion, and point mutations in PARK2 have been found in different ethnic groups [17]. In 2009, Yih-Ru Wu screened 506 Taiwan sporadic patients with age of onset below 50 years for PARK2 gene mutation and identified a novel IVS9 insertion (c.1084intron+) [18]. The c.1084intron+ was due to a G > A polymorphism at position −6 of a cryptic splice acceptor site within IVS9.
In recent years, genome-wide association studies (GWAS) have identified a number of new susceptibility loci associated with PD in different ethnic groups. Of these loci, the most striking locus is PARK16, which is located in 1q23 and contains 5 genes (SLC45A3, NUCKS1, RAB7L1, SLC41A1, and PM20D1) [19]. SLC41A1 encodes a 56 kDa Mg2+ transporter consisting of 513 amino acids, it has been proposed that PD might be associated with lack of Ca2+ and Mg2+ in the brain [20]. In 2010, Arianna Tucci sequenced PARK16 in 182 patients with PD in the United Kingdom and found A350V in SLC41A1, but failed to detect it in a large series of ethnicity-matched controls (n = 483) [19]. To inspect whether this coding mutations may or may not be the disease-causing mutation, we aim to screen A350V in Chinese Han populations.
Several obtained findings suggest that inflammation also contributes to the pathogenesis of PD [21–23]. It has been reported that the G174C variant in the IL-6 promoter may influence the risk for developing PD, particularly regarding early age of onset PD [24]. Recent studies revealed that NF-κB-mediated inflammation might also play an important role in the pathogenesis of PD [25, 26]. NOD (nucleotide-binding oligomerization domain) proteins, i.e. NOD1 (a product of CARD4 gene) and NOD2 (encoded by NOD2), are intracellular signaling molecules that recognize bacterial components, mediate the activation of NF-κB and induce or enhance apoptosis [27]. In 2007, Monika Bialecka et al. screened 308 Portland patients for 3 variants (R702W, G908R, and 1007fs) in NOD2 which were associated with Crohn’s disease (CD) in Europeans. They found that NOD2 might be associated with susceptibility to PD [28]. Although inflammatory response has long been considered as one of the factors for PD development, it has not yet been reported that R702W, G908R, 1007fs and P268S in NOD2 is associated with Chinese PD patients.
In this study, we screened 237 patients with sporadic PD and 190 controls for the 9 variants, E46K (rs104893875), G2385R (rs34778348), A1442P, IVS9, A350V, R702W (rs2066844), G908R (rs2066845), 1007fs (rs2066847) and P268S (rs2066842), in 5 genes which are possibly found to be associated with Chinese PD patients, including SNCA, LRRK2, PARK2, SLC41A1, and NOD2. These variants included both rare mutations (e.g. E46K and A1442P) and SNPs, so we also screened them in a pedigree with two PD cases. In this way, we aimed to provide references for the study of disease-causing or susceptibility gene for PD. In addition, using the overlap extension polymerase chain reaction (PCR)-based site-directed mutagenesis, we constructed wild-type and homozygous mutant plasmids for these variants, which were used as controls to determine variants in this study.
Material and methods
Study subjects
A total of 237 sporadic patients with PD (94 women and 143 men) and a pedigree with two PD cases were recruited from Xiamen First Hospital, Fujian Province, China. The average age of enrollment was 60.3 ± 11.3, ranging from 25 to 83 years. Their average age of onset was 56.4 ± 10.8 years, ranging from 23 to 80 years. Of these patients, 51 (21.5%) had early-onset PD (EOPD), defined as the age at onset <50 years; 186 (78.5%) cases had late-onset PD (LOPD), with age of onset ≥ 50 years. Patients were independently diagnosed by two neurologists. Diagnostic criteria were taken from the United Kingdom PD Brain Bank [29]. The severity of the disease was determined using the Unified Parkinson’s Disease Rating Scale (UPDRS). The control group consisted of 190 individuals (75 women and 115 men), with an average age of 51.7 ± 9.8 years (ranging from 23 to 80 years). The cases and controls were matched with respect to age, gender, and place of residence. This study was approved by the Xiamen First Hospital Ethics Committee. Informed consent was obtained from all participants.
Genomic DNA extraction
Five milliliters of peripheral blood with ethylene diamine tetraacetic acid (EDTA) anticoagulant was collected from each participant. Genomic DNA was extracted using the MagCore Genomic DNA Whole Blood Kit and HF-16 extractor (Cat. No. MGB400-04, RBC Bioscience Taiwan) according to the manufacturer’s instructions and stored all samples at −20°C before use.
