
Abstract
No one likes to feel like they have been manipulated, but in the case of cytomegalovirus (CMV) immune manipulation, we do not really have much choice. Whether you call it CMV immune modulation, manipulation, or evasion, the bottom line is that CMV alters the immune response in such a way to allow the establishment of latency with lifelong shedding. With millions of years of coevolution within their hosts, CMVs, like other herpesviruses, encode numerous proteins that can broadly influence the magnitude and quality of both innate and adaptive immune responses. These viral proteins include both homologues of host proteins, such as MHC class I or chemokine homologues, and proteins with little similarity to any other known proteins, such as the chemokine binding protein. Although a strong immune response is launched against CMV, these virally encoded proteins can interfere with the host's ability to efficiently recognize and clear virus, while others induce or alter specific immune responses to benefit viral replication or spread within the host. Modulation of host immunity allows survival of both the virus and the host. One way of describing it would be a kind of "mutually assured survival" (as opposed to MAD, Mutually Assured Destruction). Evaluation of this relationship provides important insights into the life cycle of CMV as well as a greater understanding of the complexity of the immune response to pathogens in general.
Similar content being viewed by others
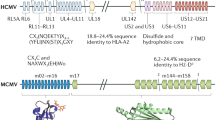

Introduction
After an initial primary infection herpesviruses establish latency for the life of the host with periodic and spontaneous reactivation. The coevolution of herpesviruses along with their host allowed these viruses to evolve mechanisms to modulate the host immune response. While some virally encoded proteins facilitate immune evasion, interfering with the host's ability to efficiently recognize and clear virus, others induce or alter specific immune responses to benefit the viral life cycle. Among the herpesviruses, cytomegaloviruses (CMVs) encode the greatest number of genes committed to altering both innate and adaptive immune responses (Additional files 1 and 2).
Innate Immune Responses
Complement Cascade
The complement system is composed of a number of plasma proteins that induce inflammatory mediators, opsonization of pathogens, and direct lysis of pathogens and infected cells. The binding of complement proteins to antibody-antigen complexes activates the classical complement pathway, bridging innate and humoral immune responses. The binding of complement proteins directly to the surface of pathogens or infected cells initiates the alternative pathway. Both pathways lead to a common activation of the C3 convertase and subsequent elicitation of chemoattractants (e.g. C5a), opsonizing factors (e.g. C3b) and the membrane attack complex. Antibody-mediated complement lysis is an important mechanism for elimination of virus-infected cells. Therefore, a number of herpesviruses encode complement regulatory proteins. Herpes simplex virus 1 (HSV-1) and Epstein Barr virus (EBV) both have homologues of regulators of complement activation (RCA) proteins, such as CD46 and CD55 [1]. Complement control proteins including CD46, CD55, and CD59 regulate and also inhibit various stages within the complement cascade [2]. While no CMV encoded RCA homologues have been identified, CMVs have evolved mechanisms for limiting complement activity. HCMV infected fibroblasts are resistant to complement lysis when treated with CMV-specific antibodies, which would be expected to induce the classical complement pathway [3]. Human CMV (HCMV) incorporates cellular CD55 and CD59 into its virion. The importance of these complement inhibitory proteins was demonstrated when antibodies against CD55 decreased HCMV replication in fibroblasts following incubation with complement components [4]. The decrease in viral titers in the presence of these antibodies showed that inhibition of complement activation (i.e. interfering with inhibitory CD55) is important for HCMV replication. HCMV also upregulates the expression of CD46 and CD55 on the surface of infected cells, which decreases the accumulation of C3 convertases. This in turn, protects the cells from complement mediated lysis [5]. Although the mechanisms differ from other herpesviruses, HCMV is able to inactivate the complement cascade, increasing virus replication/survival.
Mouse CMV (MCMV) also induces CD46 expression on infected fibroblasts. This upregulation was mapped to a CMV responsive element within the CD46 promoter [6]. Mice typically express the complement regulator CRRY instead of CD46 [7]. However, MCMV infected cells resist complement lysis and CD46 expression is associated with this decrease [6]. The viral proteins that interfere with this specific, antibody-mediated complement activity during MCMV infection have not been identified.
Fc Receptor Homologues
Clinical evidence supports a role for antibodies in limiting cytomegalovirus infection [8] and modulation of antibody-mediated immunity would be beneficial for CMV survival. CMV infected cells can bind immunoglobulins in serum of seronegative individuals [9–12]. This phenomenon was linked to the expression of Fc binding proteins (FcBPs) on the surface of infected cells that are specific for IgG but not other isotypes [13–15]. Both MCMV and HCMV encode FcBPs. MCMV encodes one FcBP, fcr-1 or m138. Although, m138 has no homology to the HCMV Fcγ BP it has conserved structural features with cellular Fcγ receptors (FcγRs) and the viral FcγR of HSV (gE-gI) [16]. Normally the FcRs from the host interact with the Fc region of Igs and mediate a number of effector functions including phagocytosis, release of inflammatory cytokines, and natural killer (NK) cell-mediated antibody dependent cellular cytotoxicity (ADCC)[17]. It has been speculated that the Fc region of anti-CMV antibodies bind the viral FcγR instead of host FcγRs and block antiviral activities. m138 is important for MCMV replication in vivo. However, both wild-type and B cell deficient mice infected with a Δm138 MCMV mutant had lower viral titers in several organs compared to mice infected with wild-type MCMV. Thus impaired growth of MCMV is not due to an enhanced humoral immune response in the absence of m138 (i.e. similar titers were found in the presence or absence of humoral responses) [18, 19]. Interestingly, m138 downregulates expression of H60 and MULT-1, two ligands of the activating NK receptor, NKG2D. The importance of this downregulation in vivo was demonstrated when the Δm138 MCMV was found at two logs reduced titer compared to wt MCMV. Both replicated equivalently in NK cell depleted mice. The downregulation of MULT-1, but not H60, is due to defective cellular trafficking that results in degradation of the protein [20]. m138 also decreases cell surface trafficking of the costimulatory molecule B7-1 on dendritic cells (DCs), interfering with the ability of these cells to activate antigen specific T cells [21]. Thus the MCMV FcγR should broadly affect both NK cell recognition and perhaps presentation to the adaptive immune response, although in a manner different from HSV-1 gE-gI.
Two HCMV FcγRs have been identified, gp34 and gp68, which are encoded by the genes TRL11/IRL11 and UL119-UL118, respectively [22, 23]. Although the function of the HCMV-encoded FcγRs has not been determined, initially it was assumed they would function like the viral FcγR from HSV-1 (gE-gI), which protects infected cells from ADCC and complement activation [24, 25]. Recently it was shown that gp68 binds the same IgG domain and with the same stoichiometry as HSV-1 gE-gI. However, the two proteins have different pH requirements for binding which may relate to different mechanisms following IgG engagement, although this possibility requires additional study [26].
Interferon-Mediated Immunity
While interferons (IFNs) are induced early during CMV infection [27–32], CMVs encode a number of proteins that mitigate the effects of IFN activation [33–35]. The type I interferons IFNα and IFNβ are part of the innate immune response to viral infections [36]. The binding of IFNα and IFNβ to the IFN receptor induces Jak/Stat signalling leading to the rapid upregulation of interferon-stimulated genes (ISGs), such as MHC class I molecules and various cytokines [37]. Two HCMV immediate early gene products, 72 kDa immediate early 1 (IE1) protein (IE1-p72) and IE2 protein (IE86), interfere with IFN signalling. IE1-p72 binds STAT2 inhibiting the ISG transcriptional activator, ISGF3, from binding to the ISG promoter [38, 39]. IE86 blocks binding of the transcription factor NFκB to the IFNβ promoter and leads to the downregulation of IFNβ expression in HCMV infected fibroblasts [40, 41].
The HCMV tegument protein pp65 (UL83) has been implicated in blocking interferon activity early during infection prior to transcription of the immediate early gene products [30]. Microarray analysis using wt HCMV and a UL83 deletion mutant revealed pp65 downregulates numerous ISGs although the mechanism for this is unclear [42]. Conflicting reports have attributed this decrease to interference with either NFκB activity or another transcriptional regulator, IRF-3, although both authors suggest a pp65-mediated defect in nuclear localization of the transcription factors likely contributes to the decreased activity [42, 43]. However, a UL83 mutant virus like that used in these previous studies was also shown to have decreased IE86 expression. In contrast, cells infected with an HCMV mutant in which stop codons were used to disrupt pp65 expression, preserving IE86 expression, showed no difference in IFN expression compared to wt HCMV [44]. This suggests IE86 activity is crucial in mitigating IFN-mediated events following infection. Nevertheless, the conservation of UL83 within CMV genomes and the attenuation of an MCMV lacking expression of the murine pp65 homolog [45] points to the importance of pp65 for CMV survival in vivo.
The MCMV encoded protein, M27, interferes with both type I and type II (IFNγ) interferon activity by downregulating the signalling molecule, STAT2 [46, 47]. M27 decreases transcription of constituents of the immunoproteasome [48]. This IFNγ inducible proteasome enhances MHC class I processing of peptides for presentation. Therefore, M27 may not only protect MCMV from the direct antiviral effects of interferon induction but also interfere with the presentation of viral peptides to CD8+ T cells, a common theme to be discussed below.
MCMV and HCMV also encode proteins that interact with interferon induced gene products. The MCMV gene products m142 and m143 decrease activation of protein kinase R (PKR) [49], an enzyme that blocks protein synthesis in infected cells leading to impaired viral replication. PKR is activated via dsRNA, which is produced during transcription of complementary strands of the CMV genome. Both m142 and m143 have dsRNA binding domains and may interfere with the binding of PKR with dsRNA, preventing its activation [50]. The HCMV encoded proteins TRS1 and IRS1 are capable of binding dsRNA and can substitute for the vaccinia virus (VV) RNA binding protein [51, 52]. However, the role of these proteins in the context of HMCV infection has not been evaluated [53].
