
Abstract
Background
Fine particulate air pollution (PM2.5) is a global health concern, as exposure to PM2.5 has consistently been found to be associated with increased cardiovascular morbidity and mortality. Although adult exposure to traffic related PM2.5, which is largely derived from diesel exhaust (DE), has been associated with increased cardiac hypertrophy, there are limited investigations into the potential effect of in utero and early life exposure on adult susceptibility to heart disease. In this study, we investigate the effect of in utero and early life exposure to DE on adult susceptibility to heart failure.
Methods
Female C57BL/6 J mice were exposed to either filtered air (FA) or DE for 3 weeks (≈300 μg/m3 PM2.5 for 6 hours/day, 5 days/week) and then introduced to male breeders for timed matings. Female mice were exposed to either FA or DE throughout pregnancy and until offspring were 3 weeks of age. Offspring were then transferred to either FA or DE for an additional 8 weeks of exposure. At 12 weeks of age, male offspring underwent a baseline echocardiographic assessment, followed by a sham or transverse aortic constriction (TAC) surgery to induce pressure overload. Following sacrifice three weeks post surgery, ventricles were processed for histology to assess myocardial fibrosis and individual cardiomyocyte hypertrophy. mRNA from lung tissue was isolated to measure expression of inflammatory cytokines IL6 and TNF α.
Results
We observed that mice exposed to DE during in utero and early life development have significantly increased susceptibility to cardiac hypertrophy, systolic failure, myocardial fibrosis, and pulmonary congestion following TAC surgery compared to FA control, or adult DE exposed mice. In utero and early life DE exposure also strongly modified the inflammatory cytokine response in the adult lung.
Conclusions
We conclude that exposure to diesel exhaust air pollution during in utero and early life development in mice increases adult susceptibility to heart failure. The results of this study may imply that the effects of air pollution on cardiovascular disease in human populations may be strongly mediated through a ‘fetal origins’ of adult disease pathway. Further investigations on this potential pathway of disease are warranted.
Similar content being viewed by others


Background
Epidemiological investigations have consistently found that exposure to fine particulate matter air pollution (ambient particles with diameter of <2.5 μm, PM2.5) is associated with an increased risk of cardiovascular morbidity and mortality [1]. In the most recent Global Burden of Disease study [2], ambient particulate matter pollution was found to be the 9th cause of disease worldwide, reaching as high as the 4th cause of disease in East Asia. In addition, household air pollution from solid fuel combustion is considered the 4th cause of disease worldwide, and the 1st cause of disease in South Asia [2]. These predictions of relative cause of disease are largely based upon our current understanding of associations between adult PM exposures and cardiovascular disease, where a rich body of epidemiology literature has consistently found strong associations between PM exposure and risk of cardiovascular morbidity and mortality [3], including evidence to suggest exposure to traffic related air pollution accelerates the progression of cardiac hypertrophy [4, 5]. Cardiac hypertrophy is a critical indicator of future risk of heart failure, and a recent meta-analysis of air pollution and risk of heart failure corroborates these observations, where risk of heart failure hospitalization and death is positively associated with PM2.5 exposure [6].
A large number of laboratory investigations utilizing animal models and controlled exposure facilities have widely proved biological plausibility behind these epidemiological observations where studies have observed that both acute and chronic exposures to PM2.5 impair vascular reactivity [7–10], incite vascular inflammation and accelerate the progression of atherosclerosis [11–14], and promote the progression of cardiac hypertrophy as well as systolic and diastolic dysfunction [15, 16]. Although there is overwhelming evidence that adult exposure to particulate air pollution has deleterious cardiovascular effects, the effects of in utero and early life exposure to PM2.5 on adult susceptibility to cardiovascular disease has currently not been investigated.
Work by Barker and colleagues has demonstrated that in utero and early life development is a critical window in programming adult susceptibility to cardiovascular disease [17–23]. In particular, impaired fetal growth is associated with adult hypertension and risk of mortality [17]. Recently, there has been a growing consensus that exposure to PM2.5 during pregnancy impairs fetal development, resulting in reduced birth weight in human populations [24]. In many urban regions diesel exhaust (DE) is an important component of PM2.5 [25], and recent work using mouse models has observed that exposure to DE during pregnancy incites fetal inflammation, which has been demonstrated to promote offspring to be susceptible to obesity as well as altered pulmonary responses to subsequent ozone exposure [26, 27]. From these previous observations, we hypothesized that exposure to DE during in utero and early life development will promote an increase in susceptibility to adult cardiovascular disease. In this report, we have tested the effect of in utero and early life exposure to DE versus adult exposure to DE on susceptibility to heart failure in adult mice.
Results
In utero and early life or adult exposure to DE does not affect baseline cardiac function
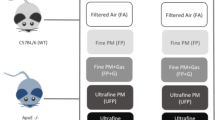
To assess the effect of developmental exposure to DE, mice were exposed to filtered air (FA) or DE in four different paradigms as shown (Figure 1) and as described in Materials and Methods. To characterize the effect of these DE exposures on adult cardiac function, we performed an echocardiographic assessment on all male mice at 12 weeks of age. We did not observe either developmental, adult, or a combined exposure to DE to have any significant effect on hypertrophy, as measured by calculated left ventricular (LV) mass (Figure 2A), or cardiac function, as measured by percentage ejection fraction (%EF) (Figure 2B).

Schematic diagram indicating diesel exhaust (DE) exposure paradigm. Four exposure groups tested the effect of developmental or adult exposure to DE or filtered air (FA) exposures on adult cardiac function. Brown color in horizontal arrows represents DE exposure (300 μg/m3, 6 hours per day, 5 days per week), while white color represents FA exposure.

Echocardiographic assessment of cardiac hypertrophy and function at 12 week of age, prior to TAC/Sham surgeries. (A) Calculated LV Mass normalized to body weight and (B) percentage ejection fraction (%EF) in FA/FA (n = 21), FA/DE (n = 7), DE/FA (n = 14), and DE/DE (n = 12) groups.
