
Abstract
Background
The flagellotropic phage 7-7-1 infects motile cells of Agrobacterium sp H13-3 by attaching to and traveling along the rotating flagellar filament to the secondary receptor at the base, where it injects its DNA into the host cell. Here we describe the complete genomic sequence of 69,391 base pairs of this unusual bacteriophage.
Methods
The sequence of the 7-7-1 genome was determined by pyro(454)sequencing to a coverage of 378-fold. It was annotated using MyRAST and a variety of internet resources. The structural proteome was analyzed by SDS-PAGE coupled electrospray ionization-tandem mass spectrometry (MS/MS).
Results
Sequence annotation and a structural proteome analysis revealed 127 open reading frames, 84 of which are unique. In six cases 7-7-1 proteins showed sequence similarity to proteins from the virulent Burkholderia myovirus BcepB1A. Unique features of the 7-7-1 genome are the physical separation of the genes encoding the small (orf100) and large (orf112) subunits of the DNA packaging complex and the apparent lack of a holin-lysin cassette. Proteomic analysis revealed the presence of 24 structural proteins, five of which were identified as baseplate (orf7), putative tail fibre (orf102), portal (orf113), major capsid (orf115) and tail sheath (orf126) proteins. In the latter case, the N-terminus was removed during capsid maturation, probably by a putative prohead protease (orf114).
Similar content being viewed by others


Background
Bacteriophage 7-7-1 is known to infect motile cells of Agrobacterium sp H13-3 (formerly Rhizobium lupini[1]), and as such is termed flagellotropic. Using electron microscopy, Lotz et al. [2] demonstrated translocation of phage 7-7-1 along flagellar filaments. Filament associated phage particles initially possess DNA-filled heads, which are subsequently found emptied when attached to the phage receptor at the flagellar base. This bimodal mechanism of adsorption dramatically increases the chance for finding the receptor at the cell surface, because (i) swimming bacteria with their flagella spread out act as a five- to 10-fold expanded target for the phage and, (ii) once attached, phage particles are directed to the receptor by a one-dimensional walk along the flagellum (instead of a random ‘search’ by three-dimensional diffusion). In no case has the process of phage translocation along the flagellum been visualized. Based on circumstantial evidence, Samuel et al. [3] have estimated that the flagellotropic phage χ of Salmonella needs < 1 s to reach the flagellar base. These authors have also provided evidence for a ‘nut and bolt’ mechanism by which phage χ moves along the filament. They argue that the long tail fiber fits the right-handed grooves between helical rows of flagellin subunits and that the counter-clockwise (CCW) rotation of the flagellum forces the phage to follow the grooves as a nut follows the threads of a bolt.
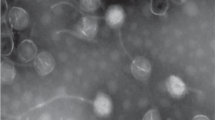
However, such conditions are not met by the ‘complex’ flagella of Agrobacterium sp H13-3. In fact, complex filaments exhibit a prominent pattern of right-handed helical ridges and grooves recommending itself as convenient ‘threads’, but the sense of flagellar rotation is exclusively clockwise (CW; [4–6]). Hence, ‘nut and bolt’ mechanics would force an attached phage particle to the distal end rather than to the flagellar base. Thus, the observed movement of 7-7-1 to the flagellar base demands a different, yet unknown mode of translocation. Differences between the two flagellotropic phages are also reflected by their distinct morphologies: electron micrographs of phage χ show a single long (200–220 nm) tail fiber wrapped around the ‘plain’ filament of Salmonella[7], whereas phage 7-7-1 exhibits five short (16 nm) tail fibers with splayed tips. Figure 1B shows a scale diagram of phage 7-7-1 as deduced from high-resolution electron micrographs (Figure 1A).

High-resolution electron micrograph (A) and scale diagram (B) of bacteriophage 7-7-1. A 14-nm collar connects the icosahedral head with the contractile tail that exhibits a surface structure of helical rows running at an angle of 50°. Five 16-nm tail fibers with splayed tips probably lead the phage along the flagellar filament to the cell surface, where they act as specific adsorption organelles that attach the phage to its final receptor. Details of the tail fine structure were uncovered by optical diffraction [8] of highly resolved electron micrographs.