Natural Killer Cells
Natural killer (NK) cells are cytotoxic cells of the innate immune response that play an important role in eliminating virus-infected cells early during infection. Signalling induced via activating receptors in absence of inhibitory receptor signalling regulates the cytotoxic activity of NK cells. Inhibitory receptors recognize specific MHC class I alleles whereas a number of ligands can bind activating receptors to trigger NK cell mediated killing. NK cells are important for controlling CMV infections both in mice and humans [54]. Therefore, it is not surprising the CMVs encode numerous proteins that interfere with NK cell activity [55].
Some HCMV encoded proteins alter expression of NK cell receptor ligands. The UL16 encoded glycoprotein, gpUL16, binds a number of ligands of the NK cell activating receptor, NKG2D. gpUL16 binds members of the UL16-binding protein (ULBP) and MIC family of proteins, which are stress induced NK ligands [56]. In particular, gpUL16 binds to ULBPs 1 and 2 [57, 58] and MICB [59]. gpUL16 downregulates these proteins, retaining them intracellularly in the ER or Golgi network [60–62]. The interaction of gpUL16 with these ligands blocks NK cell activation in vitro [56, 57, 63, 64] and may represent a way for HCMV to inhibit NK cell activation due to replication-induced cell stress.
Like gpUL16, the recently identified HCMV microRNA (miRNA), miR-UL112, also decreases expression of the NK ligand, MICB. miRNAs bind to 3' UTRs and prevent their translation. The miR-UL112 mediated decrease in MICB helps protect HCMV-infected cells from NK cell recognition and killing [65] and suggests that control of MICB expression and avoidance of NK recognition is important to the HCMV life cycle. As the field of CMV miRNA's begins to expand, an understanding of the role that miRNAs play in immune manipulation will also expand.
MCMV also encodes three proteins that downregulate ligands of the NK cell activating receptor, NKG2D. The m145 encoded protein decreases expression of MULT-1 [66], the m155 encoded glycoprotein (m155) downregulates H60 [67], and the m152 encoded glycoprotein (gp40) downregulates proteins of the RAE-1 family [68–70]. MCMV mutants lacking any one of these genes are attenuated in vivo and NK cell depletion restores MCMV growth. These results support a role for these proteins in inhibiting NK cell mediated clearance [66, 69, 71, 72]. Although the exact mechanism of action is not known for these proteins, both m155 and gp40 act at a post-transcriptional step and likely interfere with trafficking of NK ligands to the cell membrane [67, 69, 72].
MCMV encodes two homologues of MHC class I molecules, although neither has similarity to the HCMV homolog, UL18 [73, 74]. The MCMV gene m157 has low sequence homology to MHC class I molecules but structural analysis shows it has MHC-like folds [75]. m157 is expressed on infected cells and is tethered to the membrane with a glycosylphosphatidylinositol (GPI) anchor [76, 77]. m157 binds to Ly49H, an activating receptor of the Ly49 family of NK receptors [75, 78]. Interestingly, the m157-Ly49H interaction activates NK cells in vitro inducing NK cell cytotoxicity and cytokine and chemokine expression [79, 80]. In the structural paper exploring m157/Ly49H interactions, Adams et al showed that m157 activation of Ly49H is sufficient to override MHC I/NK cell inhibition [81]. In vivo, MCMV mutants lacking m157 are more virulent in mice due to decreased NK cell activity [82]. Furthermore, MCMV replicates to higher titers in mouse strains that lack expression of Ly49H receptors [83, 84], demonstrating that Ly49H activation is important for immune protection against MCMV. Although Ly49H protects against MCMV in laboratory mice, MCMV isolates from wild mice have m157 variants that fail to activate NK cells, thus providing evidence of evasion of NK recognition [85]. Mutations in m157 were also shown to accumulate following multiple passages of MCMV in mice indicating selection pressure for m157 variants that fail to activate NK cells. Furthermore, m157 also binds the inhibitory NK receptor, Ly49I, of some susceptible mice strains such as 129/J [75]. This has led to the speculation that CMVs evolved m157 to interact with NK inhibitory receptors but in some strains of mice this immune manipulation "backfires" and causes NK cell activation. These data suggest that m157 activation of NK may occur less frequently in natural infection of mice in the wild.
m144 is another MCMV encoded MHC class I homolog [74, 86]. m144 is expressed on the cell surface despite its inability to bind endogenous peptides like other MHC homologues [87]. m144 can inhibit NK cell cytolysis in vitro [88], suggesting it serves as a decoy receptor to inhibit NK cell activation. Consistent with this idea, MCMV mutants without m144 expression grow poorly in mice due to enhanced NK cell activation [89]. While a flexible region within m144 that could potentially interact with host receptors has been identified [90], m144 has not been shown to bind any cellular receptors and its mechanism of function remains unknown [88]. A rat CMV (RCMV) homolog of m144, r144, has also been identified [91]. Wild type RCMV shows enhanced replication in the salivary gland and spleen of neonatal rats when compared to an r144 deletion mutant [92]. However, what affect this protein has on NK cell activity is not currently known.
HCMV encodes several proteins that interact with and alter NK cell responses. One of the earliest identified proteins, gpUL18, is encoded by the gene UL18 and is a MHC class I homologue [93, 94]. gpUL18 binds the β2-microglobulin (β2m) and unlike the MHC homologue of MCMV, m144, gpUL18 also binds endogenous peptide [95, 96]. gpUL18 binds the NK cell inhibitory receptor LIR-1 with higher affinity than host MHC class I molecules [97–100]. However, the function of gpUL18 has not been clearly defined. gpUL18 expression both inhibits and activates NK cells in vitro [101–106]. For instance, gpUL18 activates NK cells from LIR-1- mice implicating a complex mechanism for NK cell modulation. Furthermore, clinical isolates of HCMV express gpUL18 variants with different LIR-1 binding affinities [107, 108] that may have differential functions. HCMV gpUL18-deletion mutants have shown both gpUL18 dependent and independent effects on NK cell activity in different cellular systems [101, 109]. In addition to its effect on NK cells, gpUL18 was recently shown to inhibit dendritic cell maturation and migration. While dendritic cells express LIR-1, whether this receptor mediates the effect of gpUL18 on dendritic cell activity was not determined [110]. Importantly, the HCMV encoded proteins that downregulate host MHC class I molecules do not interfere with the expression of gpUL18, which provides "protection" from NK cell lysis that occurs when MHC class I is downregulated [111, 112]. Collectively, current data suggests gpUL18 may impact the function of several cell types in vivo that may partially relate to its ability to bind and activate LIR-1 [113].
Recently, it was found that the UL142 gene encodes a second HCMV MHC class I homologue [114]. Cells expressing gpUL142 are protected from NK cell lysis. However, this effect was not evaluated in the context of HCMV infection [115]. While the mechanism of this effect has yet to be determined, gpUL142 was shown to downregulate MICA, a ligand for the activating NK cell receptor, NKG2D [116].
Interestingly, HCMV increases expression of the non-classical MHC class I molecule, HLA-E, while downregulating expression of many other MHC class I alleles [117]. This upregulation is due to the expression of UL40, a HCMV encoded protein that contains a peptide sequence identical to the HLA-E leader peptide [118–120]. Therefore, UL40 increases expression of HLA-E independent of TAP mediated peptide processing, which the HCMV encoded protein, gpUS6, inhibits during infection [121]. HLA-E is a ligand for the inhibitory receptor CD94/NKG2A and the UL40 induced upregulation of HLA-E would be expected to protect HCMV infected cells from NK cell lysis. However, conflicting reports have found UL40 inhibited or had no effect on NK cell lysis of HCMV infected cells [117, 122]. Additionally, the HLA-E restricted NK-CTLs recognize the UL40 peptide and kill virally infected cells [123, 124]. Thus, whether UL40 protects HCMV infected cells or targets them for destruction requires further evaluation.
The tegument protein, pp65, also interferes with NK cell function by binding the activating receptor NKp30. pp65 mediates the dissociation of CD3ξ, a signal transducing polypeptide, from NK cells impairing their activation [125]. Since pp65 is not secreted or expressed on infected cells, it is currently unclear how this protein mediates its effects.
Finally, the protein product of UL141, gp141, decreases the expression of CD155, a ligand for NK cell activating receptors by inhibiting CD155 transport to the cell surface [126]. This downregulation inhibits NK cell activity in vitro but further information regarding its function and impact in vivo remain to be answered.
Cytokine Homologues
Interleukin-10 (IL-10) is an immunosuppressive cytokine that downregulates inflammatory cytokine synthesis and interferes with antigen presentation by decreasing expression of MHC class II on antigen presenting cells [127]. Members of the poxvirus and herpesvirus families, including HCMV, encode IL-10 homologues [128]. In contrast to other viral IL-10 homologues, such as those encoded by EBV and orf virus, a poxvirus, both of which have high amino acid similarity (80%) to their host IL-10. The HCMV IL-10 protein, cmvIL-10, has only limited homology (27%) to human IL-10 (hIL-10) [129]. cmvIL-10 binds the IL-10 receptor, hIL-10R, albeit with lower affinity than hIL-10 [129, 130]. Nevertheless, it retains the capacity to induce a strong anti-inflammatory response. cmvIL-10 downregulates expression of IFNγ and TNFα in peripheral blood mononuclear cells (PBMCs) and decreases expression of both MHC class I and II on PBMCs and DCs [130–133]. DCs exposed to cmvIL-10 inefficiently stimulated T cell proliferation [132] supporting its role in immune suppression. Furthermore, cmvIL-10 inhibits cytokine production due to the activation of the phosphotidylinositol 3-kinase pathway [134]. Interestingly, cmvIL-10 was recently shown to stimulate B cell proliferation but the impact of this on the immune response to CMV is unclear [135]. cmvIL-10 alters the function of non-leukocyte populations as well. By interfering with placental cytotrophoblast invasion, cmvIL-10 could affect placental development, which contributes to the sequelae observed following congenital CMV infections [136].
An alternatively spliced version of cmvIL-10 was originally identified during latent HCMV infection and termed latency associated cmvIL-10 (LacmvIL-10) [137]. However, this transcript is also expressed during productive infection [138]. LacmvIL-10 fails to induce signalling pathways associated with IL-10R activation, which implies LacmvIL-10 may not bind to IL-10R or, at least, differentially activates this receptor. LacmvIL-10 also decreases MHC class II expression on granulocyte-macrophage progenitors (GM-Ps) and monocytes, both sites of CMV latency, possibly limiting clearance of latently infected cells [139].