In utero and early life exposure to DE exacerbates the response to pressure overload-induced heart failure
Although our baseline echocardiographic assessment did not reveal any significant differences, we tested the effect of these exposures on pressure overload-induced heart failure by use of the transverse aortic constriction (TAC) model. We observed in utero and early life exposure to DE to strongly accelerate the progression of TAC-induced cardiac hypertrophy and systolic dysfunction as measured by increased calculated LV Mass (Figure 3B and C) and reduced%EF (Figure 4B and C). Interestingly, we did not observe any effect of adult exposure to DE on susceptibility to TAC-induced heart failure (Figures 3A and 4A). In assessing the effect of adult exposure to DE on susceptibility to heart failure, FA/DE mice responded in a nearly identical manner to the FA/FA controls (Figures 3A and 4A). Interestingly, we did not observe DE/DE mice to have a worse phenotype than observed in the DE/FA mice, but rather there were trends to suggest DE/DE mice were less susceptible to TAC-induced heart failure than the DE/FA group (Figures 3D and 4D). In our DE/FA group, 2 of 5 TAC mice had progressed into such severe heart failure at 1-week post surgery that our predetermined criteria for early sacrifice was met. No other mouse in any treatment group reached our early sacrifice criteria, potentially suggesting DE/FA mice to be particularly sensitive to TAC-induced heart failure.

Echocardiographic assessment of calculated LV Mass normalized to body weight over time following transverse aortic constriction (TAC) or sham surgeries. (A) FA/FA (n = 7 sham, n = 8 TAC) vs. FA/DE (n = 3 sham, n = 3 TAC), (B) FA/FA vs. DE/FA (n = 5 sham, n = 5 TAC), (C) FA/FA vs. DE/DE (n = 5 sham, n = 4 TAC) groups, and (D) DE/FA vs. DE/DE.

Percentage ejection fraction (%EF) determined by echocardiography over time following transverse aortic constriction (TAC) or sham surgeries. (A) FA/FA (n = 7 sham, n = 8 TAC) vs. FA/DE (n = 3 sham, n = 3 TAC), (B) FA/FA vs. DE/FA (n = 5 sham, n = 5 TAC), (C) FA/FA vs. DE/DE (n = 5 sham, n = 4 TAC) groups, and (D) DE/FA vs. DE/DE.
Following necropsy, ventricles were removed and assessed gravimetrically (Figure 5). We observed TAC-induced heart failure to increase the ventricle weight normalized to tibia length of all treatment groups (Figure 5I). We observed in utero and early life exposure to DE, in combination with adult exposure to DE (DE/DE) to result in significantly elevated ventricle weights in comparison to FA mice. In utero and early life exposed mice (DE/FA) showed a trend of increased ventricle weights normalized to tibia length (p = 0.11), but significance was not achieved. When the effect of TAC on increased ventricle weight is normalized to sham controls, we observe DE/FA and DE/DE mice to have a response to TAC that is significantly greater than FA/FA (Figure 5J). In accordance with our echocardiography data, we did not observe adult exposure to DE alone (FA/DE) to have any effect on ventricle weights (Figure 5I and J).

Gravimetric analysis of ventricle weight at necropsy. Representative images of sagittal sections of hearts from mice three weeks post sham or transverse aortic constriction (TAC) surgeries (A-H), scale bar = 1 mm. Gravimetric analysis of ventricles normalized to tibia length (VW/tibia length) in FA/FA (n = 7 sham, n = 8 TAC), FA/DE (n = 3 sham, n = 3 TAC), DE/FA (n = 5 sham, n = 5 TAC) and DE/DE (n = 5 sham, n = 4 TAC) groups (I). Effect of TAC by exposure group was also represented as a fold effect of TAC on VW/tibia length normalized to sham mice (J).
In utero and early life exposure to DE promotes cardiac fibrosis in pressure overload-induced heart failure
To assess the potential effect of DE on development of cardiac fibrosis after pressure overload, we measured the extent of fibrosis within the myocardium by Masson’s Trichrome staining. In sham-operated mice, we did not observe either developmental, adult, or combined exposure to DE to have any effect on cardiac fibrosis (Figure 6A-D). In mice that underwent TAC, we observed an increase in myocardial fibrosis in all exposure groups (Figure 6E-I) compared to sham-operated controls. Interestingly, we observed an increased fibrotic response in the in utero and early life DE exposure cohort (DE/FA) in comparison to FA controls (Figure 6E-I). In FA/FA mice, we observed TAC-induced heart failure to result in largely perivascular fibrosis that is limited in extent (Figure 6E), whereas in utero and early life exposed mice reveal extensive interstitial fibrosis that is not limited to a perivascular distribution (Figure 6G and H). DE/DE mice also showed extensive myocardial fibrosis that exceeded that found in FA/FA or FA/DE mice (Figure 6H). We observed a trend where DE/DE mice appeared to have less extensive myocardial fibrosis than that observed in our in utero and early life exposed group (DE/FA), but this trend did not reach significance.

Assessment of cardiac fibrosis by Masson’s Trichrome staining. Representative images (4X objective) of sagittal sections of left ventricle free wall from mice three weeks post sham or transverse aortic constriction (TAC) surgeries (A-H), scale bar = 0.5 mm. Fibrotic regions of cardiac tissues are determined by blue staining in FA/FA (n = 5 sham, n = 5 TAC), FA/DE (n = 3 sham, n = 3 TAC), DE/FA (n = 5 sham, n = 5 TAC) and DE/DE (n = 5 sham, n = 4 TAC) groups. Assessment of percentage highly fibrotic regions was quantified (I).
In utero and early life exposure to DE does not increase individual cardiomyocyte cross sectional area
As in utero and early life exposure to DE appears to accelerate TAC-induced cardiac hypertrophy and increase ventricle weight, we measured individual cardiomyocyte area within the left ventricle to determine if individual cardiomyocyte hypertrophy is responsible for these observations. TAC promoted increased individual cardiomyocyte area in all treatment groups (Figure 7), but we did not observe either in utero and early life, adult, or combined DE exposure to have any additional effect on individual cardiomyocyte area (Figure 7E-I). Interestingly, in utero and early life DE exposure was associated with a significant reduction in individual cardiomyocyte area in sham-operated mice (Figure 7C and I), suggesting a potential baseline atrophic cardiomyopathy. This effect was not observed in sham DE/DE mice, although there was a trend towards a reduced myocyte area (FA/FA Sham vs DE/DE Sham T-Test, p = 0.29).

Measurement of individual cardiomyocyte hypertrophy. Representative images (40X objective) of sagittal sections of left ventricle free wall from mice three weeks post sham or transverse aortic constriction (TAC) surgeries (A-H), scale bar = 50 μm. Individual cardiomyocyte areas were traced, quantified, and analyzed in FA/FA (n = 5 sham, n = 5 TAC), FA/DE (n = 3 sham, n = 3 TAC), DE/FA (n = 5 sham, n = 5 TAC) and DE/DE (n = 5 sham, n = 4 TAC) groups (I).
