
Abstract
Transient receptor potential vanilloid type 1 (TRPV1) receptor is a non selective ligand-gated cation channel activated by capsaicin, heat, protons and endogenous lipids termed endovanilloids. As well as peripheral primary afferent neurons and dorsal root ganglia, TRPV1 receptor is also expressed in spinal and supraspinal structures such as those belonging to the endogenous antinociceptive descending pathway which is a circuitry of the supraspinal central nervous system whose task is to counteract pain. It includes periaqueductal grey (PAG) and rostral ventromedial medulla (RVM) whose activation leads to analgesia. Such an effect is associated with a glutamate increase and the activation of OFF and inhibition of ON cell population in the rostral ventromedial medulla (RVM). Activation of the antinociceptive descending pathway via TPRV1 receptor stimulation in the PAG may be a novel strategy for producing analgesia in chronic pain. This review will summarize the more recent insights into the role of TRPV1 receptor within the antinociceptive descending pathway and its possible exploitation as a target for new pain-killer agents in chronic pain conditions, with particular emphasis on the most untreatable pain state: neuropathic pain.
Similar content being viewed by others

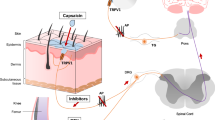

TRPV1 receptor: a member of TRP family channels
TRP ion channels described for first time in Drosophila melanogaster [1] are ion channels that respond to mechanical, thermal, chemical (i.e. acid, lipids) and many other stimuli coming from the extra and intracellular milieu [2–5]. The TRP channel family contains seven divisions: TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPA (ankyrin), TRPP (policystin) and TRPML (mucolipin) [2, 6–8]. TRPV1, however, remains the most studied and best characterized TRP family member, due to the fact that it has been implicated in a wide variety of cellular and physiological processes, including noxious physical and chemical stimuli detection, making it a promising target for pain-relieving drugs acting exactly where pain originates.
The TRPV1 channel consists of six transmembrane domains assembled as homo or hetero-tetramers with each sub-unit contributing to the cation channel structure [9–11]. It is activated by capsaicin, the pungent ingredient found in the hot chilli pepper [12], resiniferatoxin (RTX), a highly irritant diterpene ester isolated from Euphorbia resinifera [13], noxious heat (> 43°C), low pH (5.2) [12, 14], voltage [15, 16] and various endogenous lipids such as anandamide which also activates cannabinoid type 1 (CB1) receptors, 12-hydroperoxy-eicosatetraenoic acid (12-HPETE) and N-arachidonoyl dopamine (NADA) [17–19]. Other natural compounds activating TRPV1 receptor are piperine found in black pepper, eugenol in cloves and zingerone in horseradish, allicin present in garlic and onion, gingerols present in raw ginger and shogaols, which are dehydration products of gingerols present in steamed ginger [20–26]. All these compounds are lipophilic and therefore bind to the intracellular surface of TRPV1 receptor [26]. Camphor is a natural compound that activates heterologously-expressed TRPV1 channels and potentiates TRPV1 currents in dorsal root ganglia (DRG) neurons at higher doses and at a different site from capsaicin. Camphor is used as a topical analgesic since it completely desensitizes the TRPV1 channel, through a vanilloid-independent mechanism and more rapidly than capsaicin [27]. TRPV1 is directly gated by noxious heat (> 43°C), which produces a sensation of pain through direct activation or through the efferent release of pro-inflammatory neuropeptides (neurogenic inflammation) [28]. Its expression on free nerve terminals in the skin allows us to detect nociceptive temperatures and facilitates its exposition to several modulators produced in response to inflammatory conditions or tissue damage that potentiate the channel's response to temperature. Thus under certain cellular conditions, such as inflammation and ischemia, TRPV1 receptor activation leads to pain under physiological temperature. The sensitivity of TRPV1 receptor also depends on membrane potential since the channel can open in the absence of capsaicin at room temperature (23°C) at depolarized potentials [29]. In addition, TRPV1 receptor is activated and sensitized by acidic pH; a condition that leads to pain during inflammation and ischemia [30, 31].
Peripheral and spinal TRPV1 receptor distribution
TRPV1 receptor has been found in both the peripheral and central nervous system within centres known for their role in pain detection, transmission and regulation, consistent with its key role in pain. Indeed, TRPV1 receptor is expressed in all sensory ganglia (DRG, TG, Vagal) and in small sensory C and Aδ fibers, which may contain various neuropeptides including substance P (SP) and/or calcitonin gene-related peptide (CGRP) [12, 32–41]. These fibers terminate predominantly in lamina I and II of the superficial dorsal horn [42, 43]. TRPV1 receptor is also expressed postsynaptically on lamina II cell bodies [43] and within the lateral collateral path, where the majority of visceral afferents terminate [44]. Spinal cord TRPV1 receptor labelling has mainly been found in the lumbar segment L4-L6 and preferentially expressed by visceral afferents [44]. TRPV1 receptor is also expressed in glial cells in lamina I and II dorsal horn of the spinal cord [45]. Capsaicin stimulates excitatory and inhibitory transmission at dorsal horn level [46, 47]. In particular, TRPV1 receptor stimulation induces the release of substance P, which in turn excites inhibitory neurons in laminae I, III and IV resulting in the enhancement of GABA- and glycinergic inhibitory post synaptic currents (IPSCs)[48–50].
TRPV1 receptor and inflammatory pain
Phosphorylation and dephosphorylation reactions regulate TRPV1 receptor activity and proved to be crucial in promoting inflammatory pain. Activation of TRPV1 receptor during inflammation appears to be a dynamic process that is produced by the combination of endogenous ligands, phosphorylation, low pH and body temperature. Pro-inflammatory agents such as nerve growth factor (NGF), ATP, bradykinins, serotonin, histamine, proteases and chemokines lead to TRPV1 sensitization through phospholipase C activation. PLC-induced sensitization reduces the threshold for detecting painful stimuli during inflammatory pain [51–54].
TRPV1-/- mice have shown markedly decreased thermal hyperalgesia in inflammatory pain models, confirming that the sensitization of these channels is likely to be involved in this phenomenon [55, 56]. TRPV1 receptor activity is also modulated by phosphatidylinositol-4,5-bisphosphate (PIP2) via the activation of phospholipases like PLC. Two opposing effects have been reported on TRPV1 receptor activity by PIP2. It can both activate and inhibit TRPV1 receptor depending on the experimental conditions, or more specifically on the level of stimulation of the channel. PIP2 plays a role in desensitization of TRPV1 receptor; a condition that causes TRPV1 activation-induced currents decay during prolonged activation. Calcium flowing through TRPV1 channel activates a Ca2+-sensitive PLC, which hydrolyzes PIP2 and leads to its depletion, which in turn results in diminished channel activity. TRPV1 channel currents require PIP2 or other phosphoinositides since they may be inhibited by scavenging endogenous phosphoinositides with polylysine and reactivated by the application of PIP2 or other phosphoinositides [57, 58]. Nonetheless, it has also been discovered that after exposure of TRPV1 receptor to high capsaicin concentrations, the ensuing Ca2+ influx activates PLC, which results in the depletion of PIP2 and PtdIns(4)P, which in turn reduces channel activity leading to desensitization [57]. It has been suggested that the balance between the inhibitory and facilitatory effects of PIP2 on TRPV1 receptor depends on the stimulation level of the channel, since during sensitization PLC-coupled agonists induce a moderate depletion of PIP2, removing its inhibitory effect, although not producing low enough lipid levels to inhibit channel activity. In contrast, high capsaicin concentrations induce a severe depletion of PIP2 that limits channel activity and leads to desensitization [57]. On this subject, TRPV1 receptor regulation by lipids seems to be anything but simple. Moreover, it has recently been suggested that ethanol enhances TRPV1 mediated responses via PIP2-TRPV1 receptor interaction [58]. Phosphoinositide 3-kinase also modulates TRPV1 activity by facilitating TRPV1 receptor trafficking to the plasma-membrane in response to NGF released during inflammation, leading to hyperalgesia (see forward section) [59]. Other membrane-derived lipids also regulate TRPV1 receptor activity. Prostaglandin E2 (PGE2) and prostacyclin I2 (PGI2) enhance capsaicin-induced current in DRG [60] and reduce the temperature threshold for TRPV1 receptor activation [61]. PGE2 increases cAMP levels and therefore activates PKA, which directly phosphorylates the channel [62] within Thr 144, Thr 370, and Ser 502 residues which have been implicated in sensitization of heat-evoked TRPV1 receptor responses, suggesting a role for PKA in the development of thermal hyperalgesia [63]. Oleylethanolamide (OEA), a natural analogue of the endogenous cannabinoid anandamide, anandamide itself and some lipoxygenase products such as 12- and 15-HPETEs (hydroperoxyeicosatetraenoic acids) and 5- and 15-HETEs (hydroxyeicosatetraenoic acids) all positively modulate TRPV1 receptor function [17, 64].