7-7-1 is the first flagellotropic phage shown to infect a soil bacterium driven by the uni-directional CW rotation of its complex flagella, a pattern clearly different from the CCW-CW bias of the plain flagella driving Salmonella[9]. This departure from the well-studied enterobacterial paradigm and the rare phage morphology prompted us to analyze the genome and the structural proteome of 7-7-1.
Results
Genome
Electron micrographs of platinum/iridium-stained 7-7-1 DNA revealed mostly linear and a few circular molecules of approximately 25 μm contour lengths (mass of ≅73.5 kb; data not shown) suggesting DNA circularization by cohesive ends. These single-stranded termini are not covered by 454 sequencing. The 454 sequence data revealed that the genome of the phage was 69,391 bp (52.4 mol%G + C). Following automated annotation using MyRAST the genome was manually curated revealing 127 ORFs and no tRNAs. The majority (84, 65.6%) of the ORFs showed no homology to any protein in the current NCBI databases. A minority showed similarity to prophage (28, 21.9%) or phage proteins (16, 12.5%). In the latter case 7-7-1 gp20-26 were collinear to a set of genes from Burkholderia phage BcepB1A [10] which is also a virulent myovirus. Phage 7-7-1 displays a number of unique features including the physical separation of the genes encoding the small (orf100) and large (orf112) subunits of the terminase complex. In addition, there is no evidence for a holin-lysin cassette (Figure 2; Additional file 1, Table S1).

Genetic map of 7-7-1 showing genes encoding hypothetical proteins in black; conserved hypothetical proteins, blue; structural proteins, red; regulatory proteins, green; DNA and nucleotide metabolism, purple; terminase subunits, brown. Putative promoters are indicated with black arrows on stalks, while predicted rho-independent terminators are indicated with white circle on stalks, and stem-loop structures are indicated with black circle on stalks.
DNA replication
DNA replication of this phage involves a helicase (orf23) and a polymerase (orf17). The latter shows greatest sequence similarity to the DNA polymerases of Pseudomonas phage 73 (YP_001293433) and Burkholderia phage BcepGomr (YP_001210246) which are members of the Siphoviridae, and Burkholderia phage BcepB1A (YP_024903) which, like 7-7-1, is a myovirus. An InterProScan shows it to be a member of the DNA/RNA polymerases superfamily (SUPERFAMILY SSF56672) with the motif located between residues 318 and 480. Two other proteins potentially involved in replication are the products of genes 28 and 33. Gp28 is a 255 amino acid protein possessing ParB-like nuclease motifs (Pfam PF02195 ParBc; SMART SM00470 ParB-like nuclease domain and SUPERFAMILY [11] SSF110849 ParB/Sulfiredoxin) as well as ParB-like partition TIGRFAMs [12] protein motif TIGR00180 parB_part: ParB-like partition proteins. This type of protein has also been found in myoviruses such as Burkholderia ambifaria phage BcepF1 (YP_001039693), Mycobacterium phage Pio (AER49600) and enterobacterial phage P1 (AAQ14139). Gp33 contains a N-(deoxy)ribosyltransferase-like superfamily (SUPERFAMILY SSF52309) motif.
Transcription
Based upon the assumption that the genome circularizes via cohesive termini (not identified), there are two large transcriptional units encompassing orf 22–13 and orf 23–127, 1–12. Since another member of the class α-proteobacteria, Rhizobium etli, possesses rpoD-dependent promoters which closely resemble the Escherichia coli consensus sequence (TTGACA[N15-17]TATAAT) [13] we assumed that this phage might contain recognizable promoters. We identified five potential promoter sequences, including divergent promoters between the two transcription units (Additional file 2, Table S2). In addition four rho-independent terminators were identified and two high ΔG stem-loop structures. Interestingly, no bidirectional terminators were discovered between orf12 and orf13 (Additional file 2, Table S2). No evidence was found as to how transcription is temporally regulated in this virus.
