
Abstract
Background
Persons with cystic fibrosis (CF) are at-risk for health effects from ambient air pollution but little is known about the interaction of nanoparticles (NP) with CF lungs. Here we study the distribution of inhaled NP in a murine CF model and aim to reveal mechanisms contributing to adverse effects of inhaled particles in susceptible populations.
Methods
Chloride channel defective CftrTgH (neoim) Hgu mice were used to analyze lung function, lung distribution and whole body biokinetics of inhaled NP, and inflammatory responses after intratracheal administration of NP. Distribution of 20-nm titanium dioxide NP in lungs was assessed on ultrathin sections immediately and 24 h after a one-hour NP inhalation. NP biokinetics was deduced from total and regional lung deposition and from whole body translocation of inhaled 30-nm iridium NP within 24 h after aerosol inhalation. Inflammatory responses were assessed within 7 days after carbon NP instillation.
Results
Cftr mutant females had moderately reduced lung compliance and slightly increased airway resistance compared to wild type mice. We found no genotype dependent differences in total, regional and head deposition or in secondary-organ translocation of inhaled iridium NP. Titanium dioxide inhalation resulted in higher NP uptake by alveolar epithelial cells in Cftr mutants. Instillation of carbon NP induced a comparable acute and transient inflammatory response in both genotypes. The twofold increase of bronchoalveolar lavage (BAL) neutrophils in Cftr mutant compared to wild type mice at day 3 but not at days 1 and 7, indicated an impaired capacity in inflammation resolution in Cftr mutants. Concomitant to the delayed decline of neutrophils, BAL granulocyte-colony stimulating factor was augmented in Cftr mutant mice. Anti-inflammatory 15-hydroxyeicosatetraenoic acid was generally significantly lower in BAL of Cftr mutant than in wild type mice.
Conclusions
Despite lacking alterations in lung deposition and biokinetics of inhaled NP, and absence of significant differences in lung function, higher uptake of NP by alveolar epithelial cells and prolonged, acute inflammatory responses to NP exposure indicate a moderately increased susceptibility of lungs to adverse effects of inhaled NP in Cftr mutant mice and provides potential mechanisms for the increased susceptibility of CF patients to air pollution.
Similar content being viewed by others


Background
Air pollution continues to be a major threat to human health, accounting for an estimated 3.7 million premature deaths in 2012 [1]. The association between exposure to air pollution, especially to airborne particulate matter (PM), and a wide range of adverse health effects, particularly respiratory and cardiovascular effects, has been established by plentiful epidemiologic evidence, supported by mechanistic in vivo animal studies [2–4]. The susceptible populations are children, women in the reproductive age, the elderly and persons with pre-existing respiratory and cardiovascular diseases. While many studies have focused on the interaction of PM with asthma or chronic obstructive pulmonary disease (COPD) [5], relatively little is known about PM effects in patients with cystic fibrosis (CF).
Cystic fibrosis (CF) is caused by mutations in the gene encoding the CF transmembrane conductance regulator (CFTR) and results in altered ion transport regulation across epithelial membranes [6]. In lungs, defective CFTR leads to hyperviscous, adhesive airway mucus and ultimate failure of mucociliary clearance [7]. Recurrent infections with CF-specific bacteria with intense inflammatory responses contribute to irreversible lung injury [8]. The fine pulmonary structure of newborn CF infants is assumed to be normal. However, it has been suggested that the beginning of lung disease might precede the colonization with CF-related pathogens [9]. Furthermore, a significant heterogeneity in the severity of lung disease is observed in patients carrying the same mutation; the basis for the divergence between genotype and lung phenotype has been attributed to modifier genes, but also to environmental factors [10, 11]. Evidence for a relationship between PM10 and PM2.5 pollution and pulmonary disease in CF was found in a cohort study with higher rates of pulmonary exacerbations in CF patients [12]. Potential mechanisms discussed are changes in the breathing pattern, which result in increased particle deposition in CF patients [13], or alterations of mucociliary and/or macrophage-mediated particle clearance [14, 15].
Beside PM10 and PM2.5, ultrafine or nanoparticles (UFP, NP, particles with diameters < 100 nm) are of increasing concern [16]. Adverse health effects occur at current levels of exposure, and no threshold concentration has been identified below which air pollution poses no risk for human health [17, 18]. In addition, nanotechnology industry generates daily new NP, which may become airborne at some stage of their life cycle and thus pose additional health risks.
There is evidence for decreased NP clearance from lungs of patients with chronic obstructive pulmonary disease (COPD) [19] and for impaired NP uptake by macrophages in COPD mice [20]. Hence, there are several factors which may contribute to an enhanced susceptibility of CF lungs to adverse effects of inhaled NP.
To evaluate individual factors and multifactorial aspects that may lead to enhanced susceptibility in CF lungs (Figure 1A), we investigated in a comprehensive approach (Figure 1B):
-
1.
The distribution and clearance of inhaled 20-nm titanium dioxide NP (TiO2NP) at the individual particle level using analytical transmission electron microscopy (TEM) in chloride channel defective Cftr TgH (neoim) Hgu mice in comparison to wild type (WT) littermates [21].
-
2.
Inhaled radio-labeled iridium NP (IrNP) served to assess the biokinetics of NP.
-
3.
We examined lung function, since its alteration may affect intrapulmonary particle deposition.
-
4.
Lung exposure to carbon NP (CNP), an abundant component of urban, particulate air pollution served to evaluate the inflammatory responses as biological outcome.

Study motivation and experimental design. A) Individual factors and multifactorial aspects for enhanced susceptibility of CF lungs to PM exposure. B) Time frame for lung function measurements, treatments with NP and subsequent analyses. CNP-Instillation: intratracheal instillation of carbon nanoparticles; BALF: Bronchoalveolar lavage fluid.
Results
Lung physiology and fine pulmonary structures
Lung function data are summarized in Table 1. In general, body weight (BW) was lower in females than in males, but comparable among Cftr mutant and WT mice. In accordance with morphological studies [22, 23], the mutation had no effect on lung size (TLC, total lung capacity), both, for absolute values and when expressed in relation to BW (TLC/BW). Similarly, the conducting airway volume (VD, series dead space volume) and its relative proportion to lung size (VD/TLC), as well as the gas exchanging capacity across the alveolar capillary membrane (DCO, diffusing capacity for carbon monoxide CO) were not altered, the latter also not when accounting for the alveolar volume (VA, DCO/VA). However, female Cftr mutants showed about 20% reduced respiratory system (CRS) and lung compliance (CL) values and an increased respiratory system resistance (R) compared to WT mice. These differences remained statistically significant when related to lungs size (CRS/TLC, CL/TLC, and specific respiratory system resistance, sR (R × TLC), respectively). Thus, the lungs of 3 months old female Cftr mutants are somewhat less distensible and put more resistance to airflow during breathing, which results in an increased work of breathing. Lung function of heterozygous females was not significantly affected, although there was a trend to limited compliance and resistance values. In male animals, differences between WT and Cftr mutant mice were noticeably for lung compliance (CL) and resistance (R), but they were of lower extent (10%) than in females and failed to be statistically significant.