Chronic pain-induced sensitization also intensifies the expression of TRPV1 receptor in sensory neurons through transcriptional and translational regulation, post-translational changes and altered trafficking, contributing to the development of pathological pain states in which increased sensitivity to noxious stimuli occurs [65]. NGF retrograde transport increases the translation of TRPV1 receptor in the cell body during inflammation resulting in an increased translation of TRPV1 receptor to the cell body [66]. NGF also activates phosphoinositide 3-kinase (PI3K)-SRC kinase signalling pathway which phosphorylates intracellular stores of TRPV1 leading to its assembly within the cellular membrane [67].
TRPV1 receptors and neuropathic pain
The nerve growth factor (NGF), which plays a major role in the development and maintenance of neuropathic pain following peripheral nerve injury [68] has been proven to up-regulate TRPV1 receptor [69] and be involved in the phenotypic changes which lead to the development of neuropathic pain. TRPV1 receptor is up-regulated in undamaged neurons and down-regulated in damaged ones in several models of neuropathic pain [70] and, interestingly, the increased expression of TRPV1 receptor is related not only to the C-fibers but also to the myelinated A-fibers, which justifies the effectiveness of TRPV1 receptor agonist/antagonists in mechanical allodynia, apart from thermal hyperalgesia. In the tight ligation of the L5 spinal nerve, another model of neuropathic pain, the expression of TRPV1 receptors in the injured L5 dorsal root neurons decreased, whereas it increased in the uninjured L4 dorsal root neurons [70]. The up-regulation of the TRPV1 receptor at peripheral and central nervous system level in neuropathic pain conditions provides morphological evidence that the sensitivity of the vanilloid system in this painful condition is increased (conversely to the opioid system), therefore making the TRPV1 channel a suitable candidate for the future development of novel neuropathic pain relieving agents. TRPV1 receptor up-regulation also appears to occur at central sites leading to an enhancement of glutamatergic signalling in the spinal cord [71]. Similarly to peripheral sites, sensitization associated with an up-regulation of spinal TRPV1 receptor is thought to be at the base of the development of mechanical allodynia in the chronic constriction injury of the sciatic nerve [72].
The increased expression of TRPV1 receptor in pathological pain conditions [73] and its desensitization property associated with stimulation presents a valid strategy for pain relief and opens up the perspective of curing currently untreatable conditions such as neuropathic pain. Topical application of capsaicin proved effective in different neuropathic pain conditions, including post-herpetic neuralgia [74] and surgical neuropathic pain [75] but ineffective in chronic distal painful polyneuropathy [76]. However, topical capsaicin treatment such as creams, lotions or patches is associated with irritation, discomfort and pain due to the activation of sensory neurons expressing TRPV1 receptors in humans. This factor has driven chemical synthesis towards novel vanilloids with an improved desensitization/pungency ratio. The mechanisms at the basis of the anti-hyperalgesic action of topically applied capsaicin in neuropathic pain induced by partial sciatic nerve injury have been investigated. The novel expression of TRPV1 receptors on neonatal capsaicin-insensitive fibers confirmed by immunohistochemistry accounted for the anti-hyperalgesic action of topical capsaicin [77]. TRPV1-/- mice show normal responses to noxious mechanical stimuli but exhibited no vanilloid-evoked pain behaviour, reduced detection of painful heat and limited thermal hypersensitivity in the setting of inflammation. TRPV1 channels appear to be essential to the selective detection of pain sensations and to thermal hyperalgesia associated with inflammatory pain [55, 56]. Vanilloid application on sensory neurons causes pain symptoms such as thermal hyperalgesia and mechanical allodynia similar to those associated with neuropathic pain [78, 79]. Indeed TRPV1 receptor blockers should prove to be analgesics in neuropathic pain conditions. Early studies using capsazepine, the prototype TRPV1 antagonist, failed to produce any analgesic effect in models of acute and chronic pain in rats [80]. Subsequently, studies using capsazepine have shown that this drug inhibited noxious heat, protons and capsaicin-induced responses on cloned human [81] or guinea pig [82] TRPV1 receptors, whereas it failed to inhibit the responses to low pH on rat TRPV1 receptors, thus indicating possible species differences in the pharmacology of TRPV1 receptors. Interestingly, capsazepine reversed hind paw capsaicin-, CFA- and carrageenan-induced thermal hyperalgesia and mechanical allodynia in models of inflammatory and neuropathic pain in the guinea pig [83]. Capsazepine was also effective in reversing partial sciatic nerve ligation-induced mechanical hyperalgesia in such rodents while it exhibited little analgesic effect in mice or rats with neuropathic and inflammatory pain. This species-specificity in capsazepine pharmacological action has led to the development of new, potent and selective TRPV1 antagonists. Pharmaceutical companies have been expressing a great interest in the development of potent and selective TRPV1 antagonists. A potent, selective, and orally bioavailable antagonist of rat TRPV1, the N-(4-tertiarybutylphenyl)-4-(3-chloropyridin-2-yl)tetrahydropyrazine-1(2H)-carbox-amide, BCTC, has been developed by Valenzano et al. [84] and tested in models of chronic pain in rats. BCTC exhibited pain relief in mechanical and thermal hyperalgesia induced by intraplantar injection of capsaicin or CFA. BCTC also reduced already established mechanical hyperalgesia and tactile allodynia 2 weeks after partial sciatic nerve injury, and did so with a safe side effect profile [85]. Another potent and selective antagonist of both human and rat TRPV1 receptors, the 1-isoquinolin-5-yl-3-(4-trifluoromethyl-benzyl)urea, A-425619, proved dose dependently effective in several models of inflammatory and postoperative pain. A-425619 showed efficacy after either oral and intrathecal administration or local injection into the inflamed paw. Furthermore, A-425619 also showed partial efficacy in models of neuropathic pain without altering motor performance [86]. Similarly, (E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1, 4]dioxin-6-yl)acrylamide, AMG 9810, a competitive antagonist of TRPV1 receptors, has been shown to reverse thermal and mechanical hyperalgesia associated with CFA-induced inflammatory pain without any significant effect on motor function [87]. Two further potent TRPV1 receptor antagonists, one with high and the other with low CNS penetration (the 1-[3-(trifluoromethyl)pyridin-2-yl]-N-[4-(trifluoromethylsulfo-nyl)phenyl]-1,2,3,6-tetrahydropyridine-4-carboxamide, A-784168, and the N-1H-indazol-4-yl-N'-[(1R)-5-piperidin-1-yl-2,3-dihydro-1H-inden-1-yl]urea, A-795614, respectively), delivered through different administration routes (oral, intrathecal or intracerebroventricular), clarified that peripheral, spinal and supraspinal TRPV1 receptors are all involved in analgesic actions in inflammatory pain conditions. Moreover, centrally penetrating TRPV1 receptor antagonists proved more effective compared to peripheral restricted agents with the same pharmacokinetic and pharmacological profile [88], underlining an involvement of central TRPV1 receptor blockade in the analgesic action. A piperazinylpyrimidine analogous, AMG517, was shown to reverse inflammation-induced pain behaviour in rats, target central TRPV1 receptor and have a long half-life that may be amenable to a one-a-week administration [88, 89]. However, during a double blind, placebo controlled, randomized, parallel group, multicenter study for the management of pain following molar extraction, TRPV1 receptor antagonists caused prolonged hyperthermia (> 40°C) after taking a 2 mg dose, thus revealing the importance of TRPV1 receptor in central core temperature regulation [90]
TRPV1 receptor antagonists and body temperature
Most TRPV1 receptor antagonists described to date cause modest dose-limited increases in body temperature in preclinical studies [91]. Studies using TRPV1 knockout mice have unequivocally demonstrated that TRPV1 receptor antagonist-elicited hyperthermia is TRPV1 mediated [92]. Moreover, the magnitude and duration of temperature elevation caused by TRPV1 antagonists proved to be dependent on the specific properties (pharmacokinetic profile, specific modality of TRPV1 blockade) of the individual TRPV1 receptor antagonists [93, 94]. Indeed, the temperature elevation caused by the clinical candidate TRPV1 receptor antagonist ABT-102 was modest (0.6°C) and transient [94] compared to that observed with AMG-517 (1.6°C) [93]. However, the fact that numerous structurally distinct compounds blocking TRPV1 receptor cause hyperthermia indicates that TRPV1 receptor is tonically active in controlling body temperature in non-pathological states [91, 93]. Several efforts have been made in order to eliminate hyperthermia-related side effects from TRPV1 receptor blockade. Such efforts have led to the development of profile C modulators such as the (R,E)-N-(2-hydroxy-2,3-dihydro-1H-inden-4-yl)-3-(2-(piperidin-1-yl)-4 (trifluoromethyl)phenyl)acrylamide, AMG8562. AMG8562 blocks capsaicin activation of TRPV1 receptor, does not affect heat activation of TRPV1 receptor, potentiates pH 5 activation of TRPV1 receptor in vitro, and does not cause hyperthermia in vivo in rats. AMG8562 has been found to significantly block capsaicin-induced flinching behaviour, produces statistically significant efficacy in complete Freund's adjuvant- and skin incision-induced thermal hyperalgesia, and acetic acid-induced writhing model, with no profound effects on motor activity [95]. The hurdle of TRPV1 antagonist-hyperthermia therefore seems to be at a turning point.