The genome of phage 7-7-1 encodes for two proteins involved in DNA synthesis – a helicase (gp23) and a polymerase (gp17). The polymerase displayed no conserved motifs, and is distantly related to gp43 homologs from cyanomyoviruses. The helicase contained a high scoring (E-value: 1.01e-41) COG1061, DNA or RNA helicases of superfamily II protein motif (SSL2); and, homology to helicases from Burkholderia phage BcepB1A [10], and Vibrio phages VP16C and VT16T [14].
PSI-BLAST analysis of Gp3 against the NCBI virus database resulted in hits described as tail/DNA circulation protein (Salmonella phage ST64B [15], Enterobacteria phage SfV [16], Pseudomonas phage DVM-2008, and Burkholderia phage KS10 [17]. This protein possesses two protein motifs: COG4228, Mu-like prophage DNA circulation protein, and pfam07157, DNA circulation protein N-terminus (DNA_circ_N) which are conserved protein domains of indeterminate function. Gp4 contains two inconsistent overlapping motifs: COG4379, Mu-like prophage tail protein gpP (E-value: 2.99e-22), and, pfam05954, phage late control gene D protein (Phage_GPD; E-value: 1.76e-17). The homologs include tail proteins from Mu, D108, SfV and ST64B. These results, coupled with the genome location and the observation that Gp3 is a structural protein (see next section), suggest that both of these proteins are involved in the sequence/assembly of the phage tail.
Virion structural proteins
BLAST analysis revealed several proteins as being involved in phage morphogenesis including baseplate protein (gp7), tail fibre (gp102), portal (gp113), prohead protease (gp114), major capsid (gp115) and tail sheath (gp126). HHpred [18, 19] analysis on other proteins in the morphogenesis cassette was used to identify three other proteins - gp5, gp6 and gp10. Gp10 which we had termed a conserved hypothetical membrane protein shows structural similarity (Probability = 91.01; E-value = 0.11) to RCSB Protein Data Bank [20] 3BKH, the lytic transglycosylase (gp144) of Pseudomonas phage φKZ which is probably the endolysin for this virus [21]. Gp6 is related (Probability = 83.90; E-value = 0.63) to 2IA7 – a putative tail lysozyme (T4 gp5 analog); while gp5 is a structural analog of 3AJQ, phage P2 protein V which is the tailspike protein (Probability = 96.23; E-value = 0.021) [22].
Proteomics
Electrospray ionization-MS/MS analysis of the structural phage proteins separated by SDS-PAGE led to the experimental identification of 24 virion proteins with sequence coverage from 8.4 to 85.7% (Table 1/Figure 3). Although only phage proteins with a minimum number of two unique peptides were considered, the identification of gp124 by a single peptide hit was approved based on a reliable proline spectrum [23]. Electrophoretic mobilities of the identified peptides were consistent with their predicted molecular masses, and seven of the nine visible protein bands on the gel could be unambiguously identified (Figure 3). Moreover, traces of the capsid (gp115) and the tail sheath protein (gp126) were identified throughout the gel, which can be explained by aspecific retention and partial degradation of these abundant proteins.

SDS-PAGE analysis of the purified structural phage proteins (lane B) on a 12% SDS-PAGE separation gel alongside a PageRuler™ prestained protein ladder (Fermentas) (lane A). The entire lane was cut into numbered slices (i). Subsequently the origin of the visible protein bands were identified by ESI-MS/MS analysis (ii, showing Gps).
Although the major capsid protein gp115 is clearly the most abundant protein, only peptides of its C-terminus were found. This suggests that the N-terminal part is cleaved off during maturation of the capsid. Indeed, similarity searches indicate that the C-terminal part of gp115 has high similarity with the major capsid protein of the HK97 family and that gp114 is similar to various prohead proteases. As the N-terminal part of the HK97 capsid is cleaved off by a prohead protease encoded by the upstream gene [24, 25], the protein band with a molecular weight of approximately 33 kDa refers to the mature major capsid protein.