PCR amplification
Primers (Table 1) were designed using the software Primer Premier 5 according to the change of restriction sites, caused by the mutation. Primers were synthesized by Sangon Biotech (Shanghai, China).
Restriction endonuclease analysis
Target variants were detected using the PCR-RFLP (restriction fragment length polymorphism) method. The digestion reaction and restriction enzyme digestion conditions were taken from the manuals included with the restriction enzymes purchased from the New England BioLab (United States). Then 20 μl of digestion products were loaded on 2% agarose gel containing ethidium bromide, electrophoresed under 100 V for 30 min in 1× TAE, and imaged using the gel imager. Digestion results of homozygous mutants, heterozygous mutants, and wild-type are shown in Table 1.
DNA sequencing
Based on the results from PCR-RFLP and site-directed mutagenesis, the target fragments were re-amplified using PCR. PCR products were purified using a DNA gel purification kit, sequenced using the dideoxy four-color fluorescence method on an automatic sequencer (ABI, U.S.), and compared using GenBank data to confirm the mutation sites.
Capillary electrophoresis (CE)
In this study, we initially established the test method using CE to detect gene polymorphisms for PD. Briefly, based on the susceptibility gene polymorphisms of PD identified from PCR-RFLP, restriction fragments were detected using capillary electrophoresis (BIOptics Qsep100 dna-CE, Taiwan), which used a molten molding silica capillary column with an inner diameter of 75 μl and column length of 150 mm and Mops-Tris buffer of pH 7.55. Then 0.5 μl of sample was electrically injected in 4kV × 15 s at 25°C, electrophoresed at a constant voltage of 8 Kv, and detected for the laser-induced ethidium bromide emission wavelength at 590 nm. The gel electrophoresis profiles were compared to determine the genotypes.
Statistical analysis
SPSS 18.0 (Statistical Package for the Social Sciences, version 18.0 for Windows) was used for statistical analysis. Allele and genotype frequencies were compared between cases and controls using the Fisher’s exact test. To avoid multiple easily false, false discovery rate was used to correct p values. Odds ratios and 95% confidence intervals were calculated. A two-sided P value ≤0.05 was considered statistically significant. Power was calculated by Power and Sample Size Calculations Version 3.0, 2009.
Results
Pedigree
Figure 1 shows the pedigree (2 generations) of the family that we studied. The age of PD onset for the affected individuals 3 and 4 in the kindred is 45 and 40, respectively. We failed to discover any mutations of the 9 genetic variants, including rare mutations E46K and A1442P originally found in the kindred. It may be caused by low family members to detect the association. Further studies using a larger Chinese pedigree with PD patients are required to determine this.

Pedigree of Parkinson’s disease (PD). Circle, woman; square, man; filled square, affected.
Case–control study
We analyzed 9 variants, E46K, G2385R, A1442P, IVS9, A350V, P268S, R702W, G908R, and 1007fs in 237 sporadic patients with PD and 190 controls using the PCR-RFLP and CE. All the genotypic and allelic distributions analyzed in this study were in accordance with the Hardy–Weinberg equilibrium. No heterozygosity or homozygosity of E46K, A1442P, A350V, R702W, G908R, or 1007fs was found. Heterozygosity of G2385R, IVS9, and P268S were found in the case groups and controls (Figure 2). The genotype frequency, allele frequency, odds ratios, and confidence interval of these variants are shown in Table 2.

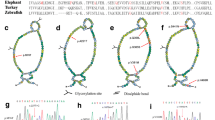
Capillary electrophoresis (CE) results of restriction fragments of 3 variants. a, c, e and b, d, f are the electropherograms and gel-views of IVS9, P268S, and G2385R after restriction digestion, respectively. The green arrows indicate markers (20 bp, 1000 bp) and black arrows indicate the restriction fragments. Restriction fragment sizes are shown in the Table 1: No mutation (wild-type/negative control), 2: Negative results in the control group, 3: Positive results (heterozygote) in the case group, 4: Homozygous mutation (homozygote/positive control). Because the signals are too strong for IVS9-4, P268S-1, P268S-2, G2385R-1, and G2385-2, 10 times markers are used to enhance the signal.