Also in agreement with the above mentioned morphological studies [22, 23], we found no statistically significant differences in any of the lung structure parameters analyzed between Cftr mutant and WT mice. The lung tissue content in Cftr mutant mice was 15.0 ± 4.2% (range 12.4 – 19.8%). This pulmonary tissue consisted of 40.2 ± 6.7% connective tissue (range 33.3 – 46.8%), 33.4 ± 12.6% capillaries (range 19.1 – 43.3%) and 26.4 ± 9.2% epithelial cells (range 16.2 – 34.1%). In WT mice, 15.3 ± 2.3% was lung tissue (range 12.7 – 17.1%), which was composed of 27.8 ± 4.6% connective tissue (range 22.9 – 31.9%), 42.3 ± 6.6% capillaries (range 34.8 – 46.4%) and 29.9 ± 4.3% epithelial cells (range 25.0 – 33.3%).
Cellular uptake of inhaled TiO2NP
A total of 469 - 659 hexagonal fields (equal to a volume of 6100 – 8600 μm3) on ultrathin lung sections were analyzed per animal for the presence of TiO2NP. Particles were first identified and morphologically localized by energy filtering transmission electron microscopy (EFTEM). NP uptake was further assessed by quantitative morphometry.
In Cftr mutants, we found a total of 8 TiO2 particles intracellularly, namely 5 NP in alveolar epithelial type 2 cells and 3 NP in macrophages, while in WT mice 7 particles were found, all in macrophages. Additional subsampling and analysis of 83 - 110 macrophages (on 2208 – 3260 hexagonal fields) per animal revealed 11 TiO2NP in Cftr mutants and 11 in WT mice. Hence, TiO2NP were found in alveolar type 2 epithelial cells as well as in macrophages in Cftr mutants and in macrophages only in WT mice (Table 2).
In animals of both genotypes, TiO2NP were found in vesicles largely exceeding NP size (Figure 2, Table 2). Thus, the ratio of particle to vesicle size RV/P, which allows discrimination of endocytic uptake mechanisms [14, 24], largely exceeded RV/P ~ 1, i.e. the value representing phagocytic particle uptake. There were no differences observed in the diameters of NP-containing vesicles (dv) and internalized TiO2NP (dNP) between Cftr mutant and WT mice (Table 2). The distance of the particles to the vesicle membrane (dP-M), however, was with 28.2 ± 38.2 nm significantly (p = 0.026) smaller in macrophages of Cftr mutants compared to 58.1 ± 76.2 nm in WT mice. There were no significant differences in any of the parameters analyzed between NP in lung epithelial cells and in macrophages.

Ultrastructural localization of TiO 2 NP in lung tissue. TiO2NP (arrows): A) in a vesicle of a macrophage that is closely associated with the alveolar epithelium (arrow heads); a) high magnification micrograph of the NP-containing vesicle. B) in large vesicle of macrophage containing lamellar structures (surfactant); b) high magnification micrograph of the NP-containing phagolysosome. C) and D), in vesicles of alveolar type 2 epithelial cells. Note the close association of the TiO2NP to the vesicle membrane.
There was no particulate titanium found in control mice, which had been exposed to clean, filtered air.
Biokinetics of IrNP
We found no significant differences in total, regional and head deposition of inhaled IrNP between Cftr mutant and WT mice (Figures 3 and 4). Inhaled NP translocation from lungs into systemic circulation and secondary target organs was also not different between the two genotypes, immediately (0 h) as well as 24 h after exposure to the NP aerosol (Figure 5).

Deposition of inhaled IrNP. A) Total deposition determined either immediately (0 h) or 24 h after a 1-h inhalation exposure; given relative to the estimated inhaled aerosol. B) Head deposition determined immediately (0 h) after a 1-h inhalation exposure. No significant difference of total and head deposition between the two animal groups.

Regional deposition of inhaled IrNP determined either immediately (0 h) or 24 h after a 1-h inhalation exposure. The fast cleared fraction (A) estimates the airway deposition fraction including head airways. Regional deposition is given relative to the totally deposited aerosol. The difference between 0 h and 24 h relates to the fact that fast clearance is not finished after the 1-h inhalation. Therefore, only the 24-h values are to be considered. No significant difference between the two animal groups. Peripheral lung retention (B) represents long-term retained IrNP. The difference between 0 h and 24 h relates to the fact that fast clearance is not finished after the 1-h inhalation, adding to lung retention. Therefore, only the 24-h values are to be considered for IrNP retention in the peripheral lungs. No significant difference between the two animal groups.

Translocation of inhaled IrNP towards secondary target organs determined 24 h after the 1-h aerosol inhalation. No significant difference between the two animal groups.
Inflammatory response to CNP
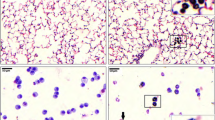
Histopathological examination of the lungs revealed focal accumulation of particle-loaded macrophages and granulocyte infiltrates with debris (alveolitis), which was most pronounced at day 1 after CNP instillation. There were no differences observed between Cftr mutant and WT mice.
Total BAL cell numbers in untreated controls were comparable in both genotypes, i.e. 3.4 ± 0.6 × 105 in Cftr mutants and 5.0 ± 1.6 × 105 in WT mice. Intratracheal instillation of 20 μg CNP induced an acute BAL inflammatory cell infiltration dominated by neutrophils at day 1, which was equivalent in both animal groups (Figure 6). The subsequent resolution of the inflammation, analyzed at days 3 and 7 after CNP instillation, however, differed significantly between the two animal groups. BAL neutrophils decreased fivefold from 35% at day 1 to 7% at day 3 in WT mice, compared to a twofold decrease from 27% to 13% in Cftr mutants. Despite of this difference at day 3, neutrophils decreased to almost baseline levels, i.e. to 3% at day 7 in both animal groups. BAL lymphocyte and macrophage numbers increased after CNP instillation as well, though not statistically different between the two animal groups. In line with the lung response, peripheral blood monocytes were significantly increased at days 1 and 3 in Cftr mutant compared to WT mice, and levelled off at day 7 as well (data not shown).

BAL cells after CNP instillation. Exposure to CNP led to a twofold increase of BAL neutrophils in Cftr mutants at day 3, but not at days 1 and 7. *: significant difference to WT mice, p < 0.05. CC: cage control (untreated animals); Sham: instillation of 50 μL pyrogene-free distilled water.
The analysis of CNP-induced production of BAL cytokines (IL-1α, -6, -12p40, -12p70, G-CSF, CXCL-1, -5, CCL-3, -5) reflected the inflammatory response observed at the leukocyte level, whereby most cytokine levels were comparable between Cftr mutant and WT mice (Table 3, Figure 7A). At day 3 after CNP instillation, however, the inflammatory mediators G-CSF, CXCL5, CCL5, osteopontin (OPN/SPP1) and lipocalin-2 (LCN2) were significantly increased in Cftr mutant compared to WT mice (Table 3, Figure 7A/B). Most strikingly, G-CSF, which is critical for the survival of granulocytes, was 13 times higher in Cftr mutant compared to WT mice. This difference matches well with the prolonged high number of BAL neutrophils and indicates increased survival and poor clearance of BAL neutrophils in Cftr mutant mice. Additional immunohistological analysis indicated focal expression of osteopontin, a marker for inflammatory macrophages [25], at the site of accumulation of particle-loaded macrophages and accompanying cell debris.