Towards hybrid compounds targeting TRPV1 and cannabinoid receptors
Several studies are emerging which highlight the fact that the analgesic effects of compounds that interact with the endocannabinoid system are also mediated by TRPV1 channels. This is due to the fact that some endovanilloids/endocannabinoids such as anandamide may activate TRPV1 and cannabinoid receptors. It is not surprising then that the effectiveness of AM404, an inhibitor of endocannabinoid cellular uptake, in alleviating neuropathic pain has been attributed (at least in part) to TRPV1 receptor stimulation [96]. Cannabidiol is a major component of Cannabis Sativa and its analgesic effect was prevented by capsazepine in a model of neuropathic pain [97]. The inhibition of the metabolism of endocannabinoids by blocking the enzyme fatty acid amide hydrolase, FAAH, proved to be effective in neuropathic pain [98]. FAAH inhibition, by elevating the levels of anandamide and other N-acylethanolamine besides cannabinoid involves TRPV1 receptors. It is worth noting that in a study carried out by Maione et al. [99], N-arachidonoyl-serotonin (AA-5-HT), a dual FAAH inhibitor and TRPV1 receptor blocker, proved to be analgesic after repeated administration in rats in the sciatic nerve ligation model of neuropathic pain. When compared to much more potent FAAH inhibitors (URB597 and OL135), AA-5-HT showed similar or even greater effectiveness, hence confirming the role of TRPV1 receptor blockade in alleviating symptoms of neuropathic pain. Therefore by simultaneously targeting FAAH enzyme and TRPV1 receptors, two different targets controlling nociception in distinct ways, this hybrid molecule could represent an alternative approach to be used in the treatment of neuropathic pain. Such a strategy might even solve the problems of the hyperthermia caused by some "pure" TRPV1 antagonists, since AA-5-HT does not cause such side effect (possibly because indirect activation of CB1 receptors might be the cause of hypothermia).
Nociceptive fiber deletion by TRPV1 activation
As an alternative to TRPV1 receptor antagonism, therapeutic nociceptive cell deletion, exploiting the enriched TRPV1 receptor expression in nerve terminals of dorsal root or trigeminal ganglia, has been proposed as a strategic approach to the management of severe pain. In fact persistent activation of TRPV1 receptors induces a strong and prolonged increase in intracellular Ca2+, leading to excitotoxicity which compromises and deletes TRPV1-expressing cells [100]. Over-stimulation of TRPV1 receptor would prove useful in deleting TRPV1 receptor-positive neurons, thereby eliminating any sensitivity to nociceptive stimuli in hyperalgesic conditions such as inflammatory or neuropathic pain, without affecting normal sensory transmission involving fibers that do not express TRPV1 receptors. RTX application to dorsal root or trigeminal ganglia selectively ablates vanilloid-sensitive nociceptive neurons, while leaving other adjacent neurons unaffected. Such treatment blocked experimental inflammatory hyperalgesia and neurogenic inflammation in rats and naturally occurring cancer and debilitating arthritic pain in dogs. Interestingly, sensations of touch, proprioreception and high threshold mechanonociception were unaffected [101]. Similarly, in rats perineural RTX application to the sciatic nerve inhibited inflammatory hyperalgesia in a dose- and time-dependent manner, despite leaving proprioreceptive and nociceptive sensations and motor control unaffected [102]. In rats already exhibiting neuropathic pain, RTX injection into the dorsal root ganglia of the L3, L4, L5 and L6 nerve roots increased the withdrawal threshold showing that the TRPV1-positive neurons mediate the most sensitive part of mechanical allodynia. When RTX was administrated into the ipsilateral dorsal root ganglia before nerve injury, such treatment prevented the development of tactile allodynia in 12 out of 14 rats. Immunohistochemical staining revealed that the TRPV1 receptor positive neurons were eliminated in rats that did not develop tactile allodynia, whereas they were still present in the allodynic rats. RTX injection in sensory ganglia could therefore represent an effective and broadly applicable strategy for pain management in neuropathies [103].
Towards supraspinal TRPV1 receptor and the activation of an antinociceptive descending system
TRPV1 receptors have been identified in various regions of the brain known for their role in pain transmission or modulation [104–106] such as RVM, PAG, amygdala, solitary tract nucleus, somatosensory cortex, anterior cingulated cortex and insula [107, 108]. TRPV1 receptor has been found on astrocytes, perivascular structures and neuron cell bodies and dendrites mainly on postsynaptic spines [104, 105, 109–111]. Capsaicin evokes glutamate release from slices of hypothalamus in a Ca2+-dependent way [110]. Such an effect was proven to be inhibited by capsazepine, suggesting that TRPV1 receptor may be expressed on glutamatergic neurons in the hypothalamus [110].
An earlier role of supraspinal TRPV1 receptor on pain transmission and modulation was evidenced by intracerebroventricular (ICV) capsaicin injections which decreased nociceptive threshold and reduced morphine- and stress-induced analgesia [111, 112]. Conversely, ICV capsazepine or ruthenium red, another TRPV1 receptor antagonist, attenuated nocifensive behaviour induced by an intradermal injection of capsaicin or formalin in mice [113]. An effect of capsaicin involving supraspinal structures was also observed after systemic capsaicin administration. This treatment increased the firing activity of locus coeruleus (LC) neurons, an area which is activated by painful stimuli and whose stimulation produces antinociception even after sensory nerve fiber destruction [114, 115]. Accordingly, TRPV1 receptor activation with capsaicin increased glutamatergic miniature excitatory postsynaptic currents in LC [116] and when injected into the ventral segmental area enhanced dopaminergic output to the nucleus accumbens [117].
Activation of the TRPV1 receptor also evokes glutamate release in the cortex [118]and capsaicin application to the somatosensory cortex reduced mechanically and electrically evoked potentials of anesthetized rats [119]and increased the firing rate of some neurons while depressing firing of other neurons in anterior cingulate cortex, an area involved in pain-related memory and descending modulation of nociception [120–122].
In one of our earlier studies, the intra-dorsolateral (DL) PAG microinjection of capsaicin increased the latency of the nociceptive reaction in the plantar test. This analgesic effect required glutamate release and subsequent activation of glutamate receptors such as group I metabotropic glutamate (mGlu) and N-methyl-D-aspartate (NMDA) receptors. Indeed riluzole, a voltage-dependent Na+ channel blocker that enhances the uptake of glutamate, mGlu subtypes I and 5 (mGlu1 and mGlu5) and NMDA receptor antagonists blocked the analgesic effect induced by capsaicin [123]. We proposed that capsaicin-induced antinociception was due to the activation of the descending antinociceptive pathway via a TRPV1 receptor-dependent increase in glutamate release and the downstream activation of postsynaptic mGlu1/5 and NMDA receptor. The microinjection of capsaicin within the DL PAG induced brief hyperalgesia followed by analgesia in another study where a higher concentration of capsaicin was used [124]. TRPV1 receptor activation and the consequent desensitization may have been responsible for the observed effects, due to the fact that sustained activation of TRPV1 receptor leads to its desensitization. In another of our studies intra-VL PAG administration of URB597, an inhibitor of FAAH, by raising the endocannabinoid level resulted in a dose-dependent dual effect on pain responses depending on TRPV1 or CB1 receptor stimulation [125]. Indeed, the different recruitment of CB1 or TRPV1 receptors by the increased endocannabinoid/vanilloid level may lead to hyperalgesia or analgesia. Hyperalgesia was proposed to be due to CB1 receptor stimulation which leads to inhibition of the antinociceptive PAG-RVM descending pathway. Higher doses of URB597 caused rapid analgesia blocked by capsazepine, being due to TRPV1 receptor stimulation. Therefore, by acting on either CB1 or TRPV1 receptor within the PAG, endocannabinoids lead to pronociceptive and antinociceptive effects through an inhibitory or facilitatory action on the pain descending pathway. Consistently, the same study showed that some neurons within the PAG coexpressed TRPV1 and CB1 receptors.
The intra- VL PAG microinjection of the FAAH and TRPV1 receptor blocker, AA-5-HT, induced analgesia in several pain models, such as tail flick, plantar and formalin tests. AA-5-HT also depressed the RVM OFF cell as well as ON cell activity. RVM ON and OFF cells represent an electrophysiological method for investigating the analgesic potential of drugs acting centrally. Indeed, morphine causes an increase in the activity of OFF cells (which are defined as antinociceptive) and an inhibition of the ON cells (pronociceptive). Being analgesic, AA-5-HT should have induced an increase in the OFF cell activity. The effect of AA-5-HT was mimicked by co-injecting the selective FAAH inhibitor URB597 (which blocks FAAH activity) in combination with I-RTX (which blocks TRPV1 receptors). Accordingly, analgesia was induced together with inhibition of ongoing ON and OFF cell activities. The recruitment of an ''alternative'' pathway, identified as PAG-locus coeruleus (LC)-spinal cord was attributed to the AA-5-HT-induced effect. Indeed, intra-VL PAG AA-5-HT increased LC neuron firing activities while intrathecal phentolamine or ketanserin prevented the analgesic effect of AA-5-HT. Moreover, intra-PAG AA-5-HT prevented the changes in the ON and OFF cell firing activity induced by intra-paw formalin, and it reverted the formalin-induced increase in LC adrenergic cell activity. All AA-5-HT effects were antagonized by cannabinoid CB1 and TRPV1 receptor antagonists, thus suggesting that co-localization of these receptors in the PAG could be an appropriate neural substrate for AA-5-HT-induced antinociception [126].