A number of primate CMVs encode proteins similar to IL-10 [140] but only the function of the rhesus CMV (RhCMV) homologue has been evaluated to date. RhcmvIL-10 downregulates cytokine expression in PBMCs and MHC class II molecule expression in monocytes [130]. Therefore, the function of human cmvIL-10 is conserved in RhCMV. Interestingly, chimpanzee cytomegalovirus (CCMV) and MCMV do not encode IL-10 homologues [140]. MCMV infection induces cellular IL-10 expression providing a mechanism for immune interference in the absence of a virally encoded IL-10 protein [141, 142]. Therefore, IL-10 immune suppression, whether cellular or viral in origin, likely creates an environment that supports efficient viral replication in the host.
Cytokine Receptor Homologues
The HCMV gene, UL144, encodes a protein with limited homology to the herpes simplex virus entry mediator (HVEM), a member of the tumour necrosis factor receptor (TNFR) superfamily [143]. UL144 is the only TNFR homolog identified in herpesviruses. The UL144 protein does not bind any known TNF ligands [143–145]. UL144 binds to a member of the Ig superfamily, B and T lymphocyte attenuator (BTLA) [146], also a ligand of HVEM [147]. Binding of UL144 to BTLA blocks T cell proliferation and could impair lymphocyte responses to HCMV [146]. UL144 activates TNFR-activated factor (TRAF6) leading to NFκB activation and upregulation of the chemokine CCL22 [145, 148]. CCL22 is a chemoattractant of Th2 and regulatory T cells. Activation and attraction of these cells may help HMCV evade T cell-mediated antiviral activity. UL144 is located in the UL/b' region of HCMV, a portion of the genome thought to encode potential virulence factors. Different UL144 genotypes have been identified in clinical isolates. However, all but one report has found no association between the different genotypes and CMV disease [144, 149–155]. Although there is much speculation on the role that UL144 plays in immune modulation, its role in CMV pathogenesis and survival is still unclear.
Viral Chemokine Homologues
Many CMVs also encode chemokine homologues. Chemokines are small, chemotactic cytokines that are important for leukocyte trafficking and activation. The best characterized of the CMV chemokines is the CC chemokine homolog of MCMV. Originally identified as the m131 gene product, MCK1, transcriptional analysis during MCMV infection determined that a spliced product of m131 and m129, referred to as MCK2 was the only transcript encoded from this locus. The CC chemokine domain is confined to the MCK1 coding region and is connected to a long carboxyl-terminal domain (199 amino acids) with no known homology to other proteins. This makes MCK2 considerably longer than other chemokines [156, 157]. In contrast to UL146 (see below), the DNA sequence of MCK2 is highly conserved in isolates from wild mice [158].
Initial studies in vitro showed MCK1 induced higher levels of Ca2+ mobilization in peritoneal exudates cells from MCMV infected mice than cells from uninfected mice. This demonstrated that MCK1 could activate cells recruited to the site of MCMV infection [159]. The spliced product, MCK2, is important for viral spread within the host. When a recombinant MCMV with mutations in the m131 gene (generated either via point mutations or insertions) was inoculated into mice, these recombinants showed a defect in dissemination to the salivary gland. Mice infected with these mutants developed less inflammation at the site of inoculation, reduced secondary viremia, as well as lower viral titers in the salivary glands, a site of dissemination following secondary viremia [160, 161]. This was the first direct evidence that a CMV encoded chemokine is important for the dissemination of the virus in its host. Additional work identified the MCK2-recruited cell type as a late myeloid progenitor, consistent with reports that CMV infects and can remain latent in cells of the myelomonocytic lineage [162].
Rat CMV (RCMV) encodes a CC chemokine with similarity to both m131 and m129, having N-terminal chemokine homology and a long carboxyl-terminus like MCK2 [163]. Rats infected with RCMV deletion mutants lacking pr131 expression, the product of the r131 locus, had reduced viral loads in the spleen and salivary glands. Additionally these mice had reduced swelling and macrophage infiltration at the site of virus inoculation [164]. Therefore, pr131 appears to be a functional homolog of MCK2.
Guinea pig CMV (GPCMV) contains a gene with homology to CC chemokines, though it lacks positional or sequence similarity to the m131/m129 genes of MCMV. GPCMV-MIP is most similar to the cellular chemokines, CCL3/CCL4 (MIP-1α/β) and CCL14 (HCC-1). GPCMV-MIP induced Ca2+ flux and migration of cells expressing hCCR-1 but did not elicit a response from cells expressing other CC receptors [165]. CCL14 is known to increase the proliferation of monocyte progenitors, therefore GPCMV-MIP may enhance the proliferation and/or recruitment of permissive cells consistent with the role of MCK2 [166].
HCMV encodes two ORFs with homology to CXC chemokines, UL146 and UL147 [167]. While no functional data is available for the protein product of UL147, vCXCL-2 (also called pUL147 and vCXC-2), analysis of the protein product of UL146, vCXCL-1 (also denoted pUL146 and vCXC-1), demonstrated its capacity as a functional chemokine. vCXCL-1 was shown to bind exclusively to hCXCR2 with an affinity similar to that of the host chemokine, CXCL8. Additionally, vCXCL-1 was able to induce chemotaxis and intracellular calcium release in human neutrophils, again to levels comparable to host chemokines [168]. A mutant HCMV virus with a deletion of UL146-UL147 was unable to infect neutrophils but retained its ability to infect other cells providing additional evidence these proteins facilitate an interaction of HCMV with neutrophils [169].
Chimpanzee CMV (CCMV) encodes homologues of UL146 and UL147 as well as the related gene UL146a. However, only the product of UL146 (vCXCL-1CCMV) has been evaluated thus far. vCXCL-1CCMV induces chemotaxis and calcium release in human neutrophils. Additionally, it was shown to increase expression of adhesion molecules on the surface of neutrophils and reduce apoptosis in these cells [170]. Taken together, the function of the HCMV and CCMV vCXCL-1s in vitro suggest the potential for these viral chemokines to alter the response of neutrophils in the course of CMV infection.
Due to the strict species specificity of CMVs, in vivo characterization of UL146 and UL147 has been limited to sequence analysis of clinical HCMV isolates. UL146 is highly variable, differing as much as 60% among isolates at the amino acid level [152]. It was postulated that this variability might correlate with the severity of CMV disease however no clear relationship has been identified [151, 171–173]. It was recently suggested the variability of UL146 may have arisen in early human populations and likely does not contribute to the pathogenesis of congenital infection [173]. Our lab is currently working to identify whether the vCXCL-1 isolates from different clinical strains induce functional differences in neutrophils.
HCMV also encodes genes with limited homology to CC chemokines. UL128 and UL130 both have signal sequences and conserved cysteine motifs similar to CC chemokines [174], but it is yet to be determined whether these proteins actually function as chemokines. Interestingly, the UL128-131 locus was found to be necessary for endothelial cell and leukocyte tropism based on the inability of HCMV UL128-131 deletion mutants to infect these cell types [169]. This likely explains the loss of endothelial cell tropism in lab-adapted strains of HCMV, a number of which were shown to have deletions or mutations in at least one gene from this region [174]. The UL128-131 proteins have recently been shown to interact with gH/gL complexes that mediate entry of CMV into endothelial and epithelial cells, in contrast to the gH/gL complexes that mediate virus entry into fibroblasts [175, 176].
Chemokine Receptor Homologues
In addition to viral chemokine homologues, CMVs encode proteins with homology to chemokine receptors [177, 178]. Chemokine receptors are members of the G-protein coupled receptor (GPCR) family [179]. The four chemokine receptor homologues encoded by the cytomegaloviruses have homology to the CC chemokine receptor family. Two genes, UL33 and UL78, are conserved among all sequenced CMVs, while primate CMVs encode two additional GPCRs, US27 and US28 [180].
The HCMV US28 encoded protein (pUS28) is the best characterized of the GPCR homologues. US28 has the highest sequence homology to the cellular receptor, CCR1, and binds a number of CC chemokines including CCL5 (RANTES), CCL2 (MCP-1), CCL3 (MIP-1α), and CCL4 (MIP-1β) [181, 182]. Interestingly, pUS28 binds with highest affinity to the CX3C chemokine, CX3CL1 (fractalkine) [183]. Upon binding, pUS28 induces chemokine internalization, removing chemokines from the extracellular environment [184–186]. Consistent with this role, pUS28 inhibits monocyte migration in vitro and media from US28 expressing, CMV-infected monolayers deplete CCL5 and CCL2 from the media [187]. Constitutive endocytosis and recycling of pUS28 contributes to chemokine internalization however chemokine binding is not sufficient to induce constitutive endocytosis of pUS28 [188]. By acting as a "chemokine sink", pUS28 could alter or inhibit chemokine-dependent immune responses. The related chemokine homolog, pUS27, also shows some ability to internalize chemokines [185, 189], perhaps illustrating a conservation of function between the viral GPCRs.
pUS28 elicits ligand-dependent signalling, triggering calcium mobilization upon CCL5, CCL2, CCL3 and CCL7 binding. pUS28 utilizes the G proteins Gαi and Gα16 to mediate calcium mobilization upon binding of CCL5 or CCL7 [181, 190, 191]. Interestingly, Gα16 expression is restricted to hematopoietic cells [192]. HCMV has a tropism for this cell lineage where pUS28 may alter Gα16 signalling to benefit CMV's life cycle. pUS28 also induces agonist-dependent migration of smooth muscle cells (SMCs). Migration of SMCs in response to pUS28 requires the protein tyrosine kinase (PTK) pathway, indicating pUS28 activates a number of cellular pathways in vitro [193, 194]. As with many CMV proteins, the effects of pUS28 vary depending on the system and cell types analyzed.