BAL cytokines and lipid mediators after CNP instillation. A) Heat map of cytokines. Black boxes indicate lowest and bright red highest detected levels for each cytokine (according to Table 3). Cytokines have been sorted by the response pattern. IL-1a, IL-6, and CXCL1 showed highest levels at day 1 after CNP exposure, CCL5, OPN, and IL-12p40 at day 3. Overall elevated cytokine levels were observed in Cftr mutant (−/−) as compared to WT (+/+) mice. B) 15-HETE. Increase over time after instillation treatment in WT mice, but independently of the CNP exposure; 15-HETE was generally reduced in Cftr mutant mice (hatched bars). *: significant difference to WT animals, p < 0.05. CC: cage control (untreated animals); Sham: instillation of 50 μL pyrogene-free distilled water.
BAL lipid mediator analysis revealed generally reduced levels of 15(S)-HETE, a major anti-inflammatory arachidonic acid metabolite from the 15-lipoxygenase pathway in Cftr mutants, statistically significant at days 1 and 3 after CNP instillation as well as after sham treatment (Figure 7B). The lipid mediators, LTB4, lipoxin-A4 and PGE2 remained unchanged (data not shown), except for 8-Isoprostan (8-IP), which was significantly reduced in Cftr mutants (48.6 ± 26.7 pg/mL) compared to WT mice (447.2 ± 219.5 pg/mL) at day 3.
Discussion
In a comprehensive approach respecting possible multifactorial causes for enhanced susceptibility of CF lungs to environmental PM exposure (Figure 1), we evaluated whether structural or functional parameters of the Cftr mutant lungs were affected by the mutation. Overall, we have no indications for microstructural changes of the peripheral lung, which is in agreement with our previous findings [22, 23]. While lung and airway volumes as well as gas exchange capacity were not affected by the mutation, we found moderate changes in lung resistance and compliance. Since structural changes were not detectable, an altered homeostasis of the pulmonary lining fluid may be a potential explanation. We also do not have indications for altered deposition or biokinetics of inhaled IrNP in the body.
However, despite finding an almost ‘normal lung’ in Cftr mutants, there is evidence for alterations in the cellular uptake of inhaled TiO2NP similar to findings we recently obtained for the uptake of inhaled 20-nm gold (Au) NP in Scnn1b transgenic mice (airway targeted overexpression of the epithelial Na + channel β subunit Scnn1b), which mimic key aspects of chronic obstructive lung diseases in humans [20]: (i) The uptake of a substantial part of the NP (26%) by alveolar epithelial cells in Cftr mutants points to less efficient TiO2NP uptake by surface macrophages in these mice. This may promote depth translocation of the NP, for which, however, we obtained no evidence from the data of the biokinetic study with IrNP. Enhanced interaction of NP with epithelial cells may induce or alter inflammatory responses, as we have observed -and which we discuss in detail below- in Cftr mutants after exposure to CNP. As in a previous inhalation study with 20-nm TiO2NP in rat lungs [26], we did not observe TiO2NP bound to the apical surface of epithelial cells. (ii) The localization of TiO2NP in vesicles largely exceeding NP size (RV/P > > 1) in all cell types and in both animal groups indicates endocytic uptake of the NP. This finding is in agreement with other reports on intra-vesicular particle localization, suggesting NP uptake by cells to mainly occur by endocytic processes [27]. (iii) The close localization of NP to the vesicular membrane (dNP-M) indicates that deposited NP attached to the cell surface, resulting in internalization of the material by endocytosis. This correlates with findings in the above mentioned inhalation studies in rodents with TiO2 and Au nanoparticles [26, 20]. In the latter study, fast attachment of AuNP to lung cells has been demonstrated [26]. Monocyte/macrophage expressed CFTR protein has recently been described to not only support phagolysosomal acidification but also contribute to the opsonization and phagocytosis of particles and bacteria [28]. Data showing the statistically significant reduction of dNP-M in Cftr mutant compared to WT mice in the present study support this idea. The absence of CFTR protein in the macrophage membrane may have resulted in smaller particle-protein complexes, with possible consequences for the efficacy of particle uptake in macrophages. In addition, internalized particles would appear closer to the vesicle membrane. Besides receptor binding, adhesion forces may represent an alternative mechanism for the close localization of AuNP to the vesicle membrane [29]. In this case, however, we would expect dNP-M values near 0. In addition, adhesion forces provide no explanation for the recorded difference in dNP-M between Cftr mutant and WT mice.
In addition to altered cellular uptake of TiO2NP in Cftr mutants, changes in the resolution of the inflammation in response to CNP exposure in these mice provide experimental support for the finding of an increased susceptibility of CF patients to environmental PM exposure [12]. While CNP instillation induced an acute alveolitis equally in Cftr mutant and WT mice at day 1, there were twice as much BAL neutrophils in Cftr mutant compared to WT mice at day 3. Thus, at day 3, 70% of the neutrophils were already removed in WT mice compared to 23% in Cftr mutants. The slower decline of BAL neutrophils indicates delayed or impaired resolution of the inflammation in Cftr mutant mice. This is further supported by the rate of the neutrophils’ decrease from day 3 to 7, which remained basically unchanged in Cftr mutants, whereas it was biphasic in WT mice (Figure 8A). Recruitment of macrophages and lymphocytes was apparently not affected in Cftr mutant mice, as the numbers of these cell types in BAL were similar in both animal groups.

Neutrophilic inflammation and its resolution in CF airways. A) Time course of BAL neutrophils after CNP instillation showing a slower decline of BAL neutrophils from days 1 to 7 in Cftr mutant compared to WT mice. *: significant difference to WT animals, p < 0.05. B) Mode of action for impaired inflammation resolution in CF airways. The accumulation of inflammatory cells (PMN) in Cftr mutant lungs at day 3 after particle exposure is probably supported by different pathways: Augmented CXCL5 levels, released from Cftr mutant inflammatory epithelial cells, enhance exposure-related PMN recruitment and accumulation on the airway surface. In addition, elevated G-CSF and reduced 15-HETE levels in Cftr mutant lungs contribute to prolonged PMN survival and impaired efferocytosis of apoptotic PMN by macrophages. PMN: Polymorphnuclear cells = neutrophils, AM: Airway macrophages, AEC: airway and alveolar epithelial cells, EC: endothelial cells.