The direct stimulation of TRPV1 receptors by microinjection of capsaicin into the VL PAG increased the latency of the nociceptive reaction [127] as it did in the DL-PAG [123]. The antinociceptive effect of capsaicin when microinjected into the VL PAG was accompanied by an increase in glutamate release in the RVM. Blockade of TRPV1 receptors by I-RTX prevented capsaicin-induced antinociception and the increase in RVM glutamate release. At a higher dose, I-RTX facilitated nociceptive response and lowered the release of glutamate, suggesting that within the VL PAG, TRPV1 receptors tonically stimulate glutamatergic output to the RVM, and concomitantly inhibit nociception [127]. The VL PAG shows a high density of TRPV1 receptor positive profiles with strong immunoreactivity mostly found in cell bodies. The high density of vesicular glutamate transporter 1 (VGLUT1) on nerve terminals surrounding TRPV1 receptor positive cells may indicate glutamatergic input on TRPV1-expressing cells. In the VL PAG, many fibers were also vesicular GABA transporter (VGAT) positive, and VGAT-immunoreactivity was observed around TRPV1 receptor positive cells, demonstrating that TRPV1-expressing neurons could also receive GABAergic inputs [127]. In accordance with behavioral studies, McGaraughty et al. [124] found that microinjection of capsaicin into the DL PAG produced an initial activation, followed by a decrease, in the tail flick related ON-cell burst of activity and an increase in OFF-cell spontaneous firing.
Microinjection of capsaicin into the VL PAG caused a decrease in the firing activity of the ON cells and a very rapid increase in the firing activity of the OFF cells. Conversely, intra-VL PAG I-RTX increased the firing activity of ON cells while decreasing the firing activity of the OFF cells. Intra-VL PAG capsaicin also decreased tail flick-induced ON cell peak firing and delayed the onset of OFF cell pause. Thus, stimulation of TRPV1 receptor within the DL and VL subregion of the PAG seems to lead to activation of the antinociceptive OFF cells and inhibition of the pronociceptive ON cells, consistent with the behavioural analgesia. These effects were prevented by pre-treatment with I-RTX at a dose which did not significantly change the ongoing RVM ON and OFF cell activities per se. At a higher dose, I-RTX increased the duration of the OFF cell pause and shortened its onset [127].
As a whole, this evidence shows that pharmacological manipulation of TPRV1 receptor within the PAG may be a suitable strategy to activate the antinociceptive PAG-RVM-dorsal horn circuitry for producing analgesia [see 128 for review].
Does TRPV1 receptor have a role in microglia/astrocytes-mediated synaptic plasticity?
Until recently, pain had been thought to arise primarily from the dysfunction of neurons. Recent evidence, however, suggests that neuro-immune changes might also contribute to pain following injury to the nervous system. Glial cells involved in mediating inflammatory processes are resident within the spinal cord and include both astroglia and microglia, the latter of which has been directly implicated in the initiation of peripheral injury-induced pain [129]. Moreover, microglia have been shown to express cannabinoid receptors [130] and to produce and inactivate endocannabinoids [131, 132]. In addition, it has been proposed that TRPV1 receptor activation mediates microglial cell death in vivo and in vitro via Ca2+-mediated mitochondrial damage and cytochrome C release [133] and that TRPV1 receptor in astrocytoma cells can contribute to apoptosis via Ca2+ influx and the activation of p38 [134]. Thus, besides the crucial role of TRPV1 channels in sensory neurons, where they regulate acute thermal nociception and inflammatory hyperalgesia, the investigation of the role of these receptors on glia and microglia could provide a new tool in further investigation into chronic diseases such as neuropathic pain. In fact, a contribution of TRPV1 receptors in glia and microglia activation has been demonstrated at the spinal level in mice with acute, inflammatory and neuropathic pain. In particular, it has been suggested that TRPV1 receptor-knockout mice show a higher density of astrocytes and microglia positive profiles under normal conditions than their wild-type littermates. Inflammatory or neuropathic pain induced a increase in glia and microglia in wild- type than in knockout mice, although the latter showed increased glial immunoreactivity as compared to untreated/uninjured animals. Thus, TRPV1 receptor could be involved in activating spinal glia in mice in different pain models including neuropathic pain and may have varying underlying mechanisms at different stages during the progression of the pain state [135]. However, the role of TRPV1 channels in glial and microglial phenotypical changes and at supraspinal level needs and deserves further investigation.
Conclusion
The few findings about the role of supraspinal TRPV1 receptors in controlling pain appear encouraging. The possible exploitation of TRPV1 receptor for activating the antinociceptive descending pathway at PAG level may be a strategy of interest. Indeed a direct agonist such as capsaicin, an indirect one such as URB597 or an hybrid anadamide hydrolysis and TRPV1 receptor blocker such as AA-5-HT, lead to analgesia. TRPV1 receptor stimulation is associated with increased glutamate release and activation of group I mGlu and NMDA receptors and with modulation of downstream RVM ON and OFF neurons. Immunohistochemical data strongly support the hypothesis that the activation/inhibition of TRPV1 receptor expressed on glutamatergic neurons following intra-PAG injection of capsaicin/I-RTX leads to an increase/decrease in glutamate release and behavioural analgesia/hyperalgesia. Exogenous compounds capable of activating TRPV1 channels or strategy increasing e ndovanilloids within the PAG-RVM counteract nociception. Thus, TRPV1 receptor (and hopefully other TRP channels such as TRPA1 and TRPM8 which often co-localize with TRPV1 on peripheral nerve terminals) may offer a suitable target for enhancing the activity of the endogenous antinociceptive descending pathway and producing analgesia. Moreover, hybrid molecules with a dual mechanism, such as AA-5-HT, which blocks FAAH activity and TRPV1 receptors, do not induce hyperthermia as "classic" TRPV1 antagonists do. As well as the PAG-RVM antinociceptive axis, other regions of the brain known to influence descending pain modulation, such as the amygdala, thalamus and LC show TRPV1 receptor localization, although the exact contribution of such receptors, if any, has yet to be determined. Further studies to clarify the role of the supraspinal TRPV1 receptor and in particular, within the antinociceptive descending pathway, may open the way to a novel strategy of pain relief in central nervous system disorders, such as neuropathic pain, which does not yet have an appropriate therapy.
References
Montell C, Rubin GM: Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron 1989, 2: 1313–1323. 10.1016/0896-6273(89)90069-X
Ramsey IS, Delling M, Clapham DE: An introduction to TRP channels. Annu Rev Physiol 2006, 68: 619–647. 10.1146/annurev.physiol.68.040204.100431
Nilius B, Voets T: TRP channels: a TR(I)P through a world of multifunctional cation channels. Pflugers Arch 2005, 451: 1–10. 10.1007/s00424-005-1462-y
Pedersen SF, Owsianik G, Nilius B: TRP channels: an overview. Cell Calcium 2005, 38: 233–252. 10.1016/j.ceca.2005.06.028
Montell C, Birnbaumer L, Flockerzi V: The TRP channels, a remarkably functional family. Cell 2002, 108: 595–598. 10.1016/S0092-8674(02)00670-0
Moran MM, Xu H, Clapham DE: TRP ion channels in the nervous system. Curr Opin Neurobiol 2004, 14: 362–369. 10.1016/j.conb.2004.05.003
Patapoutian A, Tate S, Woolf CJ: Transient receptor potential channels: targeting pain at the source. Nat Rev Drug Dis 2009, 8: 55–68. 10.1038/nrd2757
Montell C, Birnbaumer L, Flockerzi V, Bindels RJ, Bruford EA, Caterina MJ, Clapham DE, Harteneck C, Heller S, Julius D, Kojima I, Mori Y, Penner R, Prawitt D, Scharenberg AM, Schultz G, Shimizu N, Zhu MX: A unified nomenclature for the superfamily of TRP cation channels. Mol Cell 2002, 9: 229–231. 10.1016/S1097-2765(02)00448-3
Clapham DE, Runnels LW, Strubing C: The TRP ion channel family. Nat Rev Neurosci 2001, 2: 387–396. 10.1038/35077544
Kedei N, Szabo T, Lile JD, Treanor JJ, Olah Z, Iadarola MJ, Blumberg PM: Analysis of the native quaternary structure of vanilloid receptor 1. J Biol Chem 2001, 276: 28613–28619. 10.1074/jbc.M103272200
Tominaga M, Tominaga T: Structure and function of TRPV1. Pflugers Arch 2005, 451: 143–150. 10.1007/s00424-005-1457-8
Ferrer-Montiel A, Garcia-Martinez C, Morenilla-Palao C, Garcia-Sanz N, Fernandez-Carvajal A, Fernandez-Ballester G, Planells-Cases R: Molecular architecture of the vanilloid receptor. Insights for drug design. Eur J Biochem 2004, 271: 1820–1826. 10.1111/j.1432-1033.2004.04083.x
Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D: The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 1997, 389: 816–824. 10.1038/39807
Szallasi A, Blumberg PM: Resiniferatoxin, a phorbol-related diterpene, acts as an ultrapotent analog of capsaicin, the irritant constituent in red pepper. Neuroscience 1989, 30: 515–20. 10.1016/0306-4522(89)90269-8
Caterina MJ, Julius D: The vanilloid receptor: a molecular gateway to the pain pathway. Annu Rev Neurosci 2001, 24: 487–517. 10.1146/annurev.neuro.24.1.487
Gunthorpe MJ, Harries MH, Prinjha RK, Davis JB, Randall A: Voltage- and time-dependent properties of the recombinant rat vanilloid receptor (rVR1). J Physiol 2000, 3: 747–59. 10.1111/j.1469-7793.2000.t01-1-00747.x
Piper AS, Yeats JC, Bevan S, Docherty RJ: A study of the voltage dependence of capsaicin-activated membrane currents in rat sensory neurones before and after acute desensitization. J Physiol 1999, 518: 721–33. 10.1111/j.1469-7793.1999.0721p.x
Hwang SW, Cho H, Kwak J, Lee SY, Kang CJ, Jung J, Cho S, Min KH, Suh YG, Kim D, Oh U: Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proc Natl Acad Sci USA 2000, 97: 6155–60. 10.1073/pnas.97.11.6155
Price TJ, Patwardhan A, Akopian AN, Hargreaves KM, Flores CM: Modulation of trigeminal sensory neuron activity by the dual cannabinoid-vanilloid agonists anandamide, N-arachidonoyl-dopamine and arachidonyl-2-chloroethylamide. Br J Pharmacol 2004, 141: 1118–30. 10.1038/sj.bjp.0705711
Suh YG, Oh U: Activation and activators of TRPV1 and their pharmaceutical implication. Curr Pharm Des 2005, 11: 2687–98. 10.2174/1381612054546789
Liu L, Simon SA: Similarities and differences in the currents activated by capsaicin, piperine, and zingerone in rat trigeminal ganglion cells. J Neurophysiol 1996, 76: 1858–1869.