Like the Kaposi's sarcoma-associated herpesvirus (KSHV) GPCR homolog, ORF74 [195], pUS28 can constitutively activate several signalling pathways, including phospholipase C (PLC), NFκB, NFAT, and cAMP-responsive element (CRE)-dependent pathways [188, 191, 195–198]. CC chemokines do not enhance signalling, although CX3CL1 acts as a partial inverse agonist, decreasing the levels of pUS28 constitutive activity [191, 196]. The constitutive endocytosis of pUS28 occurs independently of its constitutive signalling and different domains of pUS28 control these two phenomena [188]. pUS28, like KSHV ORF74, can induce transformation of NIH3T3 cells and promote tumour formation in nude mice. Mice inoculated with cells expressing a mutated pUS28 that lacks constitutive activity show attenuated tumour formation demonstrating the importance of constitutive signalling in pUS28-mediated tumour formation [199].
Despite extensive research on the function of pUS28, its role in CMV pathogenesis in vivo is still unknown. pUS28 sequesters host chemokines in order to alter the immune response or activates signalling pathways that contribute to viral replication or spread. pUS28-induced SMC migration may play a role in the accelerated vascular disease associated with HCMV [200, 201]. Although HCMV nucleic acids have been isolated from certain cancers [202, 203], whether HCMV is causal in these cancers remains to be established. Perhaps pUS28 may contribute to tumour formation in these individuals.
The UL33 gene is conserved in all β herpesviruses, including HCMV (UL33), RCMV (R33), and MCMV (M33). While no ligands have been identified for either pUL33 or pR33, pM33 is activated by CCL5 [204]. All three viral proteins show constitutive activity, although they differentially activate specific signalling pathways [196, 205–207]. While not essential for viral replication in vitro, both pR33 and pM33 are important for replication of CMV in the salivary gland [208–210]. In MCMV, this defect was linked to the constitutive activity of pM33 [211] and illustrates the importance of constitutive signalling in vivo. Like pUS28, pM33 and pR33 induce SMC migration [204, 212]. Taken together, this suggests GPCRs of the UL33 family alter cellular trafficking during CMV infection, which may contribute to viral pathogenesis.
The UL78 gene family encoded proteins have only limited homology to known chemokine receptors [180]. Nevertheless, this gene is conserved in all β herpesviruses suggesting they have some role in the herpesviruses life cycle. The UL78 proteins of MCMV and RCMV, pM78 and pR78, respectively, are important for viral replication in vitro and in vivo [213–215]. The HCMV homolog, pUL78, is not required for replication of HCMV in vitro [216] thus MCMV and RCMV may be more dependent on its function for viral replication. Although, members of the UL78 family are clearly important for viral replication for some CMVs, data regarding their specific function as GPCRs is not currently available.
Viral Chemokine Binding Proteins
Chemokine binding proteins (CBPs) are virally encoded secreted proteins that competitively bind chemokines and interfere with their interactions with cellular receptors. Unlike viral chemokine and chemokine receptor homologues, which were likely acquired from the host, CBPs generally show no homology to known proteins and homology of these proteins across viruses is not conserved [217]. Until recently, no CMV species was known to encode a CBP. However, a HCMV encoded protein, p21.5, has been identified with chemokine binding properties. mRNA transcripts of the gene UL21.5 are packaged in the HCMV virion and thus may function soon after viral entry into the cell. Although many CBPs interact with multiple chemokines [218], p21.5 is unusual in that it selectively binds CCL5 (RANTES) in vitro blocking the interaction of the chemokine with its cellular receptors [219]. No data is available which evaluates the ability of p21.5 to interfere with CCL5 function in vivo and its effect on viral survival.
Apoptosis
Apoptosis is a mechanism for programmed cell death that plays an important role in the elimination of cells during development and virally infected cells as part of the host's innate immunity [220, 221]. Many viruses including CMVs have strategies to prevent apoptosis [222, 223]. HCMV encodes two proteins in particular that directly interfere with the apoptosis pathway. The HCMV UL36 gene encodes a viral inhibitor of caspase-8 induced apoptosis (vICA) and exon 1 of UL37 encodes the viral mitochondrial inhibitor of apoptosis (vMIA) [224].
vMIA protects cells from intrinsic apoptosis induced following damage to the mitochondrial membrane [225–227]. vMIA only blocks apoptosis mediated through the death receptor, Fas, in cells in which apoptosis proceeds through a mitochondrial-dependent step [228]. vMIA has not been identified in virion particles and inhibits apoptosis at later times compared to vICA [225, 229]. vMIA localizes to the mitochondria [230–232] and this localization is needed for vMIA to induce structural changes and inhibit mitochondrial release of cytochrome c, blocking downstream events in the apoptosis pathway [227, 233]. vMIA also induces the release of calcium from the ER which may play a role in the inhibition of apoptosis [234].
vMIA was initially shown to interact with adenine nucleotide translocator (ANT), a regulator of mitochondrial membrane permeability [227, 235]. However, this interaction does not correlate with vMIA function and is likely non-specific [235]. vMIA also binds the proapoptotic protein, Bax, inducing aggregation of Bax molecules at the mitochondrial membrane preventing membrane permeabilization [236, 237]. The screening of proteins that interact with the antiapoptotic domain of vMIA identified the protein, Growth Arrest and DNA Damage 45 (GADD45α). GADD45α enhances vMIA-induced apoptosis possibly protecting it from proteasomal degradation [238]. Interestingly, vMIA was also recently shown to interfere with caspase-independent cell death by blocking the activity of the serine protease, HtrA2/Omi [239]. Sequence homologues of vMIA have only been found in primate CMVs [240]. However, a recently identified positional homologue of vMIA in MCMV, m38.5, was shown to interact with Bax and inhibit apoptosis [226, 241, 242]. Therefore, vMIA functional homologues may exist in other cytomegaloviruses.
In contrast to vMIA, vICA protects cells in vitro from extrinsic apoptosis mediated through ligation of the death receptors, Fas and TNFR-1 [228]. vICA is a virion constituent [243] that inhibits caspase-8 activation [228]. Procaspase-8 is prevented from interacting with the adapter protein, Fas-associated death domain (FADD) and inhibits the processing of procaspase-8 to its active form [244]. Therefore, vICA is functionally similar, although mechanistically distinct from, cellular and viral FLICE inhibitory proteins (FLIPS), which also interfere with caspase-8 activation [245].
vICA is dispensable for replication and some HCMV lab strains have inactivating mutations in the UL36 gene [228, 243, 246]. Nevertheless, vICA homologues are conserved in the genomes of all sequenced β-herpesviruses except for guinea pig CMV (GPCMV) where no positional homologues has been identified [247]. The vICA homologues of RhCMV and MCMV, Rh36 and M36, respectively, also have antiapoptotic activities when transiently expressed in vitro [240, 248]. Recently, it was shown that a M36 deletion mutant was attenuated in the lungs and salivary glands of mice but could be rescued by expression of a dominant-negative variant of FADD [249]. This provides important evidence for the functional significance of this protein in vivo.
The MCMV M45 encoded protein has sequence, although not functional, homology to a cellular ribonucleotide reductase [250, 251]. Nevertheless, a M45 mutant virus is attenuated in SCID mice [251]. Recently M45 was shown to suppress the cell death pathway in a manner unique to viral proteins [252]. M45 interacts with the receptor-interacting protein kinase I (RIPI) via a RIP homotypic interaction motif (RHIM). By binding to RIPI, M45 protects certain cell types from caspase-independent cell death following death receptor signalling [253, 254]. This "alternative apoptosis" would likely be activated in HCMV infected cells in which vICA inhibits caspase-8 activity [255].
Adaptive Immune Response
T Cell Mediated Immunity
T cell mediated immune responses, particularly CD8+ T cell dependent responses, are integral to the clearance of cytomegalovirus infections [54]. Cytomegaloviruses, as do many herpesviruses, impair T cell activation by interfering with both MHC class I and II antigen processing and presentation [256]. Although this has been shown in vitro, Th or CTL responses are still generated during CMV infection in vivo [257–260].
The US region of the HCMV genome encodes a number of endoplasmic reticulum (ER) resident glycoproteins that alter MHC class I expression [261]. The first of these proteins to be expressed following HCMV infection is the US3 encoded immediate early protein, gpUS3, which likely interferes with T cell recognition early during viral replication [262–264]. gpUS3 binds MHC class I molecules and retains them in the ER, inhibiting antigen presentation to CD8+ T cells [265–269]. The transmembrane domains of gpUS3 are responsible for binding MHC class I [270, 271]. However, luminal regions of gpUS3 are needed for retention of MHC class I in the ER [270–273]. Interestingly, gpUS3 also binds tapasin and this interaction prevents tapasin-mediated protein loading of MHC class I molecules. Therefore, gpUS3 only retains MHC class I alleles that are tapasin dependent [274]. An alternatively spiced form of gpUS3 competitively binds tapasin and may represent a regulatory mechanism for gpUS3 activity [275].
In contrast to gpUS3, the binding of MHC class I molecules to the US2 and US11 encoded HCMV proteins, gpUS2 and gpUS11, leads to the rapid degradation of MHC class I molecules [276–279]. Both proteins bind MHC class I molecules resulting in their removal from the ER to the cytosol where they undergo proteasome-dependent degradation [266, 276, 277]. This process requires a functional ubiquitination system although only gpUS2 specifically depends on MHC class I ubiquitination for protein degradation [280–283]. In addition to the ubiquitin system, gpUS2 and gpUS11 associate with other cellular proteins that facilitate recognition and removal of proteins via the endoplasmic reticulum associated protein degradation (ERAD) pathway [284–287]. gpUS2 forms a complex with the chaperones, calnexin, BiP, and calreticulin to mediate MHC class I degradation [288]. In contrast, gpUS11-dependent protein degradation requires Derlin-1, a protein that plays a role in the removal of misfolded proteins [289].
Why HCMV encodes two different proteins that target MHC class I for degradation is unclear. gpUS2 and gpUS11 differ in their specificities for MHC class I alleles [290, 291]. While they may overlap in function, they likely mediate distinct effects on MHC class I expression [292, 293]. This difference in specificity is likely the result of different binding requirements for gpUS2 and gpUS11. The luminal portion of gpUS2 interacts with residues in the α2/α3 region of the luminal domain of MHC class I molecules [294–296]. Specifically, gpUS2-mediated degradation requires the presence of an arginine at residue 181 of MHC class I molecules. However, this residue is not sufficient for degradation of some MHC class I alleles and other residues must be important for these molecules [297]. The luminal region of gpUS11 is also important for MHC class I binding, however, gpUS11 interacts with residues in the α1/α2 domain of MHC class I molecules [298, 299].