Besides the acute response peaking at day 1, BAL cytokine profiling also indicated higher concentrations of G-CSF, CCL3, CCL5, CXCL5, LCN2, OPN and IL-12p40 at day 3 in Cftr mutant compared to WT mice. Thereby, the increases in CCL5, OPN and IL-12p40 in Cftr mutants indicate inflammatory-stimulated macrophages, which are important producers of G-CSF. Increased levels of CXCL5 and LCN2 indicate inflammatory stimulation of the epithelium attracting neutrophils. Consequently, increased G-CSF levels until day 3 supports the survival and persistence of the otherwise short-lived neutrophils in Cftr mutants. Thus, the congruent cellular and molecular data, which are summarized in Figure 8B, suggest that an impaired resolution of the inflammation causes increased neutrophil numbers at day 3 in Cftr mutant mice rather than escalating recruitment of inflammatory cells during the initiation phase. The generally significantly lower levels of BAL 15-HETE in Cftr mutant compared to WT mice further suggests an association of this lipid mediator with the compromised resolution of CNP-induced airway inflammation in Cftr mutants. The eicosanoid 15-HETE is not only expressed by leukocytes but also by the respiratory epithelium [30], and it is known as an anti-inflammatory and pro-resolving lipid mediator, which counteracts neutrophilic airway inflammation [31]. In humans, the 15-keto-PGE2–PPAR-γ system has been suggested to regulate abnormal mucus production in CF, with the unsaturated fatty acid derivate 15-keto-PGE2 and 15-HETE representing crucial PPAR-γ ligands [32].
In summary, despite lacking significant alterations in lung deposition and biokinetics of inhaled NP and absence of significant differences in lung function and structure, alterations in cellular uptake of inhaled TiO2 and the significantly enhanced, acute inflammatory responses to NP exposure indicate a moderately increased susceptibility of Cftr mutant lungs.
Conclusions
Our results lead to the conclusion that the function of the CFTR gene, to the extend as investigated here in the CftrTgH (neoim) Hgu mouse line, is dispensable for lung function development as well as to mechanisms required for nanoparticle deposition or translocation. In contrast, the absence of CFTR appeared to modify cellular uptake of NP and caused a moderate impairment of the resolution of a by particle instillation induced neutrophilic airway inflammation. While the described effects might not be alarming for a single exposure to NP under aseptic conditions, the lungs of a CFTR deficient organism may be more susceptible to repetitive or chronic exposures to inflammatory stimuli like particulate air pollution and airway infection. Under these conditions, insufficiently resolved inflammation might promote the development of chronic inflammatory lung disease.
Materials and methods
Experimental design
To resolve mechanisms contributing to enhanced susceptibility to inhaled NP in Cftr mutants (Figure 1A), we studied (i) lung function prior to NP exposure, (ii) NP distribution in lung cells immediately (0 h) and 24 h after a 1-h exposure to 20-nm TiO2 aerosol, and (iii) NP biokinetics within 24 h after exposure to radio-labeled 30-nm Ir aerosol (Figure 1B). Finally, (iv) inflammatory responses were determined at days 1, 3 and 7 after intratracheal instillation of carbon NP (CNP, mean agglomerate size 210 nm), in Cftr mutant mice with no signs of lung disease and in WT littermates.
Animals
Origin, genetics, husbandry
CftrTgH (neoim) Hgu transgenic (Cftrtm1HGU) mice created by targeted insertional mutagenesis have markedly decreased CFTR activity [21, 33]. They survive to adulthood and have a phenotype resembling that of human compound heterozygotes. Although these mice – like most other CF mouse models – do not develop lung disease spontaneously, Cftrtm1HGU mice develop pathogen-specific lung disease after bacterial challenge; they are more easily infected and develop more severe lung pathology than non-cf mice [34]. Uninfected Cftrtm1HGU mice have a normal fine pulmonary structure including the lung lining layer [22, 23].
Cftr mutant and WT mice (MF1, Charles River, Sulzfeld, Germany) were bread and reared in the animal facility of the Institute of Lung Biology and Disease at the Helmholtz-Center Munich, Neuherberg/Munich, Germany. Mice were housed in individually ventilated cages (IVC; BioZone, Ramsgate, UK) supplied with filtered air and access to food and water ad libitum. They were kept on a 12 h day/night cycle, humidity was maintained at 55% and the temperature of the control room was 22°C. Mouse progeny were genotyped by tail biopsy polymerase chain reaction (PCR) using the genotyping primers HGU-Neoplas-KO1: 5′-AAA GTG TAA AGC CTG GGG TGC-3′ and HGU-NeoAS-KO2: 5′-AGA ACT CGT CAA GAA GGC GAT AG-3′ generating a 3691 bp KO-product and HGU-Int9-WT1: 5′-GGA GCC TAG CAT AGA TGT CTC C-3′ and HGU-M10B-WT2: 5′-CTG CTG TAG TTG GCA AGC TTT GAC-3′ generating a 3274 bp WT-product (PCR conditions: 33× 94°C 30 sec, 64°C 30 sec, 72°C 2 min). Specified pathogen-free status was approved by a health certificate according to Federation of European Laboratory Animals Science Association (FELASA) guidelines.
Animals were treated humanely and with regard for alleviation of suffering. All experiments were conducted under federal guidelines for the use and care of laboratory animals and were approved by the Bavarian Animal Research Authority and by the Helmholtz Center’s Institutional Animal Care and Use Committee, as well as in accordance with the Swiss Federal Act on Animal Protection and the Swiss Animals Protection Ordinance.
Nanoparticles
We selected the three different NP types for the following technical reasons: Biokinetics of inhaled NP particles: Iridium and titanium dioxide NP, since we previously developed reliable techniques of stably radio-labeling the IrNP [35, 36] and elemental microanalysis by EFTEM for unambiguous identification of the TiO2NP [37]. The sensitivity of the radio-analysis allowed administering low doses in the μg range during a rather short 1-2-hour inhalation, thereby avoiding any toxicological side effects by the NP used. Besides the appropriate radio-analysis of both NP, identification of NP by EFTEM allowed elemental mapping of individual TiO2NP at the ultrastructural level in the tissue.
Inflammatory response of the lung: The comparative investigation of toxicological effects of NP in Cftr mutant and WT mice required applying higher NP doses, such that an acute inflammatory response was induced. Since carbon nanoparticles are most relevant for environmental exposure, pure elemental CNP were administered by intratracheal instillation [38]. The latter method allowed reproducible and well controlled NP dosing, which was important to compare toxicological responses to NP in Cftr mutant and WT mice. However, this bolus application is less physiological than inhalation. The deposition pattern of instilled NP is different from that of inhaled NP. The target tissue of instilled and inhaled NP, however, remains the same, as NP are deposited on the epithelium of airways and alveoli. Since the lungs of our animals were “sterile” prior to NP treatments, there was no pre-existing lung pathology with excess mucus production. In addition, we have previously performed comprehensive characterization of all three NP (IrNP: [35, 36]; TiO2NP: [37, 39]; CNP: [38, 40, 41]). Thereby, we have shown that the morphology of all three particle types was very similar (see also Figure 9), namely chain-aggregated/agglomerated NP consisting of primary particles in the size range of about 5 nm (CNP: 5 - 12 nm).

Morphology of nanoparticles. Chain-aggregated/agglomerated NP consisting of primary particles in the size range of about 5 nm (CNP: 5 - 12 nm). A) IrNP, B) TiO2NP and C) CNP.