McNamara FN, Randall A, Gunthorpe MJ: Effects of piperine, the pungent component of black pepper, at the human vanilloid receptor (TRPV1). Br J Pharmacol 2005, 144: 781–790. 10.1038/sj.bjp.0706040
Dedov VN, Tran VH, Duke CC, Connor M, Christie MJ, Mandadi S, Roufogalis BD: Gingerols: a novel class of vanilloid receptor (VR1) agonists. Br J Pharmacol 2002, 137: 793–798. 10.1038/sj.bjp.0704925
Witte DG, Cassar SC, Masters JN, Esbenshade T, Hancock AA: Use of a fluorescent imaging plate reader--based calcium assay to assess pharmacological differences between the human and rat vanilloid receptor. J Biomol Screen 2002, 7: 466–475. 10.1177/108705702237679
Iwasaki Y, Morita A, Iwasawa T, Kobata K, Sekiwa Y, Morimitsu Y, Kubota K, Watanabe T: : A nonpungent component of steamed ginger--[10]-shogaol--increases adrenaline secretion via the activation of TRPV1. Nutr Neurosci 2006, 9: 169–178. 10.1080/10284150600955164
Yang BH, Piao ZG, Kim YB, Lee CH, Lee JK, Park K, Kim JS, Oh SB: Activation of vanilloid receptor 1 (VR1) by eugenol. J Dent Res 2003, 82: 781–785. 10.1177/154405910308201004
Macpherson LJ, Geierstanger BH, Viswanath V, Bandell M, Eid SR, Hwang S, Patapoutian A: The pungency of garlic: activation of TRPA1 and TRPV1 in response to allicin. Curr Biol 2005, 15: 929–934. 10.1016/j.cub.2005.04.018
Xu H, Blair NT, Clapham DE: Camphor activates and strongly desensitizes the transient receptor potential vanilloid subtype 1 channel in a vanilloid-independent mechanism. J Neurosci 2005, 25: 8924–8937. 10.1523/JNEUROSCI.2574-05.2005
Holzer P: Local effector functions of capsaicin-sensitive sensory nerve endings: involvement of tachykinins, calcitonin gene-related peptide and other neuropeptides. Neuroscience 1988, 24: 739–768. 10.1016/0306-4522(88)90064-4
Oseguera AJ, Islas LD, Garcia-Villegas R, Rosenbaum T: On the mechanism of TBA block of the TRPV1 channel. Biophys J 2007, 92: 3901–3914. 10.1529/biophysj.106.102400
Ryu S, Liu B, Qin F: Low pH potentiates both capsaicin binding and channel gating of VR1 receptors. J Gen Physiol 2003, 122: 45–61. 10.1085/jgp.200308847
Reeh PW, Kress M: Molecular physiology of proton transduction in nociceptors. Curr Opin Pharmacol 2001, 1: 45–51. 10.1016/S1471-4892(01)00014-5
Szallasi A, Blumberg PM: Vanilloid (Capsaicin) receptors and mechanisms. Pharmacol Rev 1999, 51: 159–212.
Szallasi A, Nilsson S, Farkas-Szallasi T, Blumberg PM, Hokfelt T, Lundberg JM: Vanilloid (capsaicin) receptors in the rat: distribution in the brain, regional differences in the spinal cord, axonal transport to the periphery, and depletion by systemic vanilloid treatment. Brain Res 1995, 703: 175–183. 10.1016/0006-8993(95)01094-7
Szallasi A, Blumberg PM: Characterization of vanilloid receptors in the dorsal horn of pig spinal cord. Brain Res 1991, 547: 335–338. 10.1016/0006-8993(91)90982-2
Szolcsanyi J, Szallasi A, Szallasi Z, Joo F, Blumberg PM: Resiniferatoxin. An ultrapotent neurotoxin of capsaicin-sensitive primary afferent neurons. Ann N Y Acad Sci 1991, 632: 473–475. 10.1111/j.1749-6632.1991.tb33161.x
Szolcsanyi J, Szallasi A, Szallasi Z, Joo F, Blumberg PM: Resiniferatoxin: an ultrapotent selective modulator of capsaicin-sensitive primary afferent neurons. J Pharmacol Exp Ther 1990, 255: 923–928.
Fischer MJ, Reeh PW, Sauer SK: Proton-induced calcitonin gene-related peptide release from rat sciatic nerve axons, in vitro, involving TRPV1. Eur J Neurosci 2003, 18: 803–810. 10.1046/j.1460-9568.2003.02811.x
Bernardini N, Neuhuber W, Reeh PW, Sauer SK: Morphological evidence for functional capsaicin receptor expression and calcitonin gene-related peptide exocytosis in isolated peripheral nerve axons of the mouse. Neuroscience 2004, 126: 585–590. 10.1016/j.neuroscience.2004.03.017
Price TJ, Flores CM: Critical evaluation of the colocalization between calcitonin gene-related peptide, substance P, transient receptor potential vanilloid subfamily type 1 immunoreactivities, and isolectin B4 binding in primary afferent neurons of the rat and mouse. J Pain 2007, 8: 263–272. 10.1016/j.jpain.2006.09.005
Price TJ, Louria MD, Candelario-Soto D, Dussor GO, Jeske NA, Patwardhan AM, Diogenes A, Trott AA, Hargreaves KM, Flores CM: Treatment of trigeminal ganglion neurons in vitro with NGF, GDNF or BDNF: effects on neuronal survival, neurochemical properties and TRPV1-mediated neuropeptide secretion. BMC Neurosci 2005, 6: 4. 10.1186/1471-2202-6-4
Mezey E, Toth ZE, Cortright DN, Arzubi MK, Krause JE, Elde R, Guo A, Blumberg PM, Szallasi A: Distribution of mRNA for vanilloid receptor subtype 1 (VR1), and VR1-like immunoreactivity, in the central nervous system of the rat and human. Proc Natl Acad Sci USA 2000, 97: 3655–3660. 10.1073/pnas.060496197
Guo A, Vulchanova L, Wang J, Li X, Elde R: Immunocytochemical localization of the vanilloid receptor 1 (VR1): relationship to neuropeptides, the P2X3 purinoceptor and IB4 binding sites. Eur J Neurosci 1999, 11: 946–958. 10.1046/j.1460-9568.1999.00503.x
Valtschanoff JG, Rustioni A, Guo A, Hwang SJ: Vanilloid receptor VR1 is both presynaptic and postsynaptic in the superficial laminae of the rat dorsal horn. J Comp Neurol 2001, 436: 225–235. 10.1002/cne.1063
Hwang SJ, Valtschanoff JG: Vanilloid receptor VR1-positive afferents are distributed differently at different levels of the rat lumbar spinal cord. Neurosci Lett 2003, 349: 41–44. 10.1016/S0304-3940(03)00750-X
Doly S, Fischer J, Salio C, Conrath M: The vanilloid receptor-1 is expressed in rat spinal dorsal horn astrocytes. Neurosci Lett 2004, 357: 123–126. 10.1016/j.neulet.2003.12.051
Ueda M, Kuraishi Y, Sugimoto K, Satoh M: Evidence that glutamate is released from capsaicin-sensitive primary afferent fibers in rats: study with on-line continuous monitoring of glutamate. Neurosci Res 1994, 20: 231–7. 10.1016/0168-0102(94)90092-2
Zhou Y, Zhou ZS, Zhao ZQ: PKC regulates capsaicin-induced currents of dorsal root ganglion neurons in rats. Neuropharmacology 2001, 41: 601–608. 10.1016/S0028-3908(01)00106-X
Ferrini F, Salio C, Vergnano AM, Merighi A: Vanilloid receptor-1 (TRPV1)-dependent activation of inhibitory neurotransmission in spinal substantia gelatinosa neurons of mouse. Pain 2007, 129: 195–209. 10.1016/j.pain.2007.01.009
Zhou HY, Zhang HM, Chen SR, Pan HL: Increased C-fiber nociceptive input potentiates inhibitory glycinergic transmission in the spinal dorsal horn. J Pharmacol Exp Ther 2008, 324: 1000–1010. 10.1124/jpet.107.133470
Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D: Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 2001, 411: 957–962. 10.1038/35082088
Tominaga M, Wada M, Masu M: Potentiation of capsaicin receptor activity by metabotropic ATP receptors as a possible mechanism for ATP-evoked pain and hyperalgesia. Proc Natl Acad Sci USA 2001, 98: 6951–6956. 10.1073/pnas.111025298
Moriyama T, Iida T, Kobayashi K, Higashi T, Fukuoka T, Tsumura H, Leon C, Suzuki N, Inoue K, Gachet C, Noguchi K, Tominaga M: Possible involvement of P2Y2 metabotropic receptors in ATP-induced transient receptor potential vanilloid receptor 1-mediated thermal hypersensitivity. J Neurosci 2003, 23: 6058–6062.