In utero and early life exposure to DE is associated with increased lung weight after TAC and altered inflammatory cytokine expression
To assess the effect of developmental and adult exposure to DE on pulmonary congestion that develops in association with TAC-induced heart failure, lung weights were measured at necropsy and normalized to tibia length. In sham-operated mice, we did not observe in utero and early life, adult, or a combined DE exposure to have any effect on lung weights (Figure 8A). In TAC-operated mice, the lung weights of DE/FA TAC mice were significantly greater than all other groups, indicating that in utero and early life exposure to DE promotes an exacerbated lung response to TAC (Figure 8A).

Effects of DE and TAC on lung weight and inflammatory cytokine expression. (A) Lung weights as measured at necropsy were collected and analyzed in FA/FA (n = 7 sham, n = 8 TAC), FA/DE (n = 3 sham, n = 3 TAC), DE/FA (n = 5 sham, n = 5 TAC) and DE/DE (n = 5 sham, n = 4 TAC) groups. Relative real-time PCR analysis of IL6 (B) and TNFα (C) mRNA content in the lung as expressed as negative delta delta CT from FA/FA sham normalized to GAPDH.
As both acute and chronic exposure to DE has been suggested to incite a pulmonary cytokine response, we measured the relative mRNA content of the proinflammatory cytokines interleukin 6 (IL6) and tumor necrosis factor alpha (TNFα) in the lung tissue from all sham and TAC groups. In FA/FA control mice, we did not observe the induction of hypertrophy by TAC to alter IL6 or TNFα mRNA content in comparison to FA/FA sham mice (Figure 8B and C). We also did not observe adult exposure to DE in the absence of developmental DE exposure (FA/DE) to have any effect on lung IL6 or TNFα mRNA content in either sham or TAC mice. Interestingly, although in utero and early life DE exposure (DE/FA) did not alter IL6 or TNFα mRNA content in sham mice, we did observe IL6 to significantly increase in DE/FA TAC mice (−ddCT = 2.358, ~5-fold increase). Unexpectedly, we observed in utero and early life DE in addition to adult DE exposed mice (DE/DE) to have a dramatic increase in both IL6 and TNFα mRNA content in sham mice (−ddCT IL6 = 5.213, ~37-fold increase, -ddCT TNFα = 3.117, ~8-fold increase). We observed DE/DE TAC mice to have a trend toward reduced inflammatory cytokine expression in comparison to DE/DE sham mice, indicating that the inflammatory cytokine expression is independent of TAC-induced heart failure.
Discussion
In the present study, we investigated the effect of in utero and early life exposure to diesel exhaust (DE) air pollution (≈300 μg/m3 PM2.5, 6 hrs/day, 5 days/week) on adult susceptibility to pressure overload-induced heart failure in male mice. This concentration of DE was chosen as it is comparable to other studies investigating the vascular effects of DE inhalation [7, 9, 28] and is relevant to population exposures in highly polluted urban environments [29]. Our study focused on male offspring to control for cyclic hormonal variation. Our observation that in utero and early life exposure to DE promotes the development of heart failure in adult animals is consistent with the work of Barker and colleagues [17–23] and implies that adult cardiovascular disease may have more origins in developmental exposure to air pollution than is currently appreciated.
The mechanisms by which in utero and early life DE exposures promote adult cardiovascular disease are poorly understood. In rodent models, acute and chronic inhalation of PM2.5 can elicit systemic vascular inflammation and impair vascular reactivity [8, 10]. These mechanisms have been implicated to play an important role in the progression of cardiovascular disease as well as increase the risk of acute myocardial infarction [30–32] in adults. Appropriate vascular function is critical during embryogenesis and is vital in regulating placental blood flow and nutrient transfer to the developing fetus [33, 34]. Exposure to DE during pregnancy in mouse models has been shown to promote fetal inflammation and to promote effects on both pulmonary and neuronal function in the adult offspring [26, 27, 35]. In addition, human exposure to PM2.5 during pregnancy has been shown to decrease placental mitochondrial DNA [36], an indicator of oxidative stress, as well as cause placental DNA hypomethylation [37]. Similar observations have been observed with individuals who smoke tobacco during pregnancy, which has long been known to impact placental blood flow and result in intrauterine growth restriction (IUGR) [38–41]. Affected fetuses demonstrate hypoglycemia, hypoinsulinemia, acidosis, hypoxia, and decreased branched chain amino acid transfer resulting in long lasting adverse effects postnatally [42–47]. Rodent models have shown that exposure to uteroplacental insufficiency results in altered mitochondrial β-oxidation within muscle [48], altered fatty acid metabolism [49], increased visceral adipose tissue deposition [50], and decreased insulin sensitivity associated with elevated fasting plasma glucose [51]. These effects are thought to be due to epigenetic ‘reprogramming’, where changes in DNA methylation and histone modification have been observed to promote long lasting effects on gene expression that alter physiological and cellular function [42, 52].
Epigenetic linkage between prenatal environmental insults and adult cardiovascular phenotypes has been previously reported. Patterson et al. recently reported that prenatal hypoxia causes hypermethylation of the PKCϵ promoter, conferring susceptibility to ischemic injury to the adult rat heart [53, 54]. Our future investigations on the effect of prenatal exposure to air pollution on adult cardiovascular disease will focus on the potential for these exposures to elicit long-term epigenetic modifications.
The current evidence showing an effect of prenatal or early life exposure to air pollution on adult risk of disease in human populations is limited largely to adult pulmonary function [55] as there is clear epidemiological evidence that childhood exposure to air pollution impairs lung development [56]. Pulmonary inflammation has been postulated to promote cardiovascular disease [57]. Our study demonstrates that combined prenatal and early life exposure to air pollution can alter adult pulmonary cytokine expression and promote pressure overload-induce adult heart failure in mice, but such results cannot necessarily be extrapolated to human populations. The effects of prenatal or childhood air pollution exposure on adult cardiovascular risk in human populations are unknown. Nevertheless, we believe our data provides a strong impetus to investigate whether in utero and/or early life air pollution exposure affects adult risk of cardiovascular disease in human populations.