A final, remarkable finding is the identification of a small, 28 amino acid protein which originally fell below the threshold of gene prediction (i.e. 100 bp). Though the function of this polypeptide is unknown, the high ‘protein identification probability’ of 100% and the coverage of 85.7% confirmed its presence in the phage particle. This proves that proteogenomics, namely the use of proteome analysis to annotate the genome, is a powerful tool to identify missed protein-coding genes and thereby complements genome annotation.
Discussion
While a number of flagella-specific phages have been isolated – Salmonella phage χ, Caulobacter phages φCp34 [26], ϕCb13 and ϕCbK [27], and φ6 [28]; Bacillus phages AR9, 3NT, PBS1 [29], SP3 [30], and PBP1 [31]; Proteus phage PV22 [32]; Pseudomonas phage φCTX [33], Agrobacterium tumefaciens phages GS2 and GS6 [34]; Aeromonas hydrophila phage PM3 [35], and, Asticcacaulis biprosthecum φAcS2, and φAcM4 [36] – to the best of our knowledge only χ (Denyes, personal communication) and φCTX [37] have been sequenced. Using the BLASTP feature in BioEdit [38] the products of five 7-7-1 genes (13, 21, 26, 72 and 102) possessed homologs in Salmonella phage χ. Interestingly, we defined gp102 as a putative tail fibre protein; and, it shows weak sequence similarity from residues 203–300 to a similarly defined protein from phage χ. In view of the quite different tail fibre morphologies observed in phage χ and phage 7-7-1, respectively, the region of similarity may define a general motif involved in phage-flagellum interaction.
Bacteriophage 7-7-1 shows relatively little overall DNA sequence similarity to other phages. At the protein level, CoreGenes revealed eight homologs of BcepB1A proteins, restricted to TerS and a variety of hypothetical proteins. These results indicate that phage 7-7-1 is unique and deserving of recommendation to ICTV as the type phage in a new genus: the 7-7-1-like bacteriophages.
Materials and methods
Bacteria and bacteriophages
Agrobacterium sp H13-3 (formerly Rhizobium lupini H13-3) was isolated from the rhizosphere of Lupinus luteus[39]. Phage 7-7-1, which is an isolate from garden compost [40], exclusively infects Agrobacterium sp H13-3 [1].
Bacteria were grown in NY medium (8 g nutrient broth, 3 g yeast extract per liter) at 40 rpm in a gyratory shaker at 30 °C. Phage lysates up to 2x10¹¹ PFU per ml were obtained by infection of an exponentially growing culture at OD650nm = 0.1 (8 x 107 CFU per ml) with phage at an MOI of 5 x 10-3 followed by threefold dilution with pre-warmed NY and further incubation pending lysis.
Electron microscopy
Purified phage particles were spread on carbon-coated copper grids, washed once with distilled water and then negatively stained with 4% uranyl acetate, pH 4.8. Microscope magnifications were calibrated with a replica of an optical grating and micrographs were taken with a JEOL 7A (Japan Electron Optics Laboratory Co., Ltd.).
DNA isolation for sequencing
Phage DNA was isolated by phenol-chloroform extraction [41] and purified by using the Lambda DNA kit of Qiagen (Hilden, Germany). The DNA was subjected to pyrosequencing (454 technology) at the McGill University and Genome Québec Innovation Centre (Montreal, QC, Canada) to 378X coverage.
Genome annotation
The 7-7-1 sequence was initially subjected to automated annotation using MyRAST (http://blog.theseed.org/servers/presentations/t1/running-a-job-with-the-desktop-rast.html), tRNAScan-SE [42] and ARAGORN [43], following which all open reading frames (ORFs) were confirmed using Kodon (Applied Maths Inc., Austin, TX. USA). The individual proteins were screened against the nonredundant protein databases in NCBI using Batch BLAST (http://greengene.uml.edu/programs/ NCBI_Blast.html). In addition they were screened for conserved motifs using InterProScan [44], Pfam [45], TMHMM v2.0 [46] and Phobius [47].
Putative promoters were identified based upon sequence similarity to the consensus RpoD-specific E.coli promoter sequence TTGACA[N15-17]TATAAT while rho-independent terminators were identified using ARNold [48, 49] complemented with MFOLD [50].
The genome was submitted to NCBI and accorded accession number JQ312117.