The distribution of G2385R heterozygosity in LRRK2 was significantly different between cases and controls (10.1% vs. 2.1%, P = 0.009). The intron IVS9 heterozygous variant in PARK2 was found in both the case group (12.7%) and the control group (7.9%). Although this variant had a higher frequency in the case group than the control group, the difference was not statistically significant (P = 0.348).
Although only 8 out of 237 patients were found to carry the P268S heterozygous variant in NOD2, all these patients (2 women and 6 men) had age at onset of over 50 years. Since no controls were found to carry this variant, the difference between cases and controls was slightly significant (P = 0.0450). These results suggest that the variant P268S in NOD2 might be associated with late-onset PD and is one of the risk factors for sporadic PD in Chinese Han populations.
Discussion
Gene polymorphism is a risk factor of PD, and it shows different distributions in different ethnic groups and geographies. Although GWAS have identified a number of susceptibility genes and relevant variants associated with PD in different populations, few of them have been analyzed in other populations. To best of out knowledge, we first screened 8 variants that have been associated with neurodegeneration, E46K, A1442P, A350V, IVS9, P268S, R702W, G908R, and 1007fs in Chinese populations, and further verified the presence of the high-frequency variant G2385R in southern China.
In case–control study, both PD patients and controls carried the heterozygous G2385R mutation, and the genotype difference was slightly significant. This is consistent with the results of a previous study of 600 Chinese PD patients and 334 controls [30]. This variant was initially reported by Xingkai An. in 2008 among PD patients in Sichuan area [30]. However, we did not find E46K, A1442P, or A350V variants in patients with PD and in controls. All of them indicated following phenomenon: the ancestry effect; familial PD results from rare, highly penetrant pathogenic mutation (e.g. E46K, A1442P); multiple variants of low penetrant (e.g. A350V) contribute to the risk of PD and are involved in the etiology of PD.
Interestingly, none of homozygosity of these variants was found in either the case or control groups [31, 32]. It was reported that heterozygosity of autosomal recessive genes was very important in the initiation and development of PD. For example, it has been found that patients with PD who carry heterozygous PARK2 and PINK1 mutations have an age of onset between that of wild-type individuals and patients with homozygous mutations [33, 34]. Heterozygous mutation carriers indeed show preclinical changes at the metabolic, structural, or functional levels that are detectable by modern techniques of neuroimaging and electrophysiology. Heterozygosity for putative recessive mutations could lead to disease expression by at least three mechanisms which are haploinsuffciency, dominant-negative effect and novel gain-of-function [35]. Taken together, heterozygous mutations in putative recessive genes seem to increase the susceptibility to develop PD.
The new insertion mutation, c.1084intron+ in PARK2, introduces two new amino acids and a TAA stop codon, resulting in reduced levels of parkin protein [36]. This insertion mutation may be associated with the -6G > A polymorphism of intron 9 in PARK2. One case–control study found IVS9G > A to be a risk factor of PD in Taiwan. A functional study of IVS9G > A by Guey-Jen Lee-Chen showed that the shear efficiency of -6A was slower than that of -6G at the protein level [37]. 12.7% PD patients and 7.9% controls carried the heterozygous IVS9 mutation, but the allele difference and genotype difference were not statistically significant. This result is consistent with the results of a previous study of 506 patients and 508 controls in Taiwan [18]. Further investigation into the association between IVS9G > A and PD need, through larger sample size from various populations.
Inflammation is one of the suspected theories of PD. The activation of NF-κB and microglial cells around dopaminergic neurons have been analyzed in patients with PD [38]. In 2007, Monika Bialecka reported that three variants (R702W, G908R, and L1007fsinsC) of NOD2 encoding NOD2, a protein that can activate the apoptosis response, were significantly associated with PD in a Polish population [28]. It has been demonstrated that these three variants are not associated with CD in Chinese Han, Korean, and Japanese populations [39–42]. Similarly, these three common SNPs (R702W, G908R, and 1007fs) were not detected in our cohort. However, a novel variant P268S was observed, which has also been discovered to be associated with CD in Chinese population [43]. Although linkage disequilibrium was observed between P268S and the other three SNPs in NOD2, only P268S of the four variants was slightly significant in our study, which was consistent with recent study suggesting that P268S instead of 1007fs, G908R, R702W was a risk factor of Chinese Crohn’s disease population. It indicated that P268S may be one of risk factors for PD in Chinese individuals, or the P268S variant may be in linkage disequilibrium with another causal rare variant on NOD2 gene. The frequencies of minor allele 1007fs, G908R, R702W and P268S variants are much lower in Asians compare to Caucasians populations. In this study, genotype frequency of P268S (P = 0.0450) had slightly significant differences. The sample size has to be very large or odds ratio has to be very high to have enough power for detection effect of very rare variants.