The HCMV US6 gene, encodes another glycoprotein, gpUS6, that downregulates MHC class I expression [121, 300, 301]. Like the other US proteins that interfere with MHC class I expression, gpUS6 mediates retention of MHC class I via its luminal domain [302]. However, the gpUS6 mechanism is unique in that it binds to TAP-1 and TAP-2 heterodimers [303] in the transporter associated with antigen processing (TAP) complex [121, 300, 304, 305]. Through its association with TAP, gpUS6 prevents the binding of ATP to TAP [306] and subsequent TAP peptide translocation [300, 301]. gpUS6 expression is solely responsible for the inhibition of TAP in HCMV infected cells [307] and downregulates all MHC class I alleles tested [308]. In addition to impairing CD8+ T cell responses, this decrease in MHC class I expression also makes infected cells susceptible to NK cell cytotoxicity [308], which may explain why CMVs have evolved mechanisms for preventing NK lysis (NK cell section).
Two additional HCMV encoded proteins, gpUS8 and gpUS10, bind MHC class I molecules. However, neither protein downregulates cell surface MHC class I expression, although gpUS10 slows MHC class I maturation and egress from the ER. The function of these proteins in HCMV infection is currently unknown [309, 310].
Other cytomegaloviruses have homologues of some of the HCMV US family members. CCMV encodes a gpUS6 homologue that binds TAP. However, this interaction does not downregulate MHC class I expression in chimpanzee cells in vitro, making the function of this homologue unclear [305]. In contrast, RhCMV encodes homologues of gpUS2, gpUS3, gpUS6, and gpUS11 [311], each of which functions similarly to its HCMV counterpart [312]. A newly identified RhCMV protein, viral inhibitor of heavy chain expression (VIHCE), is encoded by the rh178 gene, a gene unrelated to the US6 gene family. VIHCE inhibits signal-peptide dependent MHC class I heavy chain translation, a mechanism distinct from other modulators of MHC class I expression [313]. Therefore, RhCMV may be a valuable model system for analyzing the function of these proteins during CMV infection in vivo.
MCMV encodes three proteins unrelated to those of HCMV that interfere with MHC class I expression in infected cells. m6, m152, and m4, encode gp48, gp40, and gp34 respectively. Both gp40 and gp48 inhibit antigen presentation to CD8+ T cells in vitro [314, 315]. gp40 and gp48 bind MHC class I molecules and retain them intracellularly [314–316], although each protein does so via different mechanisms. gp40 retains MHC class I molecules in the ER-Golgi intermediate compartment (ERGIC)-cis golgi network [314] whereas gp48 binds to MHC class I molecules and targets them to lysosomes for degradation [315]. Deletion of m152 restores MHC class I expression in MCMV infected cells suggesting gp40 is the main regulator of MHC class I downregulation [317, 318]. However, gp48 cooperates with gp40 to enhance MHC class I retention in vitro [319]. gp48 and gp40 also show different specificities for MHC class I alleles and thus are both required for efficient MHC class I downregulation in certain cell types [320, 321]. Surprisingly, the presence of m152 does not impact the CTL response in mice based on studies using recombinant MCMVs. Therefore, the in vivo role of m152 as well as m4 and m6 is not clear [322–324]. One possible explanation for this lack of CTL alteration in vivo is uninfected antigen presenting cells (APCs) cross-priming CD8+ T cells, which could circumvent function of gp40 in mice [324]. If these proteins cannot prevent priming of CD8+ T cells, what is their function? There are a couple of possibilities. One is that the delay in CD8+ T cell recognition is enough to allow the virus to establish a foothold within the host and eventually establish latency. Also without this initial CD8+ T cell recognition, immunopathology could be diminished allowing the host to survive and viral spread. Recently the Reddehase group has shown that the presence of these immune modulating proteins (gp40, and gp48) actually enhance priming of CD8+ T cells [325]. How this benefits CMV survival in vivo is hard to reconcile, but may represent the evolution of mouse immune responses to counter these "immunevasions." The Reddehase and Hill labs are both actively persuing answers to these questions and why CMVs would have evolved mechanisms to enhance CTL priming.
The MCMV protein gp34 also associates with MHC class I molecules [326, 327]. In contrast to gp40 and gp48, gp34 decreases the intracellular retention of MHC class I molecules [319, 326]. However, this phenomenon is seen only in the absence of m6 (gp48) suggesting gp48 expression may antagonize the function of gp34 and provide a mechanism to regulate the extent of MHC class I expression in infected cells [317, 319].
HCMV can also interfere with the presentation of MHC class II on antigen presenting cells such as macrophages [328]. gpUS3, gpUS2, and pp65 mediate this effect. gpUS3 binds and downregulates MHC class II, which subsequently decreases antigen presentation to CD4+ T cells. gpUS3 interferes with the sorting of MHC class II to lysosomes inhibiting peptide loading in these compartments and subsequent MHC class II expression at the cell surface [269]. gpUS2 also interferes with CD4+ T cell recognition by downregulating MHC class II molecules. Similar to its effect on MHC class I, gpUS2 binds the α chain of MHC class II resulting in the proteasome-dependent degradation of MHC class II molecules in a number of different cell types in vitro [269, 329–331]. Lastly, the tegument protein, pp65, also downregulates MHC class II expression. Yet another mechanism for MHC class II down regulation, pp65 mediates MHC class II trafficking to lysosomes causing their destruction [332]. MCMV infection also downregulates MHC class II expression although the viral proteins responsible for this are not currently identified [141, 333]. As observed with MHC class I downregulation, CMVs have evolved multiple modes for achieving manipulation of MHC class II host responses in order to achieve evolutionary success.
Conclusion
Whether it is the destruction of important immune molecules, a redirection of these proteins intracellularly, transcriptional or translational control, alteration of signal transduction cascades, or the production of novel interfering proteins, CMVs "push" and "pull" at the immune response in such a way as to ensure their evolutionary success. Based on the numerous viral proteins that respond to the host's defence mechanisms, the relationship of CMVs with their hosts is complex. In spite of the manipulation of both the magnitude and quality of the innate and adaptive immune responses, the host still launches a robust anti-CMV immune response but not before the virus establishes latency within the host. Therefore CMV infection does not eliminate host immunity but modulates it to allow survival of both the virus and the host, as both are important to the life cycle of the virus. With its millions of years of co evolution within us, we can use CMVs acquired "knowledge" of the immune system to uncover novel immune pathways. We can potentially exploit CMVs potential vulnerabilities for developing CMV vaccines (i.e. ones that can not establish latency) or novel therapeutics that could minimize CMV-induced damage in immune compromised hosts.
Authors' Information
MMK recently received her PhD with a dissertation exploring CMV viral chemokines from the University of Tennessee. TES is an assistant professor at the University of Tennessee whose lab focuses on CMV immune modulation.
References
Tortorella D, Gewurz BE, Furman MH, Schust DJ, Ploegh HL: Viral subversion of the immune system. Annu Rev Immunol 2000, 18: 861-926.
Hirsch RL: The complement system: its importance in the host response to viral infection. Microbiol Rev 1982, 46: 71-85.
Spiller OB, Hanna SM, Devine DV, Tufaro F: Neutralization of cytomegalovirus virions: the role of complement. J Infect Dis 1997, 176: 339-347.
Spear GT, Lurain NS, Parker CJ, Ghassemi M, Payne GH, Saifuddin M: Host cell-derived complement control proteins CD55 and CD59 are incorporated into the virions of two unrelated enveloped viruses. Human T cell leukemia/lymphoma virus type I (HTLV-I) and human cytomegalovirus (HCMV). J Immunol 1995, 155: 4376-4381.
Spiller OB, Morgan BP, Tufaro F, Devine DV: Altered expression of host-encoded complement regulators on human cytomegalovirus-infected cells. Eur J Immunol 1996, 26: 1532-1538.
Nomura M, Kurita-Taniguchi M, Kondo K, Inoue N, Matsumoto M, Yamanishi K, Okabe M, Seya T: Mechanism of host cell protection from complement in murine cytomegalovirus (CMV) infection: identification of a CMV-responsive element in the CD46 promoter region. Eur J Immunol 2002, 32: 2954-2964.
Holers VM, Kinoshita T, Molina H: The evolution of mouse and human complement C3-binding proteins: divergence of form but conservation of function. Immunol Today 1992, 13: 231-236.
Pass RF: Cytomegaloviruses. In Fields Virology. 4th edition. Edited by: David MK, Peter M Howley. Philadelphia: Lippincott Williams and Williams; 2001:2675-2703.
Westmoreland D, St Jeor S, Rapp F: The development by cytomegalovirus-infected cells of binding affinity for normal human immunoglobulin. J Immunol 1976, 116: 1566-1570.
Keller R, Peitchel R, Goldman JN, Goldman M: An IgG-Fc receptor induced in cytomegalovirus-infected human fibroblasts. J Immunol 1976, 116: 772-777.
Rahman AA, Teschner M, Sethi KK, Brandis H: Appearance of IgG (Fc) receptor(s) on cultured human fibroblasts infected with human cytomegalovirus. J Immunol 1976, 117: 253-258.
Frey J, Einsfelder B: Induction of surface IgG receptors in cytomegalovirus-infected human fibroblasts. Eur J Biochem 1984, 138: 213-216.
Furukawa T, Hornberger E, Sakuma S, Plotkin SA: Demonstration of immunoglobulin G receptors induced by human cytomegalovirus. J Clin Microbiol 1975, 2: 332-336.
Xu B, Murayama T, Ishida K, Furukawa T: Characterization of IgG Fc receptors induced by human cytomegalovirus. J Gen Virol 1989,70(Pt 4):893-900.
Murayama T, Natsuume-Sakai S, Shimokawa K, Furukawa T: Fc receptor(s) induced by human cytomegalovirus bind differentially with human immunoglobulin G subclasses. J Gen Virol 1986,67(Pt 7):1475-1478.