Zhang N, Inan S, Cowan A, Sun R, Wang JM, Rogers TJ, Caterina M, Oppenheim JJ: A proinflammatory chemokine, CCL3, sensitizes the heat- and capsaicin-gated ion channel TRPV1. Proc Natl Acad Sci USA 2005, 102: 4536–4541. 10.1073/pnas.0406030102
Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D: Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 2000, 288: 306–313. 10.1126/science.288.5464.306
Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA: Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 2000, 405: 183–187. 10.1038/35012076
Lukacs V, Thyagarajan B, Balla A, Varnai P, Balla T, Rohacs T: Dual regulation of TRPV1 by phosphoinositides. J Neurosci 2007, 27: 7070–7080. 10.1523/JNEUROSCI.1866-07.2007
Vetter I, Wyse BD, Roberts-Thomson SJ, Monteith GR, Cabot PJ: Mechanisms involved in potentiation of transient receptor potential vanilloid 1 responses by ethanol. Eur J Pain 2008, 12: 441–54. 10.1016/j.ejpain.2007.07.001
Stein AT, Ufret-Vincenty CA, Hua L, Santana LF, Gordon S: Phosphoinositide 3-Kinase Binds to TRPV1 and Mediates NGF-stimulated TRPV1 Trafficking to the Plasma Membrane. J Gen Physiol 2006, 128: 509–522. 10.1085/jgp.200609576
Pitchford S, Levine JD: Prostaglandins sensitize nociceptors in cell culture. Neurosci Lett 1991, 132: 105–8. 10.1016/0304-3940(91)90444-X
Moriyama T, Higashi T, Togashi K, Iida T, Segi E, Sugimoto Y, Tominaga T, Narumiya S, Tominaga M: Sensitization of TRPV1 by EP1 and IP reveals peripheral nociceptive mechanism of prostaglandins. Mol Pain 2005, 1: 3. 10.1186/1744-8069-1-3
Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW: cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron 2002, 35: 721–731. 10.1016/S0896-6273(02)00802-4
Rathee PK, Distler C, Obreja O, Neuhuber W, Wang GK, Wang SY, Nau C, Kress M: PKA/AKAP/VR-1 module: A common link of Gs-mediated signaling to thermal hyperalgesia. J Neurosci 2002, 22: 4740–4745.
Shin J, Cho H, Hwang SW, Jung J, Shin CY, Lee SY, Kim SH, Lee MG, Choi YH, Kim J, Haber NA, Reichling DB, Khasar S, Levine JD, Oh U: Bradykinin-12-lipoxygenase-VR1 signaling pathway for inflammatory hyperalgesia. Proc Natl Acad Sci USA 2002, 99: 10150–10155. 10.1073/pnas.152002699
Hucho T, Levine JD: Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron 2007, 55: 365–76. 10.1016/j.neuron.2007.07.008
Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ: p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron 2002, 36: 57–68. 10.1016/S0896-6273(02)00908-X
Zhang X, Huang J, McNaughton PA: NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J 2005, 24: 4211–23. 10.1038/sj.emboj.7600893
Campbell JN, Meyer RA: Mechanisms of neuropathic pain. Neuron 2006, 52: 77–92. 10.1016/j.neuron.2006.09.021
Winston J, Toma H, Shenoy M, Pasricha PJ: Nerve growth factor regulates VR-1 mRNA levels in cultures of adult dorsal root ganglion neurons. Pain 2001, 89: 181–6. 10.1016/S0304-3959(00)00370-5
Hudson LJ, Bevan S, Wotherspoon G, Gentry C, Fox A, Winter J: VR1 protein expression increases in undamaged DRG neurons after partial nerve injury. Eur J Neurosci 2001, 13: 2105–14. 10.1046/j.0953-816x.2001.01591.x
Lappin SC, Randall AD, Gunthorpe MJ, Morisset V: TRPV1 antagonist, SB-366791, inhibits glutamatergic synaptic transmission in rat spinal dorsal horn following peripheral inflammation. Eur J Pharmacol 2006, 540: 73–81. 10.1016/j.ejphar.2006.04.046
Kanai Y, Nakazato E, Fujiuchi A, Hara T, Imai A: Involvement of an increased spinal TRPV1 sensitization through its up-regulation in mechanical allodynia of CCI rats. Neuropharmacology 2005, 49: 977–84. 10.1016/j.neuropharm.2005.05.003
Vilceanu D, Honore P, Hogan QH, Stucky CL: Spinal nerve ligation in mouse upregulates TRPV1 heat function in injured IB4-positive nociceptors. J Pain 2010, 11: 88–99.
Watson CP, Evans RJ, Watt VR: Post-herpetic neuralgia and topical capsaicin. Pain 1988, 33: 333–40. 10.1016/0304-3959(88)90292-8
Ellison N, Loprinzi CL, Kugler J, Hatfield AK, Miser A, Sloan JA, Wender DB, Rowland KM, Molina R, Cascino TL, Vukov AM, Dhaliwal HS, Ghosh C: Phase III placebo-controlled trial of capsaicin cream in the management of surgical neuropathic pain in cancer patients. J Clin Oncol 1997, 15: 2974–2980.
Low PA, Opfer-Gehrking TL, Dyck PJ, Litchy WJ, O'Brien PC: Double-blind, placebo-controlled study of the application of capsaicin cream in chronic distal painful polyneuropathy. Pain 1995, 62: 163–8. 10.1016/0304-3959(94)00261-C
Rashid MH, Inoue M, Bakoshi S, Ueda H: Increased expression of vanilloid receptor 1 on myelinated primary afferent neurons contributes to the antihyperalgesic effect of capsaicin cream in diabetic neuropathic pain in mice. J Pharmacol Exp Ther 2003, 306: 709–717. 10.1124/jpet.103.050948
Simone DA, Ngeow JY, Putterman GJ, LaMotte RH: Hyperalgesia to heat after intradermal injection of capsaicin. Brain Res 1987, 418: 201–213. 10.1016/0006-8993(87)90982-6
Gilchrist HD, Allard BL, Simone DA: Enhanced withdrawal responses to heat and mechanical stimuli following intraplantar injection of capsaicin in rats. Pain 1996, 67: 179–188. 10.1016/0304-3959(96)03104-1
Perkins MN, Campbell EA: Capsazepine reversal of the antinociceptive action of capsaicin in vivo. Br J Pharmacol 1992, 107: 329–33.