NOD2 is a member of the Apaf-1 superfamily of apoptosis regulators that is expressed in monocytes and involved in the activation of NF-κB, by bacterial component muramyl dipeptide (MDP) [44]. Functional studies revealed that both normal and P268S NOD2 induced similar levels of NF-κB activation in response to MDP [45] and the G908R, R702W, and L1007fsinsC variants in the presence and absence of P268S are defective in their ability to respond to bacterial lipopolysaccharides and peptidoglycan, whereas P268S alone exhibited wild-type activity [46]. Thus, P268S was confirmed as the haplotype background of these three variants but has no influence on functions of the CD-associated variants of NOD2[47]. Notably, above results from NF-κB activation assay using human embryonic kidney (HEK) 293 cell lines, but the function activity of NOD2 in neurodegeneration field remain unclear. Additionally, Crane et al. found that carriage of the P268S variant was associated with greater disease activity and inversely associated with ulcerative colitis spondylarthritis [48]. We regarded the P268S of NOD2 behaving as a common factor of PD and CD. Crohn’s disease is a chronic inflammatory disorder of the gastrointestinal tract, which is thought to result from the effect of environmental factors in a genetically predisposed host [49]. A wealth of new information has emerged to suggest that inflammation-derived oxidative stress and cytokine dependent toxicity may contribute to nigrostriatal pathway degeneration and hasten progression of disease in humans with idiopathic PD. Both them suggest that inflammation may play a role connecting between PD and CD.
Conclusions
We analyzed the genetic loci associated with PD in the sporadic and family PD patients using PCR-RFLP and found P268S might be a risk factor of sporadic PD in the Chinese population. As it is the first study on NOD2 P268S in PD patients, the results should be treated with caution, as one cannot completely exclude that they are false positive. Further studies required to verify the association between P268S and susceptibility to sporadic Parkinson’s disease in large sample size, and explore the NOD2 functions involved in neurodegenerative diseases.
Authors’ information
QLM: MD, chief physician of the Department of Neurology of the First Affiliated Hospital of Xiamen University; senior visiting scholar at Harvard Medical School, Center of Neurological Diseases; Member of the World Stroke Organization (WSO). XKA: Resident of the Department of Neurology of Xiamen First Hospital; part of the team that first revealed the association of G2385R and the LRRK2 gene with PD in Chinese Han populations. QL: MD, associate-chief physician of the Department of Neurology of the he First Affiliated Hospital of Xiamen University. ZML: Master student, School of Pharmaceutical Sciences Xiamen University; participated in several projects on screening the disease genes for PD of Chinese Han population. CMT: PhD in Physical Biochemistry, Nuclear Science, National Tsing-Hua University, Postdoctoral Fellow in Biochemistry, Stanford University School of Medicine, School of Pharmaceutical Sciences; Executive Director Professor of Center for Translational Medicine, Xiamen University.
Abbreviations
- PD:
-
Parkinson’s disease
- SNPs:
-
Single nucleotide polymorphisms
- UPS:
-
Ubiquitin proteasome system
- PCR:
-
Polymerase chain reaction
- RFLP:
-
Restriction fragment length polymorphism
- CE:
-
Capillary electrophoresis
- GWAS:
-
Genome-wide association studies
- CD:
-
Crohn’s disease
- NOD:
-
Nucleotide-binding and oligomerization domain
- EOPD:
-
Early-onset PD
- LOPD:
-
Late-onset PD
- UPDRS:
-
Unified parkinson’s disease rating scale
- EDTA:
-
Ethylene diamine tetraacetic acid
- SPSS:
-
Statistical package for the social sciences
- OR:
-
Odds ratios
- CI:
-
Confidence intervals
- MDP:
-
Muramyl dipeptide
- NFAT:
-
Nuclear factor of activated T-cells.
References
Zhang ZX, Roman GC, Hong Z, Wu CB, Qu QM, Huang JB, Zhou B, Geng ZP, Wu JX, Wen HB: Parkinson’s disease in China: prevalence in Beijing, Xian, and Shanghai. Lancet. 2005, 365 (9459): 595-597.