Comparative genomics
This phage was compared at the DNA and protein levels to other related phages using progressiveMauve [51] and CoreGenes [52, 53].
Proteomics
Structural phage proteins were purified as described by Moak and Molineux [54]. Briefly, a solution of CsCl-purified phage particles (1011 PFU) was mixed with methanol and chloroform (1:1:0.75 by volume). After agitation and centrifugation, the upper layer was discarded and an equal volume of methanol was added. The protein pellet obtained by centrifugation at 14, 000 rpm for 6 min, was dried and resuspended in 12.5 mM NH4HCO3. Subsequently, the heat denatured sample (95 °C, 5 min) was loaded on a 12% SDS-PAGE gel. The Coomassie-stained gel (Simply Blue Safestain; Invitrogen) was cut into slices, which were subjected to trypsin digestion [55]. Peptides were analyzed using electrospray ionization-tandem mass spectrometry (MS/MS) as described previously by Lavigne et al. [56]. The obtained spectra were screened against a database containing all ‘stop-to-stop’ protein sequences in all six frames. Generally, the identification parameters were a ‘protein identification probability’ of at least 99.8% and a ‘best peptide identification probability’ of 95%.
Abbreviations
- BLAST:
-
Basic Local Alignment Search Tool
- ESI-MS/MS:
-
electrospray ionization tandem mass spectrometry
- Gp:
-
: Gene product
- HHpred:
-
Homology detection & structure prediction by HMM-HMM comparison
- MOI:
-
: Multiplicity of Infection, ratio of infective phage particles to vulnerable hosts
- NY medium:
-
Difco nutrient broth plus yeast extract
- PFU:
-
: Plaque Forming Unit, a measure of the number of viable viral particles
- SDS-PAGE:
-
denaturing (sodium dodecyl sulfate) polyacrylamide gel electrophoresis
- TMHMM:
-
TransMembrane prediction using Hidden Markov Models.
References
Wibberg D, Blom J, Jaenicke S, Kollin F, Rupp O, Scharf B, Schneiker-Bekel S, Sczcepanowski R, Goesmann A, Setubal JC, Schmitt R, Pühler A, Schlüter A: Complete genome sequencing of Agrobacterium sp. H13-3, the former Rhizobium lupini H13-3, reveals a tripartite genome consisting of a circular and a linear chromosome and an accessory plasmid but lacking a tumor-inducing Ti-plasmid. J Biotechnol 2011, 155: 50-62. 10.1016/j.jbiotec.2011.01.010
Lotz W, Acker G, Schmitt R: Bacteriophage 7-7-1 adsorbs to the complex flagella of Rhizobium lupini H13-3. J Gen Virol 1977, 34: 9-17. 10.1099/0022-1317-34-1-9
Samuel AD, Pitta TP, Ryu WS, Danese PN, Leung EC, Berg HC: Flagellar determinants of bacterial sensitivity to chi-phage. Proc Natl Acad Sci USA 1999, 96: 9863-9866. 10.1073/pnas.96.17.9863
Trachtenberg S, DeRosier DJ, Macnab RM: Three-dimensional structure of the complex flagellar filament of Rhizobium lupini and its relation to the structure of the plain filament. J Mol Biol 1987, 195: 603-620. 10.1016/0022-2836(87)90185-9
Scharf B: Real-time imaging of fluorescent flagellar filaments of Rhizobium lupini H13-3: flagellar rotation and pH-induced polymorphic transitions. J Bacteriol 2002, 184: 5979-5986. 10.1128/JB.184.21.5979-5986.2002
Schmitt R, Bamberger I, Acker G, Mayer F: Feinstrukturanalyse der komplexen Geißeln von Rhizobium lupini H13-3. Arch Mikrobiol 1974, 100: 145-162.
Schade SZ, Adler J, Ris H: How bacteriophage chi attacks motile bacteria. J Virol 1967, 1: 591-598.