Lung distribution and clearance of inhaled TiO2 aerosols
Three Cftr mutant and three WT mice (female, aged 10 - 12 weeks, body weight 17.7 - 20.8 g) were used. There were no significant differences in age or body weight between mutant and WT mice. For particle inhalation, animals were deeply anaesthetized by intraperitoneal (i.p.) injection of a mixture of medetomidine (Domitor®, Pfizer GmbH, Karlsruhe, Germany; 500 μg/Kg), midazolam (Dormicum®, Hoffmann-La Roche AG, Grenzach-Wyhlen, Germany; 5 mg/Kg) and fentanyl (Fentanyl®, Janssen-Cilag GmbH, Neuss, Germany; 50 μg/Kg). For lung fixation immediately after inhalation, anesthesia was deepened with i.p. injection of a mixture of xylazin (Rompun®, Bayer Vital GmbH, Leverkusen; 5 mg/Kg) and ketamine (Ketamin 10%, WDT eG, Garbsen, Germany; 100 mg/Kg) prior to exsanguination by cutting the abdominal aorta. For 24-h examinations, anesthesia was antagonized by subcutaneous injection of atipamezole (Antisedan®, Pfizer GmbH Karlsruhe, Germany; 2.5 mg/kg), flumazenil (Anexate®, Hoffmann-La Roche AG, Grenzach-Wyhlen, Germany; 500 μg/Kg), and naloxone (Narcanti®, Janssen Animal Health, Neuss, Germany; 1.2 mg/Kg). For lung fixation at 24 h, a mixture of xylazin and ketamine was injected prior to euthanasia by exsanguination as above.
TiO2 aerosols, particle inhalation and deposition
TiO2 aerosol generation and inhalation were performed as previously described [39, 26]. Briefly, ultrafine anatase TiO2 aerosols were generated with a Palas spark generator, quasi-neutralized with a radioactive 85Kr source, diluted and conditioned for inhalation in terms of gas composition, humidity and temperature. Particle size distribution and total number concentration were continuously monitored by a differential electrical mobility particle sizer (DMPS) and a condensation particle counter (CPC). The aerosol produced had a count median diameter (CMD) of about 20 nm (geometric standard deviation, GSD = 1.6) and a mean number concentration of 1 - 2 × 1013 particles/m3, resulting in a mass concentration of about 30 – 40 μg/m3. In addition, TiO2 aerosols were sampled for morphologic analysis on filters and on formvar-coated copper grids using a TEM particle sampler (Fachhochschule Nordwestschweiz FHNW, Windisch, Switzerland).
The deeply anesthetized mice were placed in airtight plethysmograph boxes for 1 h and inhaled the aerosols via an endotracheal tube by negative-pressure ventilation (-1.5 kPa) at a tidal volume of about 170 μl and a breathing rate of 120/min resulting in a minute ventilation of 20 cm3/min, which is 20% more than in spontaneously breathing mice. This inhalation protocol optimized NP deposition per exposure time. The deposited amount of TiO2 was calculated to be 0.1 μg or 7 × 109 NP in each Cftr mutant or WT mouse lung (assuming a 40% deposition probability).
Two additional mice (one Cftr mutant and one WT) were not subjected to aerosol inhalation and served as controls.
Organ and tissue processing for microscopic analysis
To analyze the distribution of inhaled TiO2NP in lung cells at the individual particle level, organs were fixed in situ by instillation of phosphate-buffered 2.5% glutaraldehyde (350 mOsm, pH 7.4) at a pressure of 20 - 25 cm H2O as described previously [22]. Thereafter, lungs were removed in toto from the thorax and stored for at least 24 h in fixative solution at 4°C. Lung volumes [42] were 0.70 - 0.95 mL in Cftr mutant and 0.62 - 0.82 mL in WT mice and were not significantly different between the two animal groups.
Lungs were then subjected to systematic tissue sampling, post fixation with osmium tetroxide and uranyl acetate (Sigma-Aldrich, Buchs, Switzerland), dehydration in ethanol and embedding in epon (Sigma-Aldrich) as described previously [37, 43]. Ultrathin sections (≤50 nm) were cut from 5 - 6 tissue blocks per animal, mounted onto formvar coated 600-mesh hexagonal copper grids and stained with uranyl acetate and lead citrate (Ultrastain; Leica, Glattbrugg, Switzerland).
TiO2NP were identified on ultrathin tissue sections by EFTEM using a LEO 912 TEM (Zeiss, Oberkochen, Germany) equipped with an in-column omega energy filter and operated at 120 kV as described previously [37]. Electron spectroscopic imaging (ESI) was performed with an energy slit width of 10 eV at the titanium L-edge at 464 eV using the three-window method implemented in the iTEM software (Olympus, Muenster, Germany). On each ultrathin section, we determined a random starting point from which we meander-like analyzed every fourth hexagonal field for the presence of TiO2NP and its anatomical-histological and (sub) cellular localization. In addition, on each ultrathin section all surface macrophages were sampled and analyzed for TiO2NP.
Finally, sections of ~ 1 μm thickness were cut from the sampled tissue blocks and stained with toluidine blue to evaluate the fine pulmonary structure by estimating the corresponding ratios with a point counting system.
Lung physiology
Lung function was determined in anesthetized and intubated, 14-week old male and female Cftr mutant (n = 35) and WT (n = 16) mice as described previously [44, 45]. It covers lung volumes, conducting airway volume, respiratory mechanics and gas exchange capacity. Body weight in females was significantly lower than in their male counterparts, the difference was 24% in Cftr mutants and 29% in WT mice (for details see Table 1).
Deposition and biokinetics of inhaled IrNP
Sixteen female Cftr mutant and 16 WT mice (age: 10 - 12 weeks; body weight: 20 - 22 g) were used for biokinetic studies. Radiolabeled Ir (192Ir) aerosols of 30 nm CMD (GSD = 1.6) were generated as described for TiO2 above; the mean aerosol concentration was 3 × 106 particles/cm3. Anesthetized mice inhaled the aerosol for 1 h during spontaneous breathing in a nose-only apparatus described previously for NP exposure of newborn rats [46]; about 2 × 109 particles corresponding to 0.9 μg were deposited in the lungs. Mice were then treated as described above and euthanized either immediately (0 h) or 24 h after the 1-h aerosol inhalation.
Organs were harvested and Ir was analyzed quantitatively by gamma-spectrometry in all organs and tissue samples, as well as the remaining carcass and the excretion.
Inflammation
Thirty nine Cftr mutant and 30 WT mice (female, age: 9 – 11 weeks, body weight: 19.1 - 22.3 g) were used to investigate the course of CNP induced lung inflammation.
CNP generation and instillation
CNP were generated by spark discharge from graphite electrodes (GFG1000, Palas, Karlsruhe, Germany) [47]. They mimic combustion-derived particles with low organic carbon content, large specific surface area (800 m2/g), as well as structural and compositional similarities to modern (Euro IV) diesel soot [48]. The physicochemical characteristics of CNP have been described in detail earlier [38, 40, 41]. Anesthetized mice were intubated by a nonsurgical technique and instilled with an aqueous suspension of 20 μg CNP in 50 μL pyrogen-free distilled water, followed by 100 μL of air [38, 49]. The suspension of poorly soluble CNP was sonicated on ice for 1 min prior to instillation, using a SonoPlus HD70 (Bachofer, Berlin, Germany) at a moderate energy of 20 W to ensure sufficient particle dispersion with a mean agglomerate size of 210 nm (Zetasizer Nano ZS, Malvern Instruments, Herrenberg, Germany). Control animals were either instilled with 50 μL of pyrogen-free distilled water (Sham), or left undisturbed in their cages (cage control, CC).