In our report, we found that an 8-week exposure to DE in adult mice did not have any effect on cardiac function at baseline or following TAC-induced heart failure, which is consistent with our previous study in which we investigated the effects of prolonged adult DE exposures (up to 6-months) and failed to observe any effect in two mouse models of cardiac hypertrophy [58]. Others have reported that exposure to concentrated ambient particulate matter (CAPs) potentiates the cardiac hypertrophic response to angiotensin II (AngII) stimulation [16] and promotes cardiac fibrosis [15]. We believe that CAPs have unique cardiotoxicity in comparison to DE, as discussed previously [58]. Our findings indicate that, in mice, the developing organism is much more susceptible to the cardiovascular effects of DE than the adult.
Conclusions
In summary, we have observed that exposure to diesel exhaust air pollution during in utero and early life development in mice increases adult susceptibility to pressure overload-induced heart failure. This effect was not observed in mice exposed to diesel exhaust as adults. This report suggests for the first time that developmental exposure to air pollution may be critical in mediating adult susceptibility to heart failure, and provides impetus to perform appropriate clinical studies to determine whether these findings are applicable to human populations.
Materials and methods
Diesel exhaust exposure and mice
Male and female C57Bl/6 J mice were purchased from The Jackson Laboratory (Bar Harbor, Maine, USA). All mice were housed in specific pathogen free (SPF) conditions on a 12/12-light/dark cycle. Female and male mice between the ages of 12 to 14 weeks were transferred to our Northlake Diesel Exposure Facility located near the University of Washington (UW) and housed under SPF conditions in Allentown caging systems (Allentown, NJ, USA) as previously described [59–61]. All animal experiments were approved by the UW Institutional Animal Care and Use Committee. Diesel exhaust (DE) was generated from a single cylinder Yanmar diesel engine (Model YDG5500EV-6EI) operating on 82% load. A detailed analysis of DE particulate components in this system has been previously reported [59]. DE exposures were conducted for 6 hours per day (9 am – 3 pm) five days a week (Monday – Friday) and DE concentrations were regulated to ≈ 300 μg/m3 of PM2.5. A 300 μg/m3 concentration of PM2.5 six hours/day, five days/week equates to a time weighted hourly average of 53 μg/m3. The following exposure characteristics were measured with DE generated under the same conditions within the exposure facility, though not on the days of this experiment. Oxides of nitrogen concentrations were 1800 ppb NOx and 60 ppb NO2, measured using a Thermo Scientific Model 42C analyzer. The concentration of carbon monoxide was 2 ppm, measured using a Langan analyzer, Model T15n. The concentration of carbon dioxide was 1000 ppm with infrared detection (Telaire Model 1050, Telaire Systems). The mass fraction of particle-bound polycyclic aromatic hydrocarbons (PAH) was 20 ng/μg PM2.5, measured with an Ecochem PAS 2000. The ratio of the organic carbon to elemental carbon mass concentration was 0.10, based on quartz filter samples adjusted with a concurrent dynamic blank; samples were analyzed by Sunset Laboratories using the IMPROVE A thermo optical reflectance method. The mass median aerodynamic diameter was 85 nm (GSD 1.2), which was obtained by gravimetric analysis of samples collected with a micro orifice uniform deposit impactor (MOUDI, MSP Model 110-NR). The count median thermodynamic equivalent diameter was 87 nm (GSD 3.0), measured with a P-Trak Ultrafine Particle Counter, Model 8525.
Female mice were exposed to either FA or DE for 3 weeks, and then paired with male mice for timed matings during weekend FA exposures. After observation of a vaginal plug, pregnant mice were then re-exposed to the same condition (FA or DE) for the duration of gestation and until weaning at postnatal day (PND) 21. At PND21, offspring were transferred to either FA or DE to continue exposure for 8 additional weeks, representing an “adult” exposure. Male mice exposed to FA throughout the study are designated FA/FA, those mice exposed to FA until weaning and subsequently transferred to DE are designated FA/DE, mice exposed to DE until weaning and transferred to FA are designated DE/FA, and mice exposed to DE throughout the study are designated DE/DE.
A total of 9 female mice became pregnant in FA exposures and 8 female mice became pregnant in DE exposures. Of 9 FA litters, 5 remained in FA exposures, while 4 were transferred to DE. Of 8 DE litters, 5 were transferred to FA, and 3 remained in DE. At 11 weeks of age, male offspring were transferred from the Northlake facility to the UW Medicine South Lake Union (SLU) SPF vivarium, where each mouse underwent echocardiographic assessment and surgery. A total of 54 male offspring were used for this study.
Echocardiography and transverse aortic constriction surgeries
Upon transfer of male offspring to the SLU SPF vivarium, mice underwent baseline echocardiographic assessment at 11–12 weeks of age. Under 0.5% isoflurane anesthesia, cardiac size and function were assessed using a Visual Sonics (Toronto, Canada) VEVO 770 system equipped with a 707B scan head as previously described [62, 63]. When the heart rate of the mouse had returned to normal followed anesthesia (>520 bpm), parasternal short axis views were obtained under M-mode. Cine loops collected from M-mode views were analyzed for anterior and posterior LV wall thickness as well as LV internal diameter at diastole and systole. Percentage ejection fraction (%EF) and LV Mass were calculated from Visual Sonics Standard Measurements and Calculations. One week after baseline echocardiographic assessment was completed, male mice were randomly assigned to either transverse aortic constriction (TAC) or sham surgeries. For surgeries, male mice between 12 and 14 weeks of age were anesthetized using ketamine (130 mg/kg i.p.) and xylazine (8.8 mg/kg i.p.) and subjected to transverse aortic constriction using a 27-gauge needle as described [62, 63]. To measure cardiac response to surgery, echocardiographic assessments were completed for each mouse at 1 week, 2 weeks, and 3 weeks post surgery. All echocardiographic measurements were performed by a blinded observer. In all exposure groups, we observed 30-40% mortality due to surgical complications, as defined by death within 72 hours of surgery.