Fahn S: Description of Parkinson’s disease as a clinical syndrome. Ann N Y Acad Sci. 2003, 991: 1-14.
Forno LS: Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol. 1996, 55 (3): 259-272.
Dauer W, Przedborski S: Parkinson’s disease: mechanisms and models. Neuron. 2003, 39 (6): 889-909. 10.1016/S0896-6273(03)00568-3.
Dawson TM, Dawson VL: Molecular pathways of neurodegeneration in Parkinson’s disease. Science. 2003, 302 (5646): 819-822. 10.1126/science.1087753.
Cookson MR: The biochemistry of Parkinson’s disease. Annu Rev Biochem. 2005, 74: 29-52. 10.1146/annurev.biochem.74.082803.133400.
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R: Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997, 276 (5321): 2045-2047. 10.1126/science.276.5321.2045.
Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B: The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004, 55 (2): 164-173. 10.1002/ana.10795.
Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O: Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998, 18 (2): 106-108. 10.1038/ng0298-106.
Chan DK, Mellick G, Cai H, Wang XL, Ng PW, Pang CP, Woo J, Kay R: The alpha-synuclein gene and Parkinson disease in a Chinese population. Arch Neurol. 2000, 57 (4): 501-503. 10.1001/archneur.57.4.501.
Mata IF, Wedemeyer WJ, Farrer MJ, Taylor JP, Gallo KA: LRRK2 in Parkinson’s disease: protein domains and functional insights. Trends Neurosci. 2006, 29 (5): 286-293. 10.1016/j.tins.2006.03.006.
Tan EK, Shen H, Tan LC, Farrer M, Yew K, Chua E, Jamora RD, Puvan K, Puong KY, Zhao Y: The G2019S LRRK2 mutation is uncommon in an Asian cohort of Parkinson’s disease patients. Neurosci Lett. 2005, 384 (3): 327-329. 10.1016/j.neulet.2005.04.103.
Funayama M, Li Y, Tomiyama H, Yoshino H, Imamichi Y, Yamamoto M, Murata M, Toda T, Mizuno Y, Hattori N: Leucine-rich repeat kinase 2 G2385R variant is a risk factor for Parkinson disease in Asian population. Neuroreport. 2007, 18 (3): 273-275. 10.1097/WNR.0b013e32801254b6.
Huang Y, Halliday GM, Vandebona H, Mellick GD, Mastaglia F, Stevens J, Kwok J, Garlepp M, Silburn PA, Horne MK: Prevalence and clinical features of common LRRK2 mutations in Australians with Parkinson’s disease. Mov Disord. 2007, 22 (7): 982-989. 10.1002/mds.21477.
Lucking CB, Durr A, Bonifati V, Vaughan J, Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW: Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med. 2000, 342 (21): 1560-1567. 10.1056/NEJM200005253422103.
Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K: Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000, 25 (3): 302-305. 10.1038/77060.
Mata IF, Lockhart PJ, Farrer MJ: Parkin genetics: one model for Parkinson’s disease. Hum Mol Genet. 2004, 13: R127-R133. 10.1093/hmg/ddh089. Spec No 1
Wu YR, Wu CH, Chao CY, Kuan CC, Zhang WL, Wang CK, Chang CY, Chang YC, Lee-Chen GJ, Chen CM: Genetic analysis of Parkin in early onset Parkinson’s disease (PD): Novel intron 9 g > a single nucleotide polymorphism and risk of Taiwanese PD. Am J Med Genet B Neuropsychiatr Genet. 2010, 153B (1): 229-234.
Tucci A, Nalls MA, Houlden H, Revesz T, Singleton AB, Wood NW, Hardy J, Paisan-Ruiz C: Genetic variability at the PARK16 locus. Eur J Hum Genet. 2010, 18 (12): 1356-1359. 10.1038/ejhg.2010.125.
Kolisek M, Launay P, Beck A, Sponder G, Serafini N, Brenkus M, Froschauer EM, Martens H, Fleig A, Schweigel M: SLC41A1 is a novel mammalian Mg2+ carrier. J Biol Chem. 2008, 283 (23): 16235-16247. 10.1074/jbc.M707276200.
Wyss-Coray T, Mucke L: Inflammation in neurodegenerative disease–a double-edged sword. Neuron. 2002, 35 (3): 419-432. 10.1016/S0896-6273(02)00794-8.