Sample collection and preparation for inflammation analysis
Anesthetized mice were euthanized by exsanguination at days 1, 3 and 7 after CNP instillation. Blood was withdrawn from the retro orbital plexus with a capillary and collected in EDTA coated tubes (Sarstedt, Germany) for hematological analysis (ADVIA Hematology Systems (Bayer Diagnostics). Lungs were lavaged with 10 × 1 mL phosphate buffered saline (PBS, calcium and magnesium free) as described previously [38]. The BAL fluids from the first two lavages were used for protein analysis after cell separation by centrifugation (425 × g, 20 min at room temperature). The cell pellet was resuspended in 1 mL RPMI 1640 medium (BioChrome, Berlin, Germany) supplemented with 10% fetal calf serum (Seromed, Berlin, Germany). Cell viability was determined by the Trypan blue dye exclusion test. Differential cell counts were performed on cytospin preparations (May-Grünwald-Giemsa staining; 2 × 200 cells were counted).
BAL protein analysis
Total BAL protein content was determined spectrophotometrically with an ELISA reader (Labsystems iEMSReader MF, Helsinki, Finland) at 620 nm, using the Bio-Rad Protein Assay Dye Reagent (no. 500-0006; BioRad, Munich, Germany).
Cytokine analysis in BAL fluid was performed using the Bio-Plex bead-based suspension array system and the appropriate detection kits (Pro Mouse 10Plex, Bio-Rad, Hercules CA, USA) and specific ELISAs (mouse, R&D Duo Sets) for osteopontin (OPN/SPP1, catalog number DY441), galectin-3 (DY1197), LIX/CXCL5 (DY443) and lipocalin-2 (NCL2/NGAL, DY1857) as described previously [49].
Lung concentrations of the lipid mediators 15(S)-HETE and PGE2 were assessed as follows: Lung homogenates (about 50 mg lung tissue/500 μL) were prepared by disrupting lung tissue in HEPES buffer, pH 7.4, with lysing matrix E (FastPrep FP120 cell disrupter; MP Biomedicals Germany GmbH, Eschwege, Germany). After centrifugation (3200 × g at 4°C for 10 min), aliquots of the supernatants were taken for determination of protein and lipid mediators. Aliquots of supernatants derived from cell homogenates of particle-treated cells (prepared as described above) and those from lung homogenates were deproteinized by adding an 8-fold volume of 90% methanol containing 0.5 mM EDTA and 1 mM 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl, pH 7.4 [50]. Methanol suspensions were stored at -40°C for 24 h followed by two centrifugation steps at 10,000 × g for 20 min at 4°C with a 24 h interval to remove the proteins. Aliquots of the obtained supernatants were dried in a vacuum centrifuge, dissolved in assay buffer and used for quantification of PGE2, Lipoxin-A4 and 15(S)-HETE by their specific enzyme immunoassays (Cayman Chemical Company, Ann Arbor, MI, USA) according to manufacturer’s protocol. Protein was measured at 595 nm using 5 μL of homogenate and 200 μL of 1:5 diluted BioRad solution (Bio-Rad, Munich, Germany) with bovine serum albumin as standard.
Histopathology and immunohistochemistry
Two to three mice of each group were sacrificed for histopathological and immunohistochemical examinations after CNP instillation. Lungs were fixed without lavage in phosphate buffered saline (pH 7.4) and 4% formalin at an inflation pressure of 20 cm H2O and embedded in paraffin.
Since we have previously shown that osteopontin (OPN/SPP1) expression is induced in alveolar macrophages at day 1 after CNP inhalation [25] and SPP1 concentration was found to be increased in BAL fluid, we performed immunohistochemical analysis of osteopontin in lung sections using a polyclonal antibody (R&D Systems, Wiesbaden, Germany). After staining with a biotinylated secondary antibody (Vector laboratories Inc., Burlingame, California, USA) and streptavidin-Vectastain Elite ABC peroxidase reagents (Vector Laboratories Inc.), lung sections were developed with Nova Red (Vector Laboratories Inc.) and counterstained with hematoxylin.
Statistics
Lung physiology, deposition, distribution and biokinetics of inhaled NP: The non-parametric Mann–Whitney Test was used for two group comparisons and one-way analysis of variance (ANOWA) for multiple group comparisons. Inflammation: Two-way ANOVA was used to analyze differences between control and various exposure groups. Group means of WT and Cftr mutant mice were compared using the Student’s t-test. Overall, a significance level of p < 0.05 was adopted.
Abbreviations
- BAL:
-
Bronchoalveolar lavage
- BW:
-
Body weight
- C:
-
Carbon
- CF:
-
Cystic fibrosis
- CFTR:
-
Cystic fibrosis transmembrane conductance regulator
- CCL:
-
Chemokine (C-C motif) ligand
- CL:
-
Lung compliance
- CMD:
-
Count median diameter
- CNP:
-
Carbon nanoparticles
- COPD:
-
Chronic obstructive pulmonary disease
- CPC:
-
Condensation particle counter
- CRS:
-
Respiratory system compliance
- CXCL:
-
Chemokine (C-X-C motif) ligand
- DCO:
-
Diffusing capacity for carbon monoxide
- EFTEM:
-
Energy filtering transmission electron microscopy
- ESI:
-
Electron spectroscopic imaging
- G-CSF:
-
Granulocyte-colony stimulating factor
- HETE:
-
Hydroxyeicosatetraenoic acid
- i.p.:
-
Intraperitoneal
- IL:
-
Interleukin
- Ir:
-
Iridium
- IrNP:
-
Iridium nanoparticles
- LCN:
-
Lipocalin
- LDH:
-
Lactate dehydrogenase
- LTB:
-
Leukotriene B
- NP:
-
Nanoparticle (s)
- OPN:
-
Osteopontin
- PCR:
-
Polymerase chain reaction
- PGE:
-
Prostaglandin E
- PM:
-
Particulate matter
- PPAR:
-
peroxisome proliferator-activated receptor
- R:
-
Resistance
- Scnn1b:
-
Epithelial Na + channel β subunit
- TiO2:
-
Titanium dioxide
- TiO2NP:
-
Titanium dioxide nanoparticles
- TLC:
-
Total lung capacity
- UFP:
-
Ultrafine particles
- DMPS:
-
Differential electrical mobility particle sizer
- SDg:
-
Geometric standard deviation
- VA:
-
Alveolar volume
- VD:
-
Series dead space volume
References
WHO: Burden of disease from air pollution for 2012. Geneva: World Health Organization 2014; 2014. http://www.who.int/mediacentre/news/releases/2014/air-pollution/en/
Anderson JO, Thundiyil JG, Stolbach A: Clearing the air: a review of the effects of particulate matter air pollution on human health. J Med Toxicol 2012,8(2):166–175. 10.1007/s13181-011-0203-1
Brook RD, Rajagopalan S, Pope CA 3rd, Brook JR, Bhatnagar A, Diez-Roux AV, Holguin F, Hong Y, Luepker RV, Mittleman MA, Peters A, Siscovick D, Smith SC Jr, Whitsel L, Kaufman JD: American Heart Association Council on Epidemiology and Prevention, Council on the Kidney in Cardiovascular Disease, and Council on Nutrition, Physical Activity and Metabolism: Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation 2010,121(21):2331–2378. 10.1161/CIR.0b013e3181dbece1
Rückerl R, Schneider A, Breitner S, Cyrys J, Peters A: Health effects of particulate air pollution: a review of epidemiological evidence. Inhal Toxicol 2011,23(10):555–592. 10.3109/08958378.2011.593587
Kelly FJ, Fussell JC: Air pollution and airway disease. Clin Exp Allergy 2011,41(8):1059–1071. 10.1111/j.1365-2222.2011.03776.x
Bear CE, Li C, Kartner N, Bridges RJ, Jensen TJ, Ramjeesingh M, Riordan JR: Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell 1992, 68: 809–818. 10.1016/0092-8674(92)90155-6
Donaldson SH, Boucher RC: Update on pathogenesis of cystic fibrosis lung disease. Curr Opin Pulm Med 2003, 9: 486–491. 10.1097/00063198-200311000-00007
Hauser AR, Jain M, Bar-Meir M, McColley SA: Clinical significance of microbial infection and adaptation in cystic fibrosis. Clin Microbiol Rev 2011,24(1):29–70. 10.1128/CMR.00036-10
Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW: Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med 1995, 151: 1075–1082.