Necropsy and histology
All mice were euthanized at 3 weeks post surgery, or earlier if predetermined euthanasia criteria were met (ejection fraction below 25% with overt heart failure). Mice were sacrificed by overdose with tribromoethanol (650 mg/kg i.p.) followed by exsanguination. Tissues were collected for gravimetric analysis prior to storage in liquid nitrogen or fixation with 4% paraformaldehyde and subsequent storage in 70% ethanol. Tissues used for histology underwent tissue processing and embedding in paraffin. Seven-micron thick cross sections were made of the heart, and Masson’s Trichrome as well as hematoxylin and eosin staining were performed using standard techniques. Extent of myocardial fibrosis was determined by Masson’s Trichrome stain where percentage of blue stain was quantified over total tissue area from 4X images of the left ventricle free wall using NIH Image J (Bethesda, MD, USA) as previously done [62]. Using NIH Image J, low power images of the LV free wall were assessed in a blind fashion. The border of the myocardium was traced manually to get a baseline area, then the area that would be considered ‘highly fibrotic’, where there is clear evidence of fibroblast collagen deposition and blue staining, is manually traced to get a subsequent area. The area of the myocardium that would be ‘highly fibrotic’ is expressed as a ‘percentage highly fibrotic area’. Individual cardiomyocyte area was quantified using NIH Image J, where the areas of 100 cardiomyocytes were quantified and averaged for each section as described previously [62]. Of 100 cardiomyocytes within the LV, area is averaged to get a single value that represents average myocyte area per heart. We then averaged cardiomyocyte area within exposure and surgery groups to assess individual cardiomyocyte hypertrophy.
Lung RNA isolation and qRT-PCR of IL6 and TNFα
Frozen lung tissue from the inferior right lobe was collected from each mouse, placed in RNA stabilizing solution Trizol (Invitrogen; Carlsbad, CA, USA) and homogenized with Polytron mechanical homogenizer (Kinematics; Bohemia, NY, USA). RNA isolation was carried out using Qiagen RNeasy Kit (Valencia, CA, USA) following the manufacturer’s protocol. Isolated RNA underwent further purification by standard alcohol precipitation and resuspension techniques. One microgram of RNA was reverse transcribed to cDNA using iScript cDNA synthesis kit (Bio-Rad; Hercules, CA, USA) following the manufacturer’s protocol. Quantitative Real-time PCR (qRT-PCR) was carried out using an ABI7500 Fast Real-Time PCR system (Applied Biosystems, Inc.; Foster City, CA, USA) using Power SYBR Green (Applied Biosystems, Inc.; Foster City, CA, USA) following the thermo-cycling settings of of 5 min 95°C to activate Taq DNA polymerase, followed by 40 cycles of 95°C for 15 seconds and 60°C for 60 seconds. Relative mRNA content of IL6 and TNFα were normalized to GAPDH, using the following primers: Tnf α, 5′-CCTGTAGCCCACGTCGTAG-3′ (forward) and 5′-GGGAGTAGACAAGGTACAACCC-3′ (reverse), Il6, 5′-TAGTCCTTCCTACCCCAATTTCC-3′ (forward) and 5′-TTGGTCCTTAGCCACTCCTTC-3′ (reverse), Gapdh, 5′-CTTCCGTGTTCCTACCC-3′ (forward) and 5′-ACCTGGTCCTCAGTGTAGCC-3′ (reverse). Relative mRNA content of each gene was calculated using the 2(−ddC(T) method as previously described [64] and represented as the change in delta CT between GAPDH and IL6 or TNFα from FA/FA sham to treatment group (−ddCT).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 5 for Microsoft OS X (GraphPad Software, Inc.; San Diego, CA, USA). Differences between two groups were determined by Student’s T-Test using an α-value of 0.05. When more than two groups were being compared, a one-way ANOVA followed by a Tukey’s Posthoc test was used. When statistical analyses between two groups were made following a variable over time, a repeated measure two-way ANOVA was used. In the DE/FA group, after week 1 data was collected, 2 mice met our predetermined early sacrifice criteria and were sacrificed prior to the 3-week time point. To assess significance between DE/FA and FA/FA or DE/DE, we performed a repeated measure two-way ANOVA across all 3 weeks while excluding these two data points. An additional one-way ANOVA followed by a Tukey’s posthoc test was performed at the one-week time point with all data points to confirm significance. All error bars in figures represent mean ± standard error of the mean; *, **, *** represent significant differences with p < 0.05, p < 0.01, and p < 0.001 respectively.
Authors’ information
CSW is a postdoctoral fellow and toxicologist. YL is a junior faculty member and cardiovascular scientist. YCC is a graduate student in toxicology. IOM is a clinical cardiology fellow. JRF is a postdoctoral fellow and exposure scientist. TVL is a senior faculty member, atmospheric scientist and environmental engineer. WMC is a staff research scientist. MTC is a senior faculty member, cardiovascular scientist and practicing cardiologist.
Abbreviations
- PM:
-
Particulate matter
- PM2.5:
-
Particulate matter with a diameter of 2.5 μm or less
- DE:
-
Diesel exhaust
- FA:
-
Filtered air
- TAC:
-
Transverse aortic constriction
- LV:
-
Left ventricle
- EF:
-
Ejection fraction
- bpm:
-
beats per minute
- TNFα:
-
Tumor necrosis factor alpha
- IL6:
-
Interleukin 6
- GAPDH:
-
Glyceraldehye 3-phosphate dehydrogenase
- qRT-PCR:
-
Quantitative real time polymerase chain reaction
- -ddCT:
-
Negative delta delta threshold cycle
- CAPs:
-
Concentrated ambient particulate matter
- SPF:
-
Specific pathogen free
- i.p.:
-
intraperitoneal injection
- cDNA:
-
complimentary deoxyribonucleic acid
- mRNA:
-
messenger ribonucleic acid
- ANOVA:
-
Analysis of variance.
References
Brook RD, Rajagopalan S, Pope CA, Brook JR, Bhatnagar A, Diez-Roux AV, Holguin F, Hong Y, Luepker RV, Mittleman MA, et al.: Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American heart association. Circulation 2010, 121: 2331–2378. 10.1161/CIR.0b013e3181dbece1
Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, Amann M, Anderson HR, Andrews KG, Aryee M, et al.: A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the global burden of disease study 2010. Lancet 2013, 380: 2224–2260.