Yanagida M, Boy De La Tour E, Alff-Steinberger C, Kellenberger E: Studies on the morphopoiesis of the head of bacteriophage T-even. 8. Multilayered polyheads. J Mol Biol 1970, 50: 35-58. 10.1016/0022-2836(70)90102-6
Schmitt R: Sinorhizobial chemotaxis: a departure from the enterobacterial paradigm. Microbiology 2002, 148: 627-631.
Summer EJ, Gonzalez CF, Bomer M, Carlile T, Morrison W, Embry A, Kucherka AM, Lee J, Mebane L, Morrison WC, Mark L, King MD, LiPuma MJ, Vidaver AK, Young R: Divergence and mosaicism among virulent soil phages of the Burkholderia cepacia complex. J Bacteriol 2006, 188: 255-268. 10.1128/JB.188.1.255-268.2006
Wilson D, Pethica R, Zhou Y, Talbot C, Vogel C, Madera M, Chothia C, Gough J: SUPERFAMILY–sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res 2009, 37: D380-D386. 10.1093/nar/gkn762
Haft DH, Selengut JD, White O: The TIGRFAMs database of protein families. Nucleic Acids Res 2003, 31: 371-373. 10.1093/nar/gkg128
Ramirez-Romero MA, Masulis I, Cevallos MA, Gonzalez V, Davila G: The Rhizobium etli sigma70 (SigA) factor recognizes a lax consensus promoter. Nucleic Acids Res 2006, 34: 1470-1480. 10.1093/nar/gkl023
Seguritan V, Feng IW, Rohwer F, Swift M, Segall AM: Genome sequences of two closely related Vibrio parahaemolyticus phages, VP16T and VP16C. J Bacteriol 2003, 185: 6434-6447. 10.1128/JB.185.21.6434-6447.2003
Mmolawa PT, Schmieger H, Heuzenroeder MW: Bacteriophage ST64B, a genetic mosaic of genes from diverse sources isolated from Salmonella enterica serovar typhimurium DT 64. J Bacteriol 2003, 185: 6481-6485. 10.1128/JB.185.21.6481-6485.2003
Allison GE, Angeles D, Tran-Dinh N, Verma NK: Complete genomic sequence of SfV, a serotype-converting temperate bacteriophage of Shigella flexneri. J Bacteriol 2002, 184: 1974-1987. 10.1128/JB.184.7.1974-1987.2002
Goudie AD, Lynch KH, Seed KD, Stothard P, Shrivastava S, Wishart DS, Dennis JJ: Genomic sequence and activity of KS10, a transposable phage of the Burkholderia cepacia complex. BMC Genomics 2008, 9: 615-615. 10.1186/1471-2164-9-615
Hildebrand A, Remmert M, Biegert A, Soding J: Fast and accurate automatic structure prediction with HHpred. Proteins 2009,77(Suppl 9):128-32. 128–132
Soding J, Biegert A, Lupas AN: The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 2005, 33: W244-W248. 10.1093/nar/gki408
Rose PW, Beran B, Bi C, Bluhm WF, Dimitropoulos D, Goodsell DS, Prlic A, Quesada M, Quinn GB, Westbrook JD, Young J, Yukich B, Zardecki C, Berman HM, Bourne PE: The RCSB Protein Data Bank: redesigned web site and web services. Nucleic Acids Res 2011, 39: D392-D401. 10.1093/nar/gkq1021
Fokine A, Miroshnikov KA, Shneider MM, Mesyanzhinov VV, Rossmann MG: Structure of the bacteriophage ϕKZ lytic transglycosylase gp144. J Biol Chem 2008, 283: 7242-7250. 10.1074/jbc.M709398200
Yamashita E, Nakagawa A, Takahashi J, Tsunoda K, Yamada S, Takeda S: The host-binding domain of the P2 phage tail spike reveals a trimeric iron-binding structure. Acta Crystallogr Sect F Struct Biol Cryst Commun 2011, 67: 837-841. 10.1107/S1744309111005999
Hunt DF, Yates JR III, Shabanowitz J, Winston S, Hauer CR: Protein sequencing by tandem mass spectrometry. Proc Natl Acad Sci U S A 1986, 83: 6233-6237. 10.1073/pnas.83.17.6233
Duda RL, Martincic K, Hendrix RW: Genetic basis of bacteriophage HK97 prohead assembly. J Mol Biol 1995, 247: 636-647.