McGeer PL, McGeer EG: Inflammation and neurodegeneration in Parkinson’s disease. Parkinsonism Relat Disord. 2004, 10 (Suppl 1): S3-S7.
Kim YS, Joh TH: Microglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson’s disease. Exp Mol Med. 2006, 38 (4): 333-347. 10.1038/emm.2006.40.
Hakansson A, Westberg L, Nilsson S, Buervenich S, Carmine A, Holmberg B, Sydow O, Olson L, Johnels B, Eriksson E: Interaction of polymorphisms in the genes encoding interleukin-6 and estrogen receptor beta on the susceptibility to Parkinson’s disease. Am J Med Genet B Neuropsychiatr Genet. 2005, 133B (1): 88-92. 10.1002/ajmg.b.30136.
Kaltschmidt B, Heinrich M, Kaltschmidt C: Stimulus-dependent activation of NF-kappaB specifies apoptosis or neuroprotection in cerebellar granule cells. Neuromolecular Med. 2002, 2 (3): 299-309. 10.1385/NMM:2:3:299.
Hirsch EC, Hunot S: Neuroinflammation in Parkinson’s disease: a target for neuroprotection?. Lancet Neurol. 2009, 8 (4): 382-397. 10.1016/S1474-4422(09)70062-6.
Inohara N, Nunez G: NODs: intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol. 2003, 3 (5): 371-382. 10.1038/nri1086.
Bialecka M, Kurzawski M, Klodowska-Duda G, Opala G, Juzwiak S, Kurzawski G, Tan EK, Drozdzik M: CARD15 variants in patients with sporadic Parkinson’s disease. Neurosci Res. 2007, 57 (3): 473-476. 10.1016/j.neures.2006.11.012.
Hughes AJ, Daniel SE, Kilford L, Lees AJ: Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992, 55 (3): 181-184. 10.1136/jnnp.55.3.181.
An XK, Peng R, Li T, Burgunder JM, Wu Y, Chen WJ, Zhang JH, Wang YC, Xu YM, Gou YR: LRRK2 Gly2385Arg variant is a risk factor of Parkinson’s disease among Han-Chinese from mainland China. Eur J Neurol. 2008, 15 (3): 301-305. 10.1111/j.1468-1331.2007.02052.x.
Abou-Sleiman PM, Muqit MM, McDonald NQ, Yang YX, Gandhi S, Healy DG, Harvey K, Harvey RJ, Deas E, Bhatia K: A heterozygous effect for PINK1 mutations in Parkinson’s disease?. Ann Neurol. 2006, 60 (4): 414-419. 10.1002/ana.20960.
Klein C: Implications of genetics on the diagnosis and care of patients with Parkinson disease. Arch Neurol. 2006, 63 (3): 328-334. 10.1001/archneur.63.3.328.
Hedrich K, Marder K, Harris J, Kann M, Lynch T, Meija-Santana H, Pramstaller PP, Schwinger E, Bressman SB, Fahn S: Evaluation of 50 probands with early-onset Parkinson’s disease for Parkin mutations. Neurology. 2002, 58 (8): 1239-1246. 10.1212/WNL.58.8.1239.
Sun M, Latourelle JC, Wooten GF, Lew MF, Klein C, Shill HA, Golbe LI, Mark MH, Racette BA, Perlmutter JS: Influence of heterozygosity for parkin mutation on onset age in familial Parkinson disease: the GenePD study. Arch Neurol. 2006, 63 (6): 826-832. 10.1001/archneur.63.6.826.
Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE: Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 2007, 6 (7): 652-662. 10.1016/S1474-4422(07)70174-6.
Matsuda N, Kitami T, Suzuki T, Mizuno Y, Hattori N, Tanaka K: Diverse effects of pathogenic mutations of Parkin that catalyze multiple monoubiquitylation in vitro. J Biol Chem. 2006, 281 (6): 3204-3209.
Wan-Ling Zhang C-HW, Tsu-Wei W, Lee-Chen G-J: Functional Analysis of Parkin Gene Intron 9 g > a SNP. Bio Formosa. 2010, 45 (1): 31-38.
Hunot S, Brugg B, Ricard D, Michel PP, Muriel MP, Ruberg M, Faucheux BA, Agid Y, Hirsch EC: Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with parkinson disease. Proc Natl Acad Sci USA. 1997, 94 (14): 7531-7536. 10.1073/pnas.94.14.7531.