Hoek G, Krishnan RM, Beelen R, Peters A, Ostro B, Brunekreef B, Kaufman JD: Long-term air pollution exposure and cardio- respiratory mortality: a review. Environ Health 2013, 12: 43. 10.1186/1476-069X-12-43
Van Hee VC, Adar SD, Szpiro AA, Barr RG, Bluemke DA, Diez Roux AV, Gill EA, Sheppard L, Kaufman JD: Exposure to traffic and left ventricular mass and function: the multi-ethnic study of atherosclerosis. Am J Respir Crit Care Med 2009, 179: 827–834. 10.1164/rccm.200808-1344OC
Van Hee VC, Adar SD, Szpiro AA, Barr RG, Roux AD, Bluemke DA, Sheppard L, Gill EA, Bahrami H, Wassel C, et al.: Common genetic variation, residential proximity to traffic exposure, and left ventricular mass: the multi-ethnic study of atherosclerosis. Environ Health Perspect 2010, 118: 962–969. 10.1289/ehp.0901535
Shah ASV, Langrish JP, Nair H, McAllister DA, Hunter AL, Donaldson K, Newby DE, MIlls NL: Global association of air pollution and heart failure: a systematic review and meta-analysis. Lancet 2013, 382: 1039–1048. 10.1016/S0140-6736(13)60898-3
Cherng TW, Campen MJ, Knuckles TL, Gonzalez Bosc L, Kanagy NL: Impairment of coronary endothelial cell ETB receptor function after short-term inhalation exposure to whole diesel emissions. Am J Physiol Regul Integr Comp Physiol 2009, 297: R640-R647. 10.1152/ajpregu.90899.2008
Cherng TW, Paffett ML, Jackson-Weaver O, Campen MJ, Walker BR, Kanagy NL: Mechanisms of diesel-induced endothelial nitric oxide synthase dysfunction in coronary arterioles. Environ Health Perspect 2011, 119: 98–103.
Knuckles TL, Lund AK, Lucas SN, Campen MJ: Diesel exhaust exposure enhances venoconstriction via uncoupling of eNOS. Toxicol Appl Pharmacol 2008, 230: 346–351. 10.1016/j.taap.2008.03.010
Sun Q, Yue P, Ying Z, Cardounel AJ, Brook RD, Devlin R, Hwang J-S, Zweier JL, Chen LC, Rajagopalan S: Air pollution exposure potentiates hypertension through reactive oxygen species-mediated activation of Rho/ROCK. Arterioscler Thromb Vasc Biol 2008, 28: 1760–1766. 10.1161/ATVBAHA.108.166967
Bai N, Kido T, Kavanagh TJ, Kaufman JD, Rosenfeld ME, van Breemen C, van Eeden SF: Exposure to diesel exhaust up-regulates iNOS expression in ApoE knockout mice. Toxicol Appl Pharmacol 2011, 255: 184–192. 10.1016/j.taap.2011.06.013
Bai N, Kido T, Suzuki H, Yang G, Kavanagh TJ, Kaufman JD, Rosenfeld ME, van Breemen C, van Eeden SF: Changes in atherosclerotic plaques induced by inhalation of diesel exhaust. Atherosclerosis 2011, 216: 299–306. 10.1016/j.atherosclerosis.2011.02.019
Sun Q, Wang A, Jin X, Natanzon A, Duquaine D, Brook RD, Aguinaldo J-GS, Fayad ZA, Fuster V, Lippmann M, et al.: Long-term air pollution exposure and acceleration of atherosclerosis and vascular inflammation in an animal model. JAMA 2005, 294: 3003–3010. 10.1001/jama.294.23.3003
Quan C, Sun Q, Lippmann M, Chen L-C: Comparative effects of inhaled diesel exhaust and ambient fine particles on inflammation, atherosclerosis, and vascular dysfunction. Inhal Toxicol 2010, 22: 738–753. 10.3109/08958371003728057
Wold LE, Ying Z, Hutchinson KR, Velten M, Gorr MW, Velten C, Youtz DJ, Wang A, Lucchesi PA, Sun Q, Rajagopalan S: Cardiovascular remodeling in response to long-term exposure to fine particulate matter air pollution. Circ Heart Fail 2012, 5: 452–461. 10.1161/CIRCHEARTFAILURE.112.966580
Ying Z, Yue P, Xu X, Zhong M, Sun Q, Mikolaj M, Wang A, Brook RD, Chen LC, Rajagopalan S: Air pollution and cardiac remodeling: a role for RhoA/Rho-kinase. Am J Physiol Heart Circ Physiol 2009, 296: H1540-H1550. 10.1152/ajpheart.01270.2008
Barker DJ, Osmond C, Golding J, Kuh D, Wadsworth ME: Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ (Clinical Res Ed) 1989, 298: 564–567. 10.1136/bmj.298.6673.564
Eriksson J, Forsén T, Tuomilehto J, Osmond C, Barker D: Fetal and childhood growth and hypertension in adult life. Hypertension 2000, 36: 790–794. 10.1161/01.HYP.36.5.790
Feldt K, Räikkönen K, Eriksson JG, Andersson S, Osmond C, Barker DJP, Phillips DIW, Kajantie E: Cardiovascular reactivity to psychological stressors in late adulthood is predicted by gestational age at birth. J Hum Hypertens 2007, 21: 401–410.
Godfrey KM, Barker DJ: Fetal programming and adult health. Public Health Nutr 2001, 4: 611–624.