Thale R, Lucin P, Schneider K, Eggers M, Koszinowski UH: Identification and expression of a murine cytomegalovirus early gene coding for an Fc receptor. J Virol 1994, 68: 7757-7765.
Ravetch JV, Bolland S: IgG Fc receptors. Annu Rev Immunol 2001, 19: 275-290.
Crnkovic-Mertens I, Messerle M, Milotic I, Szepan U, Kucic N, Krmpotic A, Jonjic S, Koszinowski UH: Virus attenuation after deletion of the cytomegalovirus Fc receptor gene is not due to antibody control. J Virol 1998, 72: 1377-1382.
Jonjic S, Pavic I, Polic B, Crnkovic I, Lucin P, Koszinowski UH: Antibodies are not essential for the resolution of primary cytomegalovirus infection but limit dissemination of recurrent virus. J Exp Med 1994, 179: 1713-1717.
Lenac T, Budt M, Arapovic J, Hasan M, Zimmermann A, Simic H, Krmpotic A, Messerle M, Ruzsics Z, Koszinowski UH, Hengel H, Jonjic S: The herpesviral Fc receptor fcr-1 down-regulates the NKG2D ligands MULT-1 and H60. J Exp Med 2006, 203: 1843-1850.
Mintern JD, Klemm EJ, Wagner M, Paquet ME, Napier MD, Kim YM, Koszinowski UH, Ploegh HL: Viral interference with B7-1 costimulation: a new role for murine cytomegalovirus fc receptor-1. J Immunol 2006, 177: 8422-8431.
Atalay R, Zimmermann A, Wagner M, Borst E, Benz C, Messerle M, Hengel H: Identification and expression of human cytomegalovirus transcription units coding for two distinct Fcgamma receptor homologs. J Virol 2002, 76: 8596-8608.
Lilley BN, Ploegh HL, Tirabassi RS: Human cytomegalovirus open reading frame TRL11/IRL11 encodes an immunoglobulin G Fc-binding protein. J Virol 2001, 75: 11218-11221.
Dubin G, Basu S, Mallory DL, Basu M, Tal-Singer R, Friedman HM: Characterization of domains of herpes simplex virus type 1 glycoprotein E involved in Fc binding activity for immunoglobulin G aggregates. J Virol 1994, 68: 2478-2485.
Dowler KW, Veltri RW: In vitro neutralization of HSV-2: inhibition by binding of normal IgG and purified Fc to virion Fc receptor (FcR). J Med Virol 1984, 13: 251-259.
Sprague ER, Reinhard H, Cheung EJ, Farley AH, Trujillo RD, Hengel H, Bjorkman PJ: The human cytomegalovirus Fc receptor gp68 binds the Fc CH2-CH3 interface of immunoglobulin G. J Virol 2008, 82: 3490-3499.
Boyle KA, Pietropaolo RL, Compton T: Engagement of the cellular receptor for glycoprotein B of human cytomegalovirus activates the interferon-responsive pathway. Mol Cell Biol 1999, 19: 3607-3613.
Zhu H, Cong JP, Shenk T: Use of differential display analysis to assess the effect of human cytomegalovirus infection on the accumulation of cellular RNAs: induction of interferon-responsive RNAs. Proc Natl Acad Sci USA 1997, 94: 13985-13990.
Simmen KA, Singh J, Luukkonen BG, Lopper M, Bittner A, Miller NE, Jackson MR, Compton T, Fruh K: Global modulation of cellular transcription by human cytomegalovirus is initiated by viral glycoprotein B. Proc Natl Acad Sci USA 2001, 98: 7140-7145.
Browne EP, Wing B, Coleman D, Shenk T: Altered cellular mRNA levels in human cytomegalovirus-infected fibroblasts: viral block to the accumulation of antiviral mRNAs. J Virol 2001, 75: 12319-12330.
Zhu H, Cong JP, Mamtora G, Gingeras T, Shenk T: Cellular gene expression altered by human cytomegalovirus: global monitoring with oligonucleotide arrays. Proc Natl Acad Sci USA 1998, 95: 14470-14475.
Navarro L, Mowen K, Rodems S, Weaver B, Reich N, Spector D, David M: Cytomegalovirus activates interferon immediate-early response gene expression and an interferon regulatory factor 3-containing interferon-stimulated response element-binding complex. Mol Cell Biol 1998, 18: 3796-3802.
Miller DM, Zhang Y, Rahill BM, Waldman WJ, Sedmak DD: Human cytomegalovirus inhibits IFN-alpha-stimulated antiviral and immunoregulatory responses by blocking multiple levels of IFN-alpha signal transduction. J Immunol 1999, 162: 6107-6113.
Powers C, DeFilippis V, Malouli D, Fruh K: Cytomegalovirus immune evasion. Curr Top Microbiol Immunol 2008, 325: 333-359.
DeFilippis V, Fruh K: Rhesus cytomegalovirus particles prevent activation of interferon regulatory factor 3. J Virol 2005, 79: 6419-6431.
Roy CR, Mocarski ES: Pathogen subversion of cell-intrinsic innate immunity. Nat Immunol 2007, 8: 1179-1187.
Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M: Functional role of type I and type II interferons in antiviral defense. Science 1994, 264: 1918-1921.
Paulus C, Krauss S, Nevels M: A human cytomegalovirus antagonist of type I IFN-dependent signal transducer and activator of transcription signaling. Proc Natl Acad Sci USA 2006, 103: 3840-3845.
Huh YH, Kim YE, Kim ET, Park JJ, Song MJ, Zhu H, Hayward GS, Ahn JH: Binding STAT2 by the acidic domain of human cytomegalovirus IE1 promotes viral growth and is negatively regulated by SUMO. J Virol 2008, 82: 10444-10454.
Taylor RT, Bresnahan WA: Human cytomegalovirus immediate-early 2 gene expression blocks virus-induced beta interferon production. J Virol 2005, 79: 3873-3877.
Taylor RT, Bresnahan WA: Human cytomegalovirus IE86 attenuates virus- and tumor necrosis factor alpha-induced NFkappaB-dependent gene expression. J Virol 2006, 80: 10763-10771.
Abate DA, Watanabe S, Mocarski ES: Major human cytomegalovirus structural protein pp65 (ppUL83) prevents interferon response factor 3 activation in the interferon response. J Virol 2004, 78: 10995-11006.
Browne EP, Shenk T: Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells. Proc Natl Acad Sci USA 2003, 100: 11439-11444.
Taylor RT, Bresnahan WA: Human cytomegalovirus immediate-early 2 protein IE86 blocks virus-induced chemokine expression. J Virol 2006, 80: 920-928.
Morello CS, Cranmer LD, Spector DH: In vivo replication, latency, and immunogenicity of murine cytomegalovirus mutants with deletions in the M83 and M84 genes, the putative homologs of human cytomegalovirus pp65 (UL83). J Virol 1999, 73: 7678-7693.
Abenes G, Lee M, Haghjoo E, Tong T, Zhan X, Liu F: Murine cytomegalovirus open reading frame M27 plays an important role in growth and virulence in mice. J Virol 2001, 75: 1697-1707.
Zimmermann A, Trilling M, Wagner M, Wilborn M, Bubic I, Jonjic S, Koszinowski U, Hengel H: A cytomegaloviral protein reveals a dual role for STAT2 in IFN-{gamma} signaling and antiviral responses. J Exp Med 2005, 201: 1543-1553.
Khan S, Zimmermann A, Basler M, Groettrup M, Hengel H: A cytomegalovirus inhibitor of gamma interferon signaling controls immunoproteasome induction. J Virol 2004, 78: 1831-1842.
Valchanova RS, Picard-Maureau M, Budt M, Brune W: Murine cytomegalovirus m142 and m143 are both required to block protein kinase R-mediated shutdown of protein synthesis. J Virol 2006, 80: 10181-10190.
Child SJ, Hanson LK, Brown CE, Janzen DM, Geballe AP: Double-stranded RNA binding by a heterodimeric complex of murine cytomegalovirus m142 and m143 proteins. J Virol 2006, 80: 10173-10180.
Hakki M, Geballe AP: Double-stranded RNA binding by human cytomegalovirus pTRS1. J Virol 2005, 79: 7311-7318.
Hakki M, Marshall EE, De Niro KL, Geballe AP: Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J Virol 2006, 80: 11817-11826.
Child SJ, Hakki M, De Niro KL, Geballe AP: Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J Virol 2004, 78: 197-205.
Edward S, Mocarski TS, Pass RobertF: Cytomegaloviruses. In Fields Virology. Volume 2. 5th edition. Edited by: David MK, Peter M Howley. Philadelphia: Lippincott Williams and Williams; 2007:2701-2772.
Wilkinson GW, Tomasec P, Stanton RJ, Armstrong M, Prod'homme V, Aicheler R, McSharry BP, Rickards CR, Cochrane D, Llewellyn-Lacey S, Wang EC, Griffin CA, Davison AJ: Modulation of natural killer cells by human cytomegalovirus. J Clin Virol 2008, 41: 206-212.
Sutherland CL, Chalupny NJ, Cosman D: The UL16-binding proteins, a novel family of MHC class I-related ligands for NKG2D, activate natural killer cell functions. Immunol Rev 2001, 181: 185-192.
Kubin M, Cassiano L, Chalupny J, Chin W, Cosman D, Fanslow W, Mullberg J, Rousseau AM, Ulrich D, Armitage R: ULBP1, 2, 3: novel MHC class I-related molecules that bind to human cytomegalovirus glycoprotein UL16, activate NK cells. Eur J Immunol 2001, 31: 1428-1437.
Cosman D, Mullberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, Kubin M, Chalupny NJ: ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001, 14: 123-133.
Wu J, Chalupny NJ, Manley TJ, Riddell SR, Cosman D, Spies T: Intracellular retention of the MHC class I-related chain B ligand of NKG2D by the human cytomegalovirus UL16 glycoprotein. J Immunol 2003, 170: 4196-4200.
Dunn C, Chalupny NJ, Sutherland CL, Dosch S, Sivakumar PV, Johnson DC, Cosman D: Human cytomegalovirus glycoprotein UL16 causes intracellular sequestration of NKG2D ligands, protecting against natural killer cell cytotoxicity. J Exp Med 2003, 197: 1427-1439.