Gao M, Cao Q, Luo LH, Wu ML, Hu WL, Hu WL, Si JM: [NOD2/CARD15 gene polymorphisms and susceptibility to Crohn’s disease in Chinese Han population]. Zhonghua Nei Ke Za Zhi. 2005, 44 (3): 210-212.
Croucher PJ, Mascheretti S, Hampe J, Huse K, Frenzel H, Stoll M, Lu T, Nikolaus S, Yang SK, Krawczak M: Haplotype structure and association to Crohn’s disease of CARD15 mutations in two ethnically divergent populations. Eur J Hum Genet. 2003, 11 (1): 6-16. 10.1038/sj.ejhg.5200897.
Yamazaki K, Takazoe M, Tanaka T, Kazumori T, Nakamura Y: Absence of mutation in the NOD2/CARD15 gene among 483 Japanese patients with Crohn’s disease. J Hum Genet. 2002, 47 (9): 469-472. 10.1007/s100380200067.
Leong RW, Armuzzi A, Ahmad T, Wong ML, Tse P, Jewell DP, Sung JJ: NOD2/CARD15 gene polymorphisms and Crohn’s disease in the Chinese population. Aliment Pharmacol Ther. 2003, 17 (12): 1465-1470. 10.1046/j.1365-2036.2003.01607.x.
Lv C, Yang X, Zhang Y, Zhao X, Chen Z, Long J, Zhong C, Zhi J, Yao G, Jiang B: Confirmation of three inflammatory bowel disease susceptibility loci in a Chinese cohort. Int J Colorectal Dis. 2012, 27 (11): 1465-1472. 10.1007/s00384-012-1450-6.
Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G: Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. 2001, 276 (7): 4812-4818. 10.1074/jbc.M008072200.
Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, Fukase K, Inamura S, Kusumoto S, Hashimoto M: Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003, 278 (8): 5509-5512. 10.1074/jbc.C200673200.
Bonen DK, Ogura Y, Nicolae DL, Inohara N, Saab L, Tanabe T, Chen FF, Foster SJ, Duerr RH, Brant SR: Crohn’s disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology. 2003, 124 (1): 140-146. 10.1053/gast.2003.50019.
Chamaillard M, Philpott D, Girardin SE, Zouali H, Lesage S, Chareyre F, Bui TH, Giovannini M, Zaehringer U, Penard-Lacronique V: Gene-environment interaction modulated by allelic heterogeneity in inflammatory diseases. Proc Natl Acad Sci USA. 2003, 100 (6): 3455-3460. 10.1073/pnas.0530276100.
Crane AM, Bradbury L, Van Heel DA, McGovern DP, Brophy S, Rubin L, Siminovitch KA, Wordsworth BP, Calin A, Brown MA: Role of NOD2 variants in spondylarthritis. Arthritis Rheum. 2002, 46 (6): 1629-1633. 10.1002/art.10329.
Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH: A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001, 411 (6837): 603-606. 10.1038/35079114.
Acknowledgements
We would like to thank all the patients and control subjects who participated in this study. We would like to thank Professor Chi-Meng Tzeng for his guidance and the members of our laboratory, Huiming Ye, Xiaonan Lv, Yi Li, and Mingjie Lei for their help. This study was supported by Fujian Province Natural Science Foundation Association of glucocerebrosidase gene mutations with Parkinson’s disease in Chinese Han patients (No. 2009D001), Fujian Province Health Department Youth Research Projects Association of glutamate transporter gene and patients with Parkinson’s disease (No. 2009-2-72) and Fujian Province Science and Technology Department Key Project of The Screening of Susceptibility Gene in Parkinson’s Disease and The Platform Establishment of Molecular Diagnosis (No. 2012D062).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
QLM, XKA, and QL collected the blood samples from PD patients and controls, extracted genomic DNA, and searched relevant literature. ZML performed PCR, RFLP, and CE analysis and built positive and negative control plasmids. HJZ and LLC designed the primers for PCR, cloning, and site-directed mutagenesis and performed sequence alignment. WQH and PH participated in statistical analysis and the production of tables. CMT designed all the experiments and drafted the manuscript. All authors read and approved the final manuscript.
Qilin Ma, Xingkai An, Zhiming Li contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ma, Q., An, X., Li, Z. et al. P268S in NOD2 associates with susceptibility to Parkinson’s disease in Chinese population. Behav Brain Funct 9, 19 (2013). https://doi.org/10.1186/1744-9081-9-19
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1744-9081-9-19