Law CM, de Swiet M, Osmond C, Fayers PM, Barker DJ, Cruddas AM, Fall CH: Initiation of hypertension in utero and its amplification throughout life. BMJ (Clin Res Ed) 1993, 306: 24–27. 10.1136/bmj.306.6869.24
Martyn CN, Barker DJ, Jespersen S, Greenwald S, Osmond C, Berry C: Growth in utero, adult blood pressure, and arterial compliance. Br Heart J 1995, 73: 116–121. 10.1136/hrt.73.2.116
Vijayakumar M, Fall CH, Osmond C, Barker DJ: Birth weight, weight at one year, and left ventricular mass in adult life. Br Heart J 1995, 73: 363–367. 10.1136/hrt.73.4.363
Dadvand P, Parker J, Bell ML, Bonzini M, Brauer M, Darrow LA, Gehring U, Glinianaia SV, Gouveia N, Ha E-H, et al.: Maternal exposure to particulate air pollution and term birth weight: a multi-country evaluation of effect and heterogeneity. Environ Health Perspect 2013, 121: 267–373. 10.1289/ehp.1205575
Lewtas J: Air pollution combustion emissions: characterization of causative agents and mechanisms associated with cancer, reproductive, and cardiovascular effects. Mutat Res 2007, 636: 95–133. 10.1016/j.mrrev.2007.08.003
Auten RL, Gilmour MI, Krantz QT, Potts EN, Mason SN, Foster WM: Maternal diesel inhalation increases airway hyperreactivity in ozone-exposed offspring. Am J Respir Cell Mol Biol 2012, 46: 454–460. 10.1165/rcmb.2011-0256OC
Bolton JL, Smith SH, Huff NC, Gilmour MI, Foster WM, Auten RL, Bilbo SD: Prenatal air pollution exposure induces neuroinflammation and predisposes offspring to weight gain in adulthood in a sex-specific manner. FASEB J 2012, 26: 4743–4754. 10.1096/fj.12-210989
Campen MJ, Lund AK, Knuckles TL, Conklin DJ, Bishop B, Young D, Seilkop S, Seagrave J, Reed MD, Mcdonald JD: Inhaled diesel emissions alter atherosclerotic plaque composition in ApoE(−/−) mice. Toxicol Appl Pharmacol 2010, 242: 310–317. 10.1016/j.taap.2009.10.021
Wang J-F, Hu M-G, Xu C-D, Christakos G, Zhao Y: Estimation of citywide air pollution in Beijing. PloS one 2013, 8: e53400. 10.1371/journal.pone.0053400
Miller KA, Siscovick DS, Sheppard L, Shepherd K, Sullivan JH, Anderson GL, Kaufman JD: Long-term exposure to air pollution and incidence of cardiovascular events in women. N Engl J Med 2007, 356: 447–458. 10.1056/NEJMoa054409
Mills NL, Törnqvist H, Gonzalez MC, Vink E, Robinson SD, Söderberg S, Boon NA, Donaldson K, Sandström T, Blomberg A, Newby DE: Ischemic and thrombotic effects of dilute diesel-exhaust inhalation in men with coronary heart disease. N Engl J Med 2007, 357: 1075–1082. 10.1056/NEJMoa066314
Mills NL, Törnqvist H, Robinson SD, Gonzalez M, Darnley K, MacNee W, Boon NA, Donaldson K, Blomberg A, Sandstrom T, Newby DE: Diesel exhaust inhalation causes vascular dysfunction and impaired endogenous fibrinolysis. Circulation 2005, 112: 3930–3936. 10.1161/CIRCULATIONAHA.105.588962
Arck PC, Hecher K: Fetomaternal immune cross-talk and its consequences for maternal and offspring's health. Nature medicine 2013, 19: 548–556.
Ward JM, Elmore SA, Foley JF: Pathology methods for the evaluation of embryonic and perinatal developmental defects and lethality in genetically engineered mice. Vet Pathol 2012, 49: 71–84. 10.1177/0300985811429811
Auten RL, Potts EN, Mason SN, Fischer B, Huang Y, Foster WM: Maternal exposure to particulate matter increases postnatal ozone-induced airway hyperreactivity in juvenile mice. Am J Respir Crit Care Med 2009, 180: 1218–1226. 10.1164/rccm.200901-0116OC
Janssen BG, Munters E, Pieters N, Smeets K, Cox B, Cuypers A, Fierens F, Penders J, Vangronsveld J, Gyselaers W, Nawrot TS: Placental mitochondrial DNA content and particulate air pollution during in utero life. Environ Health Perspect 2012, 120: 1346–1352. 10.1289/ehp.1104458
Janssen BG, Godderis L, Pieters N, Poels K, Kici Ski M, Cuypers A, Fierens F, Penders J, Plusquin M, Gyselaers W, Nawrot TS: Placental DNA hypomethylation in association with particulate air pollution in early life. Part Fibre Toxicol 2013, 10: 22. 10.1186/1743-8977-10-22
Andersen KV, Hermann N: Placenta flow reduction in pregnant smokers. Acta Obstet Gynecol Scand 1984, 63: 707–709. 10.3109/00016348409154668
Burton GJ, Palmer ME, Dalton KJ: Morphometric differences between the placental vasculature of non-smokers, smokers and ex-smokers. Br J Obstet Gynaecol 1989, 96: 907–915. 10.1111/j.1471-0528.1989.tb03344.x
Demir R, Demir AY, Yinanc M: Structural changes in placental barrier of smoking mother. A quantitative and ultrastructural study. Pathol Res Pract 1994, 190: 656–667. 10.1016/S0344-0338(11)80744-2
Larsen LG, Clausen HV, Jønsson L: Stereologic examination of placentas from mothers who smoke during pregnancy. Am J Obstet Gynecol 2002, 186: 531–537. 10.1067/mob.2002.120481
MacLennan NK, James SJ, Melnyk S, Piroozi A, Jernigan S, Hsu JL, Janke SM, Pham TD, Lane RH: Uteroplacental insufficiency alters DNA methylation, one-carbon metabolism, and histone acetylation in IUGR rats. Physiol Genomics 2004, 18: 43–50. 10.1152/physiolgenomics.00042.2004
Bussey ME, Finley S, LaBarbera A, Ogata ES: Hypoglycemia in the newborn growth-retarded rat: delayed phosphoenolpyruvate carboxykinase induction despite increased glucagon availability. Pediatr Res 1985, 19: 363–367. 10.1203/00006450-198519040-00010
Economides DL, Nicolaides KH: Blood glucose and oxygen tension levels in small-for-gestational-age fetuses. Am J Obstet Gynecol 1989, 160: 385–389. 10.1016/0002-9378(89)90453-5
Economides DL, Nicolaides KH, Gahl WA, Bernardini I, Bottoms S, Evans M: Cordocentesis in the diagnosis of intrauterine starvation. Am J Obstet Gynecol 1989, 161: 1004–1008. 10.1016/0002-9378(89)90772-2
Ogata ES, Bussey ME, Finley S: Altered gas exchange, limited glucose and branched chain amino acids, and hypoinsulinism retard fetal growth in the rat. Metabolism 1986, 35: 970–977. 10.1016/0026-0495(86)90064-8
Ogata ES, Bussey ME, LaBarbera A, Finley S: Altered growth, hypoglycemia, hypoalaninemia, and ketonemia in the young rat: postnatal consequences of intrauterine growth retardation. Pediatr Res 1985, 19: 32–37. 10.1203/00006450-198501000-00010
Lane RH, Kelley DE, Ritov VH, Tsirka AE, Gruetzmacher EM: Altered expression and function of mitochondrial beta-oxidation enzymes in juvenile intrauterine-growth-retarded rat skeletal muscle. Pediatr Res 2001, 50: 83–90. 10.1203/00006450-200107000-00016
Lane RH, Kelley DE, Gruetzmacher EM, Devaskar SU: Uteroplacental insufficiency alters hepatic fatty acid-metabolizing enzymes in juvenile and adult rats. Am J Physiol Regul Integr Comp Physiol 2001, 280: R183-R190.