McIntyre P, McLatchie LM, Chambers A, Phillips E, Clarke M, Savidge J, Toms C, Peacock M, Shah K, Winter J, Weerasakera N, Webb M, Rang HP, Bevan S, James IF: Pharmacological differences between the human and rat vanilloid receptor 1 (VR1). Br J Pharmacol 2001, 132: 1084–1094. 10.1038/sj.bjp.0703918
Savidge J, Davis C, Shah K, Colley S, Phillips E, Ranasinghe S, Winter J, Kotsonis P, Rang H, McIntyre P: Cloning and functional characterization of the guinea pig vanilloid receptor 1. Neuropharmacology 2002, 43: 450–456. 10.1016/S0028-3908(02)00122-3
Walker KM, Urban L, Medhurst SJ, Patel S, Panesar M, Fox AJ, McIntyre P: The VR1 antagonist capsazepine reverses mechanical hyperalgesia in models of inflammatory and neuropathic pain. J Pharmacol Exp Ther 2003, 304: 56–62. 10.1124/jpet.102.042010
Valenzano KJ, Grant ER, Wu G, Hachicha M, Schmid L, Tafesse L, Sun Q, Rotshteyn Y, Francis J, Limberis J, Malik S, Whittemore ER, Hodges D: N-(4-tertiarybutylphenyl)-4-(3-chloropyridin-2-yl)tetrahydropyrazine-1(2H)-carbox-amide (BCTC), a novel, orally effective vanilloid receptor 1 antagonist with analgesic properties: I. in vitro characterization and pharmacokinetic properties. J Pharmacol Exp Ther 2003, 306: 377–386. 10.1124/jpet.102.045674
Pomonis JD, Harrison JE, Mark L, Bristol DR, Valenzano KJ, Walker K: N-(4-Tertiarybutylphenyl)-4-(3-cholorphyridin-2-yl)tetrahydropyrazine-1(2H)-carbox-amide (BCTC), a novel, orally effective vanilloid receptor 1antagonist with analgesic properties: II. in vivo characterization in rat models of inflammatory and neuropathic pain. J Pharmacol Exp Ther 2003, 306: 387–93. 10.1124/jpet.102.046268
Honore P, Wismer CT, Mikusa J, Zhu CZ, Zhong C, Gauvin DM, Gomtsyan A, El Kouhen R, Lee CH, Marsh K, Sullivan JP, Faltynek CR, Jarvis MF: A-425619 [1-isoquinolin-5-yl-3-(4-trifluoromethyl-benzyl)-urea], a novel transient receptor potential type V1 receptor antagonist, relieves pathophysiological pain associated with inflammation and tissue injury in rats. J Pharmacol Exp Ther 2005, 314: 410–21. 10.1124/jpet.105.083915
Gavva NR, Tamir R, Qu Y, Klionsky L, Zhang TJ, Immke D, Wang J, Zhu D, Vanderah TW, Porreca F, Doherty EM, Norman MH, Wild KD, Bannon AW, Louis JC, Treanor JJ: AMG 9810 [(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)acrylamide], a novel vanilloid receptor 1 (TRPV1) antagonist with antihyperalgesic properties. J Pharmacol Exp Ther 2005, 313: 474–84. 10.1124/jpet.104.079855
Cui M, Honore P, Zhong C, Gauvin D, Mikusa J, Hernandez G, Chandran P, Gomtsyan A, Brown B, Bayburt EK, Marsh K, Bianchi B, McDonald H, Niforatos W, Neelands TR, Moreland RB, Decker MW, Lee CH, Sullivan JP, Faltynek CR: TRPV1 receptors in the CNS play a key role in broad-spectrum analgesia of TRPV1 antagonists. J Neurosci 2006, 26: 9385–93. 10.1523/JNEUROSCI.1246-06.2006
Bak A, Gore A, Yanez E, Stanton M, Tufekcic S, Syed R, Akrami A, Rose M, Surpaneni S, Bostick T, King A, Neervannan S, Ostovic D, Koparkar A: The co-crystal approach to improve the exposure of a water-insoluble compound: AMG 517 sorbic acid cocrystal characterization and pharmacokinetics. J Pharm Sci 2008, 97: 3942–56. 10.1002/jps.21280
Gavva NR, Treanor JJ, Garami A, Fang L, Surapaneni S, Akrami A, Alvarez F, Bak A, Darling M, Gore A, Jang GR, Kesslak JP, Ni L, Norman MH, Palluconi G, Rose MJ, Salfi M, Tan E, Romanovsky AA, Banfield C, Davar G: Pharmacological blockade of the vanilloid receptor TRPV1 elicits marked hyperthermia in humans. Pain 2008, 136: 202–210. 10.1016/j.pain.2008.01.024
Gavva NR: Body-temperature maintenance as the predominant function of the vanilloid receptor TRPV1. Trends Pharmacol Sci 2008, 29: 550–7. 10.1016/j.tips.2008.08.003
Steiner AA, Turek VF, Almeida MC, Burmeister JJ, Oliveira DL, Roberts JL, Bannon AW, Norman MH, Louis JC, Treanor JJ, Gavva NR, Romanovsky AA: Nonthermal activation of transient receptor potential vanilloid-1 channels in abdominal viscera tonically inhibits autonomic cold-defense effectors. J Neurosci 2007, 27: 7459–68. 10.1523/JNEUROSCI.1483-07.2007
Gavva NR, Bannon AW, Hovland DN, Lehto SG, Klionsky L, Surapeneni S, Immke DC, Henley C, Arik L, Bak A, Davis J, Ernst N, Hever G, Kuang R, Shi L, Tamir R, Wang J, Wang W, Zajic G, Zhu D, Norman MH, Louis JC, Magal E, Treanor JJ: Repeated administration of vanilloid receptor TRPV1 antagonists attenuates hyperthermia elicited by TRPV1 blockade. J Pharmacol Exp Ther 2007, 323: 128–37. 10.1124/jpet.107.125674
Honore P, Chandran P, Hernandez G, Gauvin DM, Mikusa JP, Zhong C, Joshi SK, Ghilardi JR, Sevcik MA, Fryer RM, Segreti JA, Banfor PN, Marsh K, Neelands T, Bayburt E, Daanen JF, Gomtsyan A, Lee CH, Kort ME, Reilly RM, Surowy CS, Kym PR, Mantyh PW, Sullivan JP, Jarvis MF, Faltynek CR: Repeated dosing of ABT-102, a potent and selective TRPV1 antagonist, enhances TRPV1-mediated analgesic activity in rodents, but attenuates antagonist-induced hyperthermia. Pain 2009, 142: 27–35. 10.1016/j.pain.2008.11.004
Lehto SG, Tamir R, Deng H, Klionsky L, Kuang R, Le A, Lee D, Louis JC, Magal E, Manning BH, Rubino J, Surapaneni S, Tamayo N, Wang T, Wang J, Wang J, Wang W, Youngblood B, Zhang M, Zhu D, Norman MH, Gavva NR: Antihyperalgesic effects of (R,E)-N-(2-hydroxy-2,3-dihydro-1H-inden-4-yl)-3-(2-(piperidin-1-yl)-4-(trifluoromethyl)phenyl)-acrylamide (AMG8562), a novel transient receptor potential vanilloid type 1 modulator that does not cause hyperthermia in rats. J Pharmacol Exp Ther 2008, 326: 218–29. 10.1124/jpet.107.132233
Rodella LF, Borsani E, Rezzani R, Ricci F, Buffoli B, Bianchi R: AM404, an inhibitor of anandamide reuptake decreases Fos-immunoreactivity in the spinal cord of neuropathic rats after non-noxious stimulation. Eur J Pharmacol 2005, 508: 139–46. 10.1016/j.ejphar.2004.12.031
Costa B, Giagnoni G, Franke C, Trovato AE, Colleoni M: Vanilloid TRPV1 receptor mediates the antihyperalgesic effect of the nonpsychoactive cannabinoid, cannabidiol, in a rat model of acute inflammation. Br J Pharmacol 2004, 143: 247–50. 10.1038/sj.bjp.0705920
Jhaveri MD, Richardson D, Chapman V: Endocannabinoid metabolism and uptake: novel targets for neuropathic and inflammatory pain. Br J Pharmacol 2007, 152: 624–32. 10.1038/sj.bjp.0707433
Maione S, De Petrocellis L, de Novellis V, Moriello AS, Petrosino S, Palazzo E, Rossi FS, Woodward DF, Di Marzo V: Analgesic actions of N-arachidonoyl-serotonin, a fatty acid amide hydrolase inhibitor with antagonistic activity at vanilloid TRPV1 receptors. Br J Pharmacol 2007, 150: 766–81. 10.1038/sj.bjp.0707145
Olah Z, Szabo T, Karai L, Hough C, Fields RD, Caudle RM, Blumberg PM, Iadarola MJ: Ligand-induced dynamic membrane changes and cell deletion conferred by vanilloid receptor 1. J Biol Chem 2001, 276: 11021–30. 10.1074/jbc.M008392200
Karai L, Brown DC, Mannes AJ, Connelly ST, Brown J, Gandal M, Wellisch OM, Neubert JK, Olah Z, Iadarola MJ: Deletion of vanilloid receptor 1-expressing primary afferent neurons for pain control. J Clin Invest 2004, 113: 1344–52.