Rolle A, Mousavi-Jazi M, Eriksson M, Odeberg J, Soderberg-Naucler C, Cosman D, Karre K, Cerboni C: Effects of human cytomegalovirus infection on ligands for the activating NKG2D receptor of NK cells: up-regulation of UL16-binding protein (ULBP)1 and ULBP2 is counteracted by the viral UL16 protein. J Immunol 2003, 171: 902-908.
Vales-Gomez M, Winterhalter A, Roda-Navarro P, Zimmermann A, Boyle L, Hengel H, Brooks A, Reyburn HT: The human cytomegalovirus glycoprotein UL16 traffics through the plasma membrane and the nuclear envelope. Cell Microbiol 2006, 8: 581-590.
Welte SA, Sinzger C, Lutz SZ, Singh-Jasuja H, Sampaio KL, Eknigk U, Rammensee HG, Steinle A: Selective intracellular retention of virally induced NKG2D ligands by the human cytomegalovirus UL16 glycoprotein. Eur J Immunol 2003, 33: 194-203.
Odeberg J, Browne H, Metkar S, Froelich CJ, Branden L, Cosman D, Soderberg-Naucler C: The human cytomegalovirus protein UL16 mediates increased resistance to natural killer cell cytotoxicity through resistance to cytolytic proteins. J Virol 2003, 77: 4539-4545.
Stern-Ginossar N, Elefant N, Zimmermann A, Wolf DG, Saleh N, Biton M, Horwitz E, Prokocimer Z, Prichard M, Hahn G, Goldman-Wohl D, Greenfield C, Yagel S, Hengel H, Altuvia Y, Margalit H, Mandelboim O: Host immune system gene targeting by a viral miRNA. Science 2007, 317: 376-381.
Krmpotic A, Hasan M, Loewendorf A, Saulig T, Halenius A, Lenac T, Polic B, Bubic I, Kriegeskorte A, Pernjak-Pugel E, Messerle M, Hengel H, Busch DH, Koszinowski UH, Jonjic S: NK cell activation through the NKG2D ligand MULT-1 is selectively prevented by the glycoprotein encoded by mouse cytomegalovirus gene m145. J Exp Med 2005, 201: 211-220.
Lodoen MB, Abenes G, Umamoto S, Houchins JP, Liu F, Lanier LL: The cytomegalovirus m155 gene product subverts natural killer cell antiviral protection by disruption of H60-NKG2D interactions. J Exp Med 2004, 200: 1075-1081.
Lodoen M, Ogasawara K, Hamerman JA, Arase H, Houchins JP, Mocarski ES, Lanier LL: NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J Exp Med 2003, 197: 1245-1253.
Krmpotic A, Busch DH, Bubic I, Gebhardt F, Hengel H, Hasan M, Scalzo AA, Koszinowski UH, Jonjic S: MCMV glycoprotein gp40 confers virus resistance to CD8+ T cells and NK cells in vivo. Nat Immunol 2002, 3: 529-535.
Pinto AK, Jamieson AM, Raulet DH, Hill AB: The role of NKG2D signaling in inhibition of cytotoxic T-lymphocyte lysis by the Murine cytomegalovirus immunoevasin m152/gp40. J Virol 2007, 81: 12564-12571.
Abenes G, Chan K, Lee M, Haghjoo E, Zhu J, Zhou T, Zhan X, Liu F: Murine cytomegalovirus with a transposon insertional mutation at open reading frame m155 is deficient in growth and virulence in mice. J Virol 2004, 78: 6891-6899.
Hasan M, Krmpotic A, Ruzsics Z, Bubic I, Lenac T, Halenius A, Loewendorf A, Messerle M, Hengel H, Jonjic S, Koszinowski UH: Selective down-regulation of the NKG2D ligand H60 by mouse cytomegalovirus m155 glycoprotein. J Virol 2005, 79: 2920-2930.
Cavanaugh VJ, Stenberg RM, Staley TL, Virgin HWt, MacDonald MR, Paetzold S, Farrell HE, Rawlinson WD, Campbell AE: Murine cytomegalovirus with a deletion of genes spanning HindIII-J and -I displays altered cell and tissue tropism. J Virol 1996, 70: 1365-1374.
Rawlinson WD, Farrell HE, Barrell BG: Analysis of the complete DNA sequence of murine cytomegalovirus. J Virol 1996, 70: 8833-8849.
Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL: Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 2002, 296: 1323-1326.
Davis AH, Guseva NV, Ball BL, Heusel JW: Characterization of Murine Cytomegalovirus m157 from Infected Cells and Identification of Critical Residues Mediating Recognition by the NK Cell Receptor Ly49H. J Immunol 2008, 181: 265-275.
Tripathy SK, Smith HR, Holroyd EA, Pingel JT, Yokoyama WM: Expression of m157, a murine cytomegalovirus-encoded putative major histocompatibility class I (MHC-I)-like protein, is independent of viral regulation of host MHC-I. J Virol 2006, 80: 545-550.
Kielczewska A, Kim HS, Lanier LL, Dimasi N, Vidal SM: Critical residues at the Ly49 natural killer receptor's homodimer interface determine functional recognition of m157, a mouse cytomegalovirus MHC class I-like protein. J Immunol 2007, 178: 369-377.
Smith HR, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, Iizuka K, Furukawa H, Beckman DL, Pingel JT, Scalzo AA, Fremont DH, Yokoyama WM: Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Natl Acad Sci USA 2002, 99: 8826-8831.
Dorner BG, Smith HR, French AR, Kim S, Poursine-Laurent J, Beckman DL, Pingel JT, Kroczek RA, Yokoyama WM: Coordinate expression of cytokines and chemokines by NK cells during murine cytomegalovirus infection. J Immunol 2004, 172: 3119-3131.
Adams EJ, Juo ZS, Venook RT, Boulanger MJ, Arase H, Lanier LL, Garcia KC: Structural elucidation of the m157 mouse cytomegalovirus ligand for Ly49 natural killer cell receptors. Proc Natl Acad Sci USA 2007, 104: 10128-10133.
Bubic I, Wagner M, Krmpotic A, Saulig T, Kim S, Yokoyama WM, Jonjic S, Koszinowski UH: Gain of virulence caused by loss of a gene in murine cytomegalovirus. J Virol 2004, 78: 7536-7544.
Daniels KA, Devora G, Lai WC, O'Donnell CL, Bennett M, Welsh RM: Murine cytomegalovirus is regulated by a discrete subset of natural killer cells reactive with monoclonal antibody to Ly49H. J Exp Med 2001, 194: 29-44.
Fodil-Cornu N, Lee SH, Belanger S, Makrigiannis AP, Biron CA, Buller RM, Vidal SM: Ly49h-deficient C57BL/6 mice: a new mouse cytomegalovirus-susceptible model remains resistant to unrelated pathogens controlled by the NK gene complex. J Immunol 2008, 181: 6394-6405.
Voigt V, Forbes CA, Tonkin JN, Degli-Esposti MA, Smith HR, Yokoyama WM, Scalzo AA: Murine cytomegalovirus m157 mutation and variation leads to immune evasion of natural killer cells. Proc Natl Acad Sci USA 2003, 100: 13483-13488.
Hanson LK, Dalton BL, Karabekian Z, Farrell HE, Rawlinson WD, Stenberg RM, Campbell AE: Transcriptional analysis of the murine cytomegalovirus HindIII-I region: identification of a novel immediate-early gene region. Virology 1999, 260: 156-164.
Chapman TL, Bjorkman PJ: Characterization of a murine cytomegalovirus class I major histocompatibility complex (MHC) homolog: comparison to MHC molecules and to the human cytomegalovirus MHC homolog. J Virol 1998, 72: 460-466.
Cretney E, Degli-Esposti MA, Densley EH, Farrell HE, Davis-Poynter NJ, Smyth MJ: m144, a murine cytomegalovirus (MCMV)-encoded major histocompatibility complex class I homologue, confers tumor resistance to natural killer cell-mediated rejection. J Exp Med 1999, 190: 435-444.
Farrell HE, Vally H, Lynch DM, Fleming P, Shellam GR, Scalzo AA, Davis-Poynter NJ: Inhibition of natural killer cells by a cytomegalovirus MHC class I homologue in vivo. Nature 1997, 386: 510-514.
Natarajan K, Hicks A, Mans J, Robinson H, Guan R, Mariuzza RA, Margulies DH: Crystal structure of the murine cytomegalovirus MHC-I homolog m144. J Mol Biol 2006, 358: 157-171.
Beisser PS, Kloover JS, Grauls GE, Blok MJ, Bruggeman CA, Vink C: The r144 major histocompatibility complex class I-like gene of rat cytomegalovirus is dispensable for both acute and long-term infection in the immunocompromised host. J Virol 2000, 74: 1045-1050.
Kloover JS, Grauls GE, Blok MJ, Vink C, Bruggeman CA: A rat cytomegalovirus strain with a disruption of the r144 MHC class I-like gene is attenuated in the acute phase of infection in neonatal rats. Arch Virol 2002, 147: 813-824.
Beck S, Barrell BG: Human cytomegalovirus encodes a glycoprotein homologous to MHC class-I antigens. Nature 1988, 331: 269-272.
Wagner CS, Ljunggren HG, Achour A: Immune modulation by the human cytomegalovirus-encoded molecule UL18, a mystery yet to be solved. J Immunol 2008, 180: 19-24.
Browne H, Smith G, Beck S, Minson T: A complex between the MHC class I homologue encoded by human cytomegalovirus and beta 2 microglobulin. Nature 1990, 347: 770-772.
Fahnestock ML, Johnson JL, Feldman RM, Neveu JM, Lane WS, Bjorkman PJ: The MHC class I homolog encoded by human cytomegalovirus binds endogenous peptides. Immunity 1995, 3: 583-590.
Cosman D, Fanger N, Borges L, Kubin M, Chin W, Peterson L, Hsu ML: A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity 1997, 7: 273-282.
Yang Z, Bjorkman PJ: Structure of UL18, a peptide-binding viral MHC mimic, bound to a host inhibitory receptor. Proc Natl Acad Sci USA 2008, 105: 10095-10100.