Huang RK, Khayat R, Lee KK, Gertsman I, Duda RL, Hendrix RW, Johnson JE: The Prohead-I structure of bacteriophage HK97: implications for scaffold-mediated control of particle assembly and maturation. J Mol Biol 2011, 408: 541-554. 10.1016/j.jmb.2011.01.016
Fukuda A, Miyakawa K, Iba H, Okada Y: A flagellotropic bacteriophage and flagella formation in Caulobacter. Virology 1976, 71: 583-592. 10.1016/0042-6822(76)90383-4
Guerrero-Ferreira RC, Viollier PH, Ely B, Poindexter JS, Georgieva M, Jensen GJ, Wright ER: Alternative mechanism for bacteriophage adsorption to the motile bacterium Caulobacter crescentus. Proc Natl Acad Sci USA 2011, 108: 9963-9968. 10.1073/pnas.1012388108
Jollick JD, Wright BL: A flagella specific bacteriophage for Caulobacter. J Gen Virol 1974, 22: 197-205. 10.1099/0022-1317-22-2-197
Vieira G, de Lencastre H, Archer L: Restriction analysis of PBS 1-related phages. Arch Virol 1989, 106: 121-126. 10.1007/BF01311043
Shea TB, Seaman E: SP3: a flagellotropic bacteriophage of Bacillus subtilis. J Gen Virol 1984, 65: 2073-2076. 10.1099/0022-1317-65-11-2073
Lovett PS: PBPI: a flagella specific bacteriophage mediating transduction in Bacillus pumilus. Virology 1972, 47: 743-752. 10.1016/0042-6822(72)90564-8
Zhilenkov EL, Popova VM, Popov DV, Zavalsky LY, Svetoch EA, Stern NJ, Seal BS: The ability of flagellum-specific Proteus vulgaris bacteriophage PV22 to interact with Campylobacter jejuni flagella in culture. Virol J 2006, 3: 50. 10.1186/1743-422X-3-50
Geiben-Lynn R, Sauber K, Lutz F: Flagellin inhibits Myoviridae phage ϕCTX infection of Pseudomonas aeruginosa strain GuA18: purification and mapping of binding site. Arch Microbiol 2001, 176: 339-346. 10.1007/s002030100332
Bradley DE, Douglas CJ, Peschon J: Flagella-specific bacteriophages of Agrobacterium tumefaciens: demonstration of virulence of nonmotile mutants. Can J Microbiol 1984, 30: 676-681. 10.1139/m84-101
Merino S, Camprubi S, Tomas JM: Isolation and characterization of bacteriophage PM3 from Aeromonas hydrophila the bacterial receptor for which is the monopolar flagellum. FEMS Microbiol Lett 1990, 57: 277-282.
Pate JL, Petzold SJ, Umbreit TH: Two flagellotropic phages and one pilus-specific phage active against Asticcacaulis biprosthecum. Virology 1979, 94: 24-37. 10.1016/0042-6822(79)90435-5
Nakayama K, Kanaya S, Ohnishi M, Terawaki Y, Hayashi T: The complete nucleotide sequence of ϕCTX, a cytotoxin-converting phage of Pseudomonas aeruginosa: implications for phage evolution and horizontal gene transfer via bacteriophages. Mol Microbiol 1999, 31: 399-419. 10.1046/j.1365-2958.1999.01158.x
Tippmann HF: Analysis for free: comparing programs for sequence analysis. Brief Bioinform 2004, 5: 82-87. 10.1093/bib/5.1.82
Gábor M: Transformation of streptomycin markers in rough strains of Rhizobium lupini. II. The relation between the derterminant of streptomycin dependence and those for streptomycin resistance and sensitiveness. Genetics 1965, 52: 905-913.
Lotz W, Mayer F: Electron microscopical characterization of newly isolated Rhizobium lupini bacteriophages. Can J Microbiol 1972, 18: 1271-1274. 10.1139/m72-196
Sambrook J, Russell DW: Molecular Cloning: A Laboratory Manual. Third edition edition. Cold Spring Harbor Press, Cold Spring Harbor, New York; 2001.