Boyle MP: Strategies for identifying modifier genes in cystic fibrosis. Proc Am Thorac Soc 2007,4(1):52–57. 10.1513/pats.200605-129JG
Hamosh A, Corey M: Correlation between genotype and phenotype in patients with cystic fibrosis The Cystic Fibrosis Genotype-Phenotype Consortium. N Engl J Med 1993, 329: 1308–1313.
Goss CH, Newsom SA, Schildcrout JS, Sheppard L, Kaufman JD: Effect of ambient air pollution on pulmonary exacerbations and lung function in cystic fibrosis. Am J Respir Crit Care Med 2004, 169: 816–821. 10.1164/rccm.200306-779OC
Brown JS, Bennett WD: Deposition of coarse particles in cystic fibrosis: model predictions versus experimental results. J Aerosol Med 2004,17(3):239–248. 10.1089/jam.2004.17.239
Geiser M: Update on macrophage clearance of inhaled micro- and nanoparticles. J Aerosol Med Pulm Drug Deliv 2010,23(4):207–217. 10.1089/jamp.2009.0797
Randell SH, Boucher RC: Effective mucus clearance is essential for respiratory health. Am J Respir Cell Mol Biol 2006, 35: 20–28. 10.1165/rcmb.2006-0082SF
Kreyling WG, Tuch T, Peters A, Pitz M, Heinrich J, Stolzel M, Cyrys J, Heyder J, Wichmann HE: Diverging long-term trends in ambient urban particle mass and number concentrations associated with emission changes caused by the German unification. Atmos Environ 2003, 37: 3841–3848. 10.1016/S1352-2310(03)00457-6
Landy MK, Roberts MJ, Thomas SR: The wrong questions and why. In The Environmental Protection Agency: asking the wrong questions. New York, NY: Oxford University Press; 1990.
Landy MK, Roberts MJ, Thomas SR: Good questions. In The Environmental Protection Agency: asking the wrong questions. New York, NY: Oxford University Press; 1990.
Möller W, Felten K, Sommerer K, Scheuch G, Meyer G, Meyer P, Häussinger K, Kreyling WG: Deposition, retention and translocation of ultrafine particles from the central airways and lung periphery. Am J Respir Crit Care Med 2008, 177: 426–432. 10.1164/rccm.200602-301OC
Geiser M, Quaile O, Wenk A, Wigge C, Eigeldinger-Berthou S, Hirn S, Schäffler M, Schleh C, Möller W, Mall MA, Kreyling WG: Cellular uptake and localization of inhaled gold nanoparticles in lungs of mice with chronic obstructive pulmonary disease. Particle Fibre Toxicol 2013, 10: 19. 10.1186/1743-8977-10-19
Dorin JR, Dickinson P, Alton EW, Smith SN, Geddes DM, Stevenson BJ, Kimber WL, Fleming S, Clarke AR, Hooper ML, Andersen L, Beddington RSP, Porteous DJ: Cystic fibrosis in the mouse by targeted insertional mutagenesis. Nature 1992, 359: 211–215. 10.1038/359211a0
Geiser M, Zimmermann B, Baumann M, Cruz-Orive LM: Does lack of Cftr gene lead to developmental abnormalities in the lung? Exp Lung Res 2000, 26: 551–564. 10.1080/019021400750048090
Geiser M, Bastian S: Surface-lining layer of airways in cystic fibrosis mice. Am J Physiol Lung Cell Mol Physiol 2003, 285: L1277-L1285.
Swanson JA: Shaping cups into phagosomes and macropinosomes. Nat Rev Mol Cell Biol 2008,9(8):639–649. 10.1038/nrm2447
André E, Stoeger T, Takenaka S, Bahnweg M, Ritter B, Karg E, Lentner B, Reinhard C, Schulz H, Wjst M: Inhalation of ultrafine carbon particles triggers biphasic pro-inflammatory response in the mouse lung. Eur Respir J 2006,28(2):275–285. 10.1183/09031936.06.00071205
Geiser M, Rothen-Rutishauser B, Kapp N, Schürch S, Kreyling W, Schulz H, Semmler M, Im Hof V, Heyder J, Gehr P: Ultrafine particles cross cellular membranes by non-phagocytic mechanisms in lungs and in cultured cells. Environ Health Perspect 2005, 113: 1555–1560. 10.1289/ehp.8006
Mühlfeld C, Gehr P, Rothen-Rutishauser B: Translocation and cellular entering mechanisms of nanoparticles in the respiratory tract. Swiss Med Wkly 2008, 138: 387–391.
Van de Weert-van Leeuwen PB, Van Meegen MA, Speirs JJ, Pals DJ, Rooijakkers SH, Van der Ent CK, Terheggen-Lagro SW, Arets HG, Beekman JM: Optimal complement-mediated phagocytosis of Pseudomonas aeruginosa by monocytes is cystic fibrosis transmembrane conductance regulator-dependent. Am J Respir Cell Mol Biol 2013,49(3):463–470. 10.1165/rcmb.2012-0502OC
Rimai DS, Quesnel DJ, Busnaia AA: The adhesion of dry particles in the nanometer to micrometer-size range. Colloid Surface Physicochem Eng Aspect 2000, 165: 3–10. 10.1016/S0927-7757(99)00439-2
Nadel JA, Conrad DJ, Ueki IF, Schuster A, Sigal E: Immunocytochemical localization of arachidonate 15-lipoxygenase in erythrocytes, leukocytes, and airway cells. J Clin Invest 1991,87(4):1139–1145. 10.1172/JCI115110
Profita M, Sala A, Riccobono L, Pace E, Paternò A, Zarini S, Siena L, Mirabella A, Bonsignore G, Vignola AM: 15(S)-HETE modulates LTB (4) production and neutrophil chemotaxis in chronic bronchitis. Am J Physiol Cell Physiol 2000,279(4):C1249-C1258.