Neubert JK, King C, Malphurs W, Wong F, Weaver JP, Jenkins AC, Rossi HL, Caudle RM: Characterization of mouse orofacial pain and the effects of lesioning TRPV1-expressing neurons on operant behavior. Mol Pain 2008, 4: 3–13. 10.1186/1744-8069-4-3
Tender GC, Li YY, Cui JG: Vanilloid receptor 1-positive neurons mediate thermal hyperalgesia and tactile allodynia. Spine J 2008, 8: 351–358. 10.1016/j.spinee.2007.08.005
Mezey E, Tóth ZE, Cortright DN, Arzubi MK, Krause JE, Elde R, Guo A, Blumberg PM, Szallasi A: Distribution of mRNA for vanilloid receptor subtype 1 (VR1), and VR1-like immunoreactivity, in the central nervous system of the rat and human. Proc Natl Acad Sci USA 2000, 97: 3655–3660. 10.1073/pnas.060496197
Roberts JC, Davis JB, Benham CD: [3H]Resiniferatoxin autoradiography in the CNS of wild-type and TRPV1 null mice defines TRPV1 (VR-1) protein distribution. Brain Res 2004, 995: 176–183. 10.1016/j.brainres.2003.10.001
Szabo T, Biro T, Gonzalez AF, Palkovits M, Blumberg PM: Pharmacological characterization of vanilloid receptor located in the brain. Brain Res Mol Brain Res 2002, 98: 51–57. 10.1016/S0169-328X(01)00313-8
Millan MJ: The induction of pain: an integrative review. Prog Neurobiol 1999, 57: 1–164. 10.1016/S0301-0082(98)00048-3
Millan MJ: Descending control of pain. Prog Neurobiol 2002, 66: 355–474. 10.1016/S0301-0082(02)00009-6
Liapi A, Wood JN: Extensive co-localization and heteromultimer formation of the vanilloid receptor-like protein TRPV2 and the capsaicin receptor TRPV1 in the adult rat cerebral cortex. Eur J Neurosci 2005, 22: 825–834. 10.1111/j.1460-9568.2005.04270.x
Sasamura T, Sasaki M, Tohda C, Kuraishi Y: Existence of capsaicin-sensitive glutamatergic terminals in rat hypothalamus. Neuroreport 1998, 22: 2045–2048. 10.1097/00001756-199806220-00025
Bodnar RJ, Kirchgessner A, Nilaver G, Mulhern J, Zimmerman EA: Intraventricular capsaicin: alterations in analgesic responsivity without depletion of substance P. Neuroscience 1982, 7: 631–638. 10.1016/0306-4522(82)90068-9
Bodnar RJ, Simone DA, Kordower JH, Kirchgessner AL, Nilaver G: Capsaicin treatment and stress-induced analgesia. Pharmacol Biochem Behav 1983, 18: 65–71. 10.1016/0091-3057(83)90253-8
Santos AR, Calixto JB: Ruthenium red and capsazepine antinociceptive effect in formalin and capsaicin models of pain in mice. Neurosci Lett 1997, 235: 73–76. 10.1016/S0304-3940(97)00722-2
Hajós M, Engberg G, Elam M: Reduced responsiveness of locus coeruleus neurons to cutaneous thermal stimuli in capsaicin-treated rats. Neurosci Lett 1986, 70: 382–387. 10.1016/0304-3940(86)90584-7
Hajós M, Jancsó G, Engberg G: Capsaicin-induced excitation of locus coeruleus neurons. Acta Physiol Scand 1987, 129: 415–420. 10.1111/j.1748-1716.1987.tb08086.x
Marinelli S, Pascucci T, Bernardi G, Puglisi-Allegra S, Mercuri NB: Activation of TRPV1 in the VTA excites dopaminergic neurons and increases chemical- and noxious-induced dopamine release in the nucleus accumbens. Neuropsychopharmacology 2005, 30: 864–870. 10.1038/sj.npp.1300615
Marinelli S, Vaughan CW, Christie MJ, Connor M: Capsaicin activation of glutamatergic synaptic transmission in the rat locus coeruleus in vitro. J Physiol 2002, 543: 531–540. 10.1113/jphysiol.2002.022863
Sasamura T, Kuraishi Y: Peripheral and central actions of capsaicin and VR1 receptor. Jpn J Pharmacol 1999, 80: 275–280. 10.1254/jjp.80.275
Toldi J, Joo F, Wolfe JR: Capsaicin differentially influences somatosensory cortical responses evoked by peripheral electrical or mechanical stimulation. Neuroscience 1992, 49: 135–139. 10.1016/0306-4522(92)90081-C
Calejesan AA, Kim SJ, Zhuo M: Descending facilitatory modulation of a behavioral nociceptive response by stimulation in the adult rat anterior cingulate cortex. Eur J Pain 2000, 4: 83–96. 10.1053/eujp.1999.0158
Tang J, Ko S, Ding HK, Qiu CS, Calejesan AA, Zhuo M: Pavlovian fear memory induced by activation in the anterior cingulated cortex. Mol Pain 2005, 1: 6. 10.1186/1744-8069-1-6
Steenland HW, Ko SW, Wu LJ, Zhuo M: Hot receptors in the brain. Mol Pain 2006, 2: 34–42. 10.1186/1744-8069-2-34
Palazzo E, de Novellis V, Marabese I, Cuomo D, Rossi F, Berrino L, Rossi F, Maione S: Interaction between vanilloid and glutamate receptors in the central modulation of nociception. Eur J Pharmacol. 2002, 439: 69–75.
McGaraughty S, Chu KL, Bitner RS, Martino B, El Kouhen R, Han P, Nikkel AL, Burgard EC, Faltynek CR, Jarvis M: Capsaicin infused into the PAG affects rat tail flick responses to noxious heat and alters neuronal firing in the RVM. J Neurophysiol 2003, 90: 2702–2710. 10.1152/jn.00433.2003
Maione S, Bisogno T, de Novellis V, Palazzo E, Cristino L, Valenti M, Petrosino S, Guglielmotti V, Rossi F, Di Marzo V: Elevation of endocannabinoid levels in the ventrolateral periaqueductal grey through inhibition of fatty acid amide hydrolase affects descending nociceptive pathways via both cannabinoid receptor type 1 and transient receptor potential vanilloid type-1 receptors. J Pharmacol Exp Ther 2006, 316: 969–982. 10.1124/jpet.105.093286
de Novellis V, Palazzo E, Rossi F, De Petrocellis L, Petrosino S, Guida F, Luongo L, Migliozzi A, Cristino L, Marabese I, Starowicz K, Di Marzo V, Maione S: The analgesic effect of N-arachidonoyl-serotonin, a FAAH inhibitor and TRPV1 receptor antagonist, associated with changes in rostral ventromedial medulla and locus coeruleus cell activity in rats. Neuropharmacology 2008, 55: 1105–1113. 10.1016/j.neuropharm.2008.06.023
Starowicz K, Maione S, Cristino L, Palazzo E, Marabese I, Rossi F, de Novellis V, Di Marzo V: Tonic endovanilloid facilitation of glutamate release in brainstem descending antinociceptive pathways. J Neurosci 2007, 27: 13739–13749. 10.1523/JNEUROSCI.3258-07.2007
Palazzo E, Rossi F, Maione S: Role of TRPV1 receptors in descending modulation of pain. Mol Cell Endocrinol 2008, 286: S79–83. 10.1016/j.mce.2008.01.013
Watkins LR, Milligan ED, Maier SF: Spinal cord glia: new players in pain. Pain 2001, 93: 201–5. 10.1016/S0304-3959(01)00359-1
Luongo L, Palazzo E, Tambaro S, Giordano C, Gatta L, Scafuro MA, Rossi FS, Lazzari P, Pani L, de Novellis V, Malcangio M, Maione S: 1-(2',4'-dichlorophenyl)-6- methyl-N-cyclohexylamine-1,4-dihydroindeno[1,2-c]pyrazole-3- carboxamide, a novel CB2 agonist, alleviates neuropathic pain through functional microglial changes in mice. Neurobiol Dis 2010, 37: 177–85. 10.1016/j.nbd.2009.09.021
Carrier EJ, Kearn CS, Barkmeier AJ, Breese NM, Yang W, Nithipatikom K, Pfister SL, Campbell WB, Hillard CJ: Cultured rat microglial cells synthesize the endocannabinoid 2-arachidonylglycerol, which increases proliferation via a CB2 receptor-dependent mechanism. Mol Pharmacol. 2004, 65: 999–1007.
Luongo L, Palazzo E, de Novellis V, Maione S: Role of endocannabinoid system in neuron-glia cross-talk. The Open Pain Journal 2010, 3: 29–36. 10.2174/1876386301003020029
Kim SR, Kim SU, Oh U, Jin BK: Transient receptor potential vanilloid subtype 1 mediates microglial cell death in vivo and in vitro via Ca2+-mediated mitochondrial damage and cytochrome c release. J Immunol 2006, 177: 4322–9.
Amantini C, Mosca M, Nabissi M, Lucciarini R, Caprodossi S, Arcella A, Giangaspero F, Santoni G: Capsaicin-induced apoptosis of glioma cells is mediated by TRPV1 vanilloid receptor and requires p38 MAPK activation. J Neurochem 2007, 102: 977–90. 10.1111/j.1471-4159.2007.04582.x
Chen Y, Willcockson HH, Valtschanoff JG: Influence of the vanilloid receptor TRPV1 on the activation of spinal cord glia in mouse models of pain. Exp Neurol 2009, 220: 383–90. 10.1016/j.expneurol.2009.09.030
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
EP has written the manuscript. EP and SM have conceived and conceptualized the manuscript. LL has written the paragraph on microglia. VdN, LB and FR contributed to the drafting of the paper. All authors have read and approved the final manuscript.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Palazzo, E., Luongo, L., de Novellis, V. et al. Moving towards supraspinal TRPV1 receptors for chronic pain relief. Mol Pain 6, 66 (2010). https://doi.org/10.1186/1744-8069-6-66
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1744-8069-6-66