Bagley HN, Wang Y, Campbell MS, Yu X, Lane RH, Joss-Moore LA: Maternal docosahexaenoic acid increases adiponectin and normalizes IUGR-induced changes in rat adipose deposition. J Obes 2013, 2013: 312153.
Tsirka AE, Gruetzmacher EM, Kelley DE, Ritov VH, Devaskar SU, Lane RH: Myocardial gene expression of glucose transporter 1 and glucose transporter 4 in response to uteroplacental insufficiency in the rat. J Endocrinol 2001, 169: 373–380. 10.1677/joe.0.1690373
Ke X, Lei Q, James SJ, Kelleher SL, Melnyk S, Jernigan S, Yu X, Wang L, Callaway CW, Gill G, et al.: Uteroplacental insufficiency affects epigenetic determinants of chromatin structure in brains of neonatal and juvenile IUGR rats. Physiol Genomics 2006, 25: 16–28. 10.1152/physiolgenomics.00093.2005
Patterson AJ, Chen M, Xue Q, Xiao D, Zhang L: Chronic prenatal hypoxia induces epigenetic programming of PKC{epsilon} gene repression in rat hearts. Circ Res 2010, 107: 365–373. 10.1161/CIRCRESAHA.110.221259
Patterson AJ, Xiao D, Xiong F, Dixon B, Zhang L: Hypoxia-derived oxidative stress mediates epigenetic repression of PKCϵ gene in foetal rat hearts. Cardiovasc Res 2012, 93: 302–310. 10.1093/cvr/cvr322
Proietti E, Röösli M, Frey U, Latzin P: Air pollution during pregnancy and neonatal outcome: a review. J Aerosol Med Pulm Drug Deliv 2013, 26: 9–23. 10.1089/jamp.2011.0932
Gauderman WJ, Avol E, Gilliland F, Vora H, Thomas D, Berhane K, McConnell R, Kuenzli N, Lurmann F, Rappaport E, et al.: The effect of air pollution on lung development from 10 to 18 years of age. N Engl J Med 2004, 351: 1057–1067. 10.1056/NEJMoa040610
Van Eeden S, Leipsic J, Paul Man SF, Sin DD: The relationship between lung inflammation and cardiovascular disease. Am J Respir Crit Care Med 2012, 186: 11–16. 10.1164/rccm.201203-0455PP
Liu Y, Chien W-M, Medvedev IO, Weldy CS, Luchtel DL, Rosenfeld ME, Chin MT: Inhalation of diesel exhaust does not exacerbate cardiac hypertrophy or heart failure in two mouse models of cardiac hypertrophy. Part Fibre Toxicol 2013, 10: 49. 10.1186/1743-8977-10-49
Gould T, Larson T, Stewart J, Kaufman JD, Slater D, Mcewen N: A controlled inhalation diesel exhaust exposure facility with dynamic feedback control of PM concentration. Inhal Toxicol 2008, 20: 49–52. 10.1080/08958370701758478
Weldy CS, Luttrell IP, White CC, Morgan-Stevenson V, Cox DP, Carosino CM, Larson TV, Stewart JA, Kaufman JD, Kim F, et al.: Glutathione (GSH) and the GSH synthesis gene Gclm modulate plasma redox and vascular responses to acute diesel exhaust inhalation in mice. Inhal Toxicol 2013, 25: 444–454. 10.3109/08958378.2013.801004
Yin F, Lawal A, Ricks J, Fox JR, Larson T, Navab M, Fogelman AM, Rosenfeld ME, Araujo JA: Diesel exhaust induces systemic lipid peroxidation and development of dysfunctional pro-oxidant and pro-inflammatory high-density lipoprotein. Arterioscler Thromb Vasc Biol 2013, 33: 1153–1161. 10.1161/ATVBAHA.112.300552
Liu Y, Yu M, Wu L, Chin MT: The bHLH transcription factor CHF1/Hey2 regulates susceptibility to apoptosis and heart failure after pressure overload. Am J Physiol Heart Circ Physiol 2010, 298: H2082-H2092. 10.1152/ajpheart.00747.2009
Yu M, Liu Y, Xiang F, Li Y, Cullen D, Liao R, Beyer RP, Bammler TK, Chin MT: CHF1/Hey2 promotes physiological hypertrophy in response to pressure overload through selective repression and activation of specific transcriptional pathways. OMICS 2009, 13: 501–511. 10.1089/omi.2009.0086
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods (San Diego, Calif) 2001, 25: 402–408. 10.1006/meth.2001.1262
Acknowledgements
We thank Mr. James Stewart and Dr. Timothy Gould for their expert technical assistance with diesel exhaust exposures. YL, WMC, and MTC were supported by NIEHS DISCOVER Center grant P50 ES015915, CSW was supported by the Experimental Pathology of Cardiovascular Disease Training Grant (T32HL007312), IOM was supported by the Fellowship Training Program in Cardiovascular Disease (T32HL07828) and JRF was supported by the Biostatistics, Epidemiologic and Bioinformatic Training in Environmental Health Training Grant (T32ES015459). Additional support was provided by the NIEHS Center for Ecogenetics and Environmental Health though a pilot project award funded by grant P30ES007033.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors have no conflicts to disclose.
Authors’ contributions
CSW, contributed to generating hypothesis and experimental design, conducted experiments, interpreted data, and is the primary author of the manuscript; YL, performed animal surgeries, contributed to preparation of manuscript; YCC, performed qRT-PCR experiments, contributed to preparation of manuscript; IOM, analyzed echocardiography cine loops, contributed to preparation of manuscript; JRF, performed diesel exhaust characterization experiments, contributed to preparation of manuscript; TVL, contributed to diesel exhaust characterization experiments, contributed to preparation of manuscript; WMC, assisted in animal necropsies, contributed to preparation of manuscript; MTC, contributed to generating hypothesis and experimental design, contributed to preparation of manuscript and was responsible for overall coordination of the project. All authors read and approved of the final copy of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Weldy, C.S., Liu, Y., Chang, YC. et al. In utero and early life exposure to diesel exhaust air pollution increases adult susceptibility to heart failure in mice. Part Fibre Toxicol 10, 59 (2013). https://doi.org/10.1186/1743-8977-10-59
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-8977-10-59