Harmon GS, Dumlao DS, Ng DT, Barrett KE, Dennis EA, Dong H, Glass CK: Pharmacological correction of a defect in PPAR-gamma signaling ameliorates disease severity in Cftr-deficient mice. Nat Med 2010,16(3):313–318. 10.1038/nm.2101
Dorin JR, Stevenson BJ, Fleming S, Alton EW, Dickinson P, Porteous DJ: Long term survival of the exon 10 insertional cystic fibrosis mutant mouse is a consequence of low level residual wild type Cftr gene expression. Mamm Genome 1994, 5: 465–472. 10.1007/BF00369314
Davidson DJ, Dorin JR, McLachlan G, Ranaldi V, Lamb D, Doherty C, Govan J, Porteous DJ: Lung disease in the cystic fibrosis mouse exposed to bacterial pathogens. Nat Genet 1995, 9: 351–357. 10.1038/ng0495-351
Kreyling WG, Semmler M, Erbe F, Mayer P, Takenaka S, Schulz H, Oberdörster G, Ziesenis A: Translocation of ultrafine insoluble iridium particles from lung epithelium to extrapulmonary organs is size dependent but very low. J Toxicol Environ Health-Part A 2002, 65: 1513–1530. 10.1080/00984100290071649
Szymczak W, Menzel N, Kreyling WG, Wittmaack K: TOF-SIMS characterisation of spark-generated nanoparticles made from pairs of Ir-Ir and Ir-C electrodes. Internat J Mass Spectrom 2006, 254: 70–84. 10.1016/j.ijms.2006.05.010
Kapp N, Kreyling W, Schulz H, Im Hof V, Semmler M, Gehr P, Geiser M: Identification of inhaled ultrafine titanium oxide particles by analytical electron microscopy in rat lungs. Microsc Res Techn 2004, 63: 298–305. 10.1002/jemt.20044
Stoeger T, Reinhard C, Takenaka S, Schroeppel A, Karg E, Ritter B, Heyder J, Schulz H: Instillation of six different ultrafine carbon particles indicates a surface area threshold dose for acute lung inflammation in mice. Environ Health Perspect 2006,114(3):328–333.
Kreyling WG, Biswas P, Messing ME, Gibson N, Geiser M, Wenk A, Sahu M, Deppert K, Cydzik I, Wigge C, Schmid O, Semmler-Behnke M: Generation and characterization of stable, highly concentrated titanium dioxide nanoparticle aerosols for rodent inhalation studies. J Nanopart Res 2010, 13: 511–524.
Stoeger T, Takenaka S, Frankenberger B, Ritter B, Karg E, Maier K, Schulz H, Schmid O: Deducing in vivo toxicity of combustion-derived nanoparticles from a cell-free oxidative potency assay and metabolic activation of organic compounds. Environ Health Perspect 2009,117(1):54–60.
Matuschek G, Karg E, Schröppel A, Schulz H, Schmid O: Chemical investigation of eight different types of carbonaceous particles using thermoanalytical techniques. Environ Sci Technol 2007, 41: 8406–8411. 10.1021/es062660v
Scherle W: A simple method for volumetry of organs in quantitative stereology. Mikroskopie 1970, 26: 57–60.
Im Hof V, Scheuch G, Geiser M, Gebhard J, Gehr P, Heyder J: Techniques for the determination of particle deposition in lungs of hamsters. J Aerosol Med 1989, 2: 247–259. 10.1089/jam.1989.2.247
Reinhard C, Eder G, Fuchs H, Ziesenis A, Heyder J, Schulz H: Inbred strain variation in lung function. Mamm Genome 2002, 13: 429–437. 10.1007/s00335-002-3005-6
Reinhard C, Meyer B, Fuchs H, Stoeger T, Eder G, Ruschendorf F, Heyder J, Nurnberg P, de Angelis MH, Schulz H: Genomewide linkage analysis identifies novel genetic loci for lung function in mice. Am J Respir Crit Care Med 2005, 171: 880–888. 10.1164/rccm.200409-1204OC
Semmler-Behnke M, Kreyling WG, Schulz H, Takenaka S, Butler JP, Henry FS, Tsuda A: Nanoparticle delivery in infant lungs. Proc Natl Acad Sci U S A 2012,109(13):5092–5097. 10.1073/pnas.1119339109
Roth C, Ferron GA, Karg E, Lentner B, Schumann G, Takenaka S, Heyder J: Generation of ultrafine particles by spark discharging. Aerosol Sci Techn 2004, 38: 228–235. 10.1080/02786820490247632
Su DS, Jentoft RE, Muller JO, Rothe D, Jacob E, Simpson CD, Tomović Ž, Müllen K, Messerer A, Pöschl U, Niessner R, Schlögl R: Microstructure and oxidation behaviour of Euro IV diesel engine soot: a comparative study with synthetic model soot substances. Catal Today 2004, 90: 127–132. 10.1016/j.cattod.2004.04.017
Götz AA, Vidal-Puig A, Rödel HG, de Angelis MH, Stoeger T: Carbon-nanoparticle-triggered acute lung inflammation and its resolution are not altered in PPARγ-defective (P465L) mice. Part Fibre Toxicol 2011, 8: 28. 10.1186/1743-8977-8-28
Beck-Speier I, Dayal N, Karg E, Maier KL, Schumann G, Schulz H, Semmler M, Takenaka S, Stettmaier K, Bors W, Ghio A, Samet JM, Heyder J: Oxidative stress and lipid mediators induced in alveolar macrophages by ultrafine particles. Free Radic Biol Med 2005, 38: 1080–1092. 10.1016/j.freeradbiomed.2005.01.004
Acknowledgements
We thank Barbara Kupferschmid, Barbara Krieger, Stephanie Hirtz, Claudia Zeller, Alexander Wenk and Gunter Eder for excellent technical assistance with this study. This research was funded by the Swiss National Science Foundation grants 3200B0-105419 and 310030–120763 (to M.G.) and by the EU-FP6 project Particle-Risk (012912 (NEST)) and the EU FP7 project ENPRA (NMP4-SL-2009-228789) (to W.G.K.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
The authors’ responsibilities were as follows – MG, TS, MSB, WGK and HS designed the research and were responsible for the experiments. All authors conducted specific parts of the research. MG, TS, WGK and HS wrote the manuscript. All authors critically read and approved the final manuscript.
Marianne Geiser, Tobias Stoeger contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Geiser, M., Stoeger, T., Casaulta, M. et al. Biokinetics of nanoparticles and susceptibility to particulate exposure in a murine model of cystic fibrosis. Part Fibre Toxicol 11, 19 (2014). https://doi.org/10.1186/1743-8977-11-19
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-8977-11-19