Lowe TM, Eddy SR: tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997, 25: 955-964.
Laslett D, Canback B: ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res 2004, 32: 11-16. 10.1093/nar/gkh152
Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R: InterProScan: protein domains identifier. Nucleic Acids Res 2005, 33: W116-W120. 10.1093/nar/gki442
Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer EL, Eddy SR, Bateman A: The Pfam protein families database. Nucleic Acids Res 2010, 38: D211-D222. 10.1093/nar/gkp985
Sonnhammer EL, Von HG, Krogh A: A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol 1998, 6: 175-182.
Kall L, Krogh A, Sonnhammer EL: A combined transmembrane topology and signal peptide prediction method. J Mol Biol 2004, 338: 1027-1036. 10.1016/j.jmb.2004.03.016
Gautheret D, Lambert A: Direct RNA motif definition and identification from multiple sequence alignments using secondary structure profiles. J Mol Biol 2001, 313: 1003-1011. 10.1006/jmbi.2001.5102
Macke TJ, Ecker DJ, Gutell RR, Gautheret D, Case DA, Sampath R: RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Res 2001, 29: 4724-4735. 10.1093/nar/29.22.4724
Zuker M, Zuker M: Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003, 31: 3406-3415. 10.1093/nar/gkg595
Darling AE, Mau B, Perna NT: progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 2010, 5: e11147. 10.1371/journal.pone.0011147
Kropinski AM, Borodovsky M, Carver TJ, Cerdeno-Tarraga AM, Darling A, Lomsadze A, Mahadevan P, Stothard P, Seto D, Van DG, Wishart DS: In silico identification of genes in bacteriophage DNA. Methods Mol Biol 2009, 502: 57-89. 10.1007/978-1-60327-565-1_6
Zafar N, Mazumder R, Seto D: CoreGenes: a computational tool for identifying and cataloging "core" genes in a set of small genomes. BMC Bioinformatics 2002, 3: 12. 10.1186/1471-2105-3-12
Moak M, Molineux IJ: Peptidoglycan hydrolytic activities associated with bacteriophage virions. Mol Microbiol 2004, 51: 1169-1183. 10.1046/j.1365-2958.2003.03894.x
Shevchenko A, Jensen ON, Podtelejnikov AV, Sagliocco F, Wilm M, Vorm O, Mortensen P, Shevchenko A, Boucherie H, Mann M: Linking genome and proteome by mass spectrometry: large-scale identification of yeast proteins from two dimensional gels. Proc Natl Acad Sci USA 1996, 93: 14440-14445. 10.1073/pnas.93.25.14440
Lavigne R, Noben JP, Hertveldt K, Ceyssens PJ, Briers Y, Dumont D, Roucourt B, Krylov VN, Mesyanzhinov VV, Robben J, Volckaert G: The structural proteome of Pseudomonas aeruginosa bacteriophage ϕKMV. Microbiology 2006, 152: 529-534. 10.1099/mic.0.28431-0
Acknowledgements
We wish to thank Paul Thienel for artwork and Hermine Reisner for technical assistance. A.M.K. was supported by a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada. AV holds a PhD scholarship of the FWO Vlaanderen.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors have no competing interests to disclose.
Authors’ contributions
AMK performed DNA sequence analyses and sequence annotation, AV, RL and JN analyzed the proteome, PB and RS provided phage DNA, a high-titer lysate, and the fine structure analysis of 7-7-1 particles. All authors contributed to the writing of this manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12985_2012_1924_MOESM2_ESM.doc
Additional file 2:Table S2. Putative promoters and predicted terminators found in 7-7-1. Table S3. Mass spectroscopic analysis of individual gel bands, mzML formatted files in zipped format. (DOC 33 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kropinski, A.M., Van den Bossche, A., Lavigne, R. et al. Genome and proteome analysis of 7-7-1, a flagellotropic phage infecting Agrobacterium sp H13-3. Virol J 9, 102 (2012). https://doi.org/10.1186/1743-422X-9-102
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-9-102