Occhino M, Ghiotto F, Soro S, Mortarino M, Bosi S, Maffei M, Bruno S, Nardini M, Figini M, Tramontano A, Ciccone E: Dissecting the structural determinants of the interaction between the human cytomegalovirus UL18 protein and the CD85j immune receptor. J Immunol 2008, 180: 957-968.
Chapman TL, Heikeman AP, Bjorkman PJ: The inhibitory receptor LIR-1 uses a common binding interaction to recognize class I MHC molecules and the viral homolog UL18. Immunity 1999, 11: 603-613.
Leong CC, Chapman TL, Bjorkman PJ, Formankova D, Mocarski ES, Phillips JH, Lanier LL: Modulation of natural killer cell cytotoxicity in human cytomegalovirus infection: the role of endogenous class I major histocompatibility complex and a viral class I homolog. J Exp Med 1998, 187: 1681-1687.
Reyburn HT, Mandelboim O, Vales-Gomez M, Davis DM, Pazmany L, Strominger JL: The class I MHC homologue of human cytomegalovirus inhibits attack by natural killer cells. Nature 1997, 386: 514-517.
Prod'homme V, Griffin C, Aicheler RJ, Wang EC, McSharry BP, Rickards CR, Stanton RJ, Borysiewicz LK, Lopez-Botet M, Wilkinson GW, Tomasec P: The human cytomegalovirus MHC class I homolog UL18 inhibits LIR-1+ but activates LIR-1-NK cells. J Immunol 2007, 178: 4473-4481.
Wagner CS, Riise GC, Bergstrom T, Karre K, Carbone E, Berg L: Increased expression of leukocyte Ig-like receptor-1 and activating role of UL18 in the response to cytomegalovirus infection. J Immunol 2007, 178: 3536-3543.
Kim JS, Choi SE, Yun IH, Kim JY, Ahn C, Kim SJ, Ha J, Hwang ES, Cha CY, Miyagawa S, Park CG: Human cytomegalovirus UL18 alleviated human NK-mediated swine endothelial cell lysis. Biochem Biophys Res Commun 2004, 315: 144-150.
Saverino D, Ghiotto F, Merlo A, Bruno S, Battini L, Occhino M, Maffei M, Tenca C, Pileri S, Baldi L, Fabbi M, Bachi A, De Santanna A, Grossi CE, Ciccone E: Specific recognition of the viral protein UL18 by CD85j/LIR-1/ILT2 on CD8+ T cells mediates the non-MHC-restricted lysis of human cytomegalovirus-infected cells. J Immunol 2004, 172: 5629-5637.
Garrigue I, Corte MF, Magnin N, Couzi L, Capdepont S, Rio C, Merville P, Dechanet-Merville J, Fleury H, Lafon ME: Variability of UL18, UL40, UL111a and US3 immunomodulatory genes among human cytomegalovirus clinical isolates from renal transplant recipients. J Clin Virol 2007, 40: 120-128.
Cerboni C, Achour A, Warnmark A, Mousavi-Jazi M, Sandalova T, Hsu ML, Cosman D, Karre K, Carbone E: Spontaneous mutations in the human CMV HLA class I homologue UL18 affect its binding to the inhibitory receptor LIR-1/ILT2/CD85j. Eur J Immunol 2006, 36: 732-741.
Odeberg J, Cerboni C, Browne H, Karre K, Moller E, Carbone E, Soderberg-Naucler C: Human cytomegalovirus (HCMV)-infected endothelial cells and macrophages are less susceptible to natural killer lysis independent of the downregulation of classical HLA class I molecules or expression of the HCMV class I homologue, UL18. Scand J Immunol 2002, 55: 149-161.
Wagner CS, Walther-Jallow L, Buentke E, Ljunggren HG, Achour A, Chambers BJ: Human cytomegalovirus-derived protein UL18 alters the phenotype and function of monocyte-derived dendritic cells. J Leukoc Biol 2008, 83: 56-63.
Park B, Oh H, Lee S, Song Y, Shin J, Sung YC, Hwang SY, Ahn K: The MHC class I homolog of human cytomegalovirus is resistant to down-regulation mediated by the unique short region protein (US)2, US3, US6, and US11 gene products. J Immunol 2002, 168: 3464-3469.
Kim Y, Park B, Cho S, Shin J, Cho K, Jun Y, Ahn K: Human cytomegalovirus UL18 utilizes US6 for evading the NK and T-cell responses. PLoS Pathog 2008, 4: e1000123.
Hassan-Walker AF, Cope AV, Griffiths PD, Emery VC: Transcription of the human cytomegalovirus natural killer decoy gene, UL18, in vitro and in vivo. J Gen Virol 1998,79(Pt 9):2113-2116.
Davison AJ, Dolan A, Akter P, Addison C, Dargan DJ, Alcendor DJ, McGeoch DJ, Hayward GS: The human cytomegalovirus genome revisited: comparison with the chimpanzee cytomegalovirus genome. J Gen Virol 2003, 84: 17-28.
Wills MR, Ashiru O, Reeves MB, Okecha G, Trowsdale J, Tomasec P, Wilkinson GW, Sinclair J, Sissons JG: Human cytomegalovirus encodes an MHC class I-like molecule (UL142) that functions to inhibit NK cell lysis. J Immunol 2005, 175: 7457-7465.
Chalupny NJ, Rein-Weston A, Dosch S, Cosman D: Down-regulation of the NKG2D ligand MICA by the human cytomegalovirus glycoprotein UL142. Biochem Biophys Res Commun 2006, 346: 175-181.
Wang EC, McSharry B, Retiere C, Tomasec P, Williams S, Borysiewicz LK, Braud VM, Wilkinson GW: UL40-mediated NK evasion during productive infection with human cytomegalovirus. Proc Natl Acad Sci USA 2002, 99: 7570-7575.
Tomasec P, Braud VM, Rickards C, Powell MB, McSharry BP, Gadola S, Cerundolo V, Borysiewicz LK, McMichael AJ, Wilkinson GW: Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science 2000, 287: 1031.
Ulbrecht M, Martinozzi S, Grzeschik M, Hengel H, Ellwart JW, Pla M, Weiss EH: Cutting edge: the human cytomegalovirus UL40 gene product contains a ligand for HLA-E and prevents NK cell-mediated lysis. J Immunol 2000, 164: 5019-5022.
Cerboni C, Mousavi-Jazi M, Wakiguchi H, Carbone E, Karre K, Soderstrom K: Synergistic effect of IFN-gamma and human cytomegalovirus protein UL40 in the HLA-E-dependent protection from NK cell-mediated cytotoxicity. Eur J Immunol 2001, 31: 2926-2935.
Lehner PJ, Karttunen JT, Wilkinson GW, Cresswell P: The human cytomegalovirus US6 glycoprotein inhibits transporter associated with antigen processing-dependent peptide translocation. Proc Natl Acad Sci USA 1997, 94: 6904-6909.
Falk CS, Mach M, Schendel DJ, Weiss EH, Hilgert I, Hahn G: NK cell activity during human cytomegalovirus infection is dominated by US2-11-mediated HLA class I down-regulation. J Immunol 2002, 169: 3257-3266.
Pietra G, Romagnani C, Mazzarino P, Falco M, Millo E, Moretta A, Moretta L, Mingari MC: HLA-E-restricted recognition of cytomegalovirus-derived peptides by human CD8+ cytolytic T lymphocytes. Proc Natl Acad Sci USA 2003, 100: 10896-10901.
Mazzarino P, Pietra G, Vacca P, Falco M, Colau D, Coulie P, Moretta L, Mingari MC: Identification of effector-memory CMV-specific T lymphocytes that kill CMV-infected target cells in an HLA-E-restricted fashion. Eur J Immunol 2005, 35: 3240-3247.
Arnon TI, Achdout H, Levi O, Markel G, Saleh N, Katz G, Gazit R, Gonen-Gross T, Hanna J, Nahari E, Porgador A, Honigman A, Plachter B, Mevorach D, Wolf DG, Mandelboim O: Inhibition of the NKp30 activating receptor by pp65 of human cytomegalovirus. Nat Immunol 2005, 6: 515-523.
Tomasec P, Wang EC, Davison AJ, Vojtesek B, Armstrong M, Griffin C, McSharry BP, Morris RJ, Llewellyn-Lacey S, Rickards C, Nomoto A, Sinzger C, Wilkinson GW: Downregulation of natural killer cell-activating ligand CD155 by human cytomegalovirus UL141. Nat Immunol 2005, 6: 181-188.
Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A: Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol 2001, 19: 683-765.
Redpath S, Ghazal P, Gascoigne NR: Hijacking and exploitation of IL-10 by intracellular pathogens. Trends Microbiol 2001, 9: 86-92.
Kotenko SV, Saccani S, Izotova LS, Mirochnitchenko OV, Pestka S: Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10). Proc Natl Acad Sci USA 2000, 97: 1695-1700.
Spencer JV, Lockridge KM, Barry PA, Lin G, Tsang M, Penfold ME, Schall TJ: Potent immunosuppressive activities of cytomegalovirus-encoded interleukin-10. J Virol 2002, 76: 1285-1292.
Raftery MJ, Wieland D, Gronewald S, Kraus AA, Giese T, Schonrich G: Shaping phenotype, function, and survival of dendritic cells by cytomegalovirus-encoded IL-10. J Immunol 2004, 173: 3383-3391.
Chang WL, Baumgarth N, Yu D, Barry PA: Human cytomegalovirus-encoded interleukin-10 homolog inhibits maturation of dendritic cells and alters their functionality. J Virol 2004, 78: 8720-8731.
Jones BC, Logsdon NJ, Josephson K, Cook J, Barry PA, Walter MR: Crystal structure of human cytomegalovirus IL-10 bound to soluble human IL-10R1. Proc Natl Acad Sci USA 2002, 99: 9404-9409.
Spencer JV: The cytomegalovirus homolog of interleukin-10 requires phosphatidylinositol 3-kinase activity for inhibition of cytokine synthesis in monocytes. J Virol 2007, 81: 2083-2086.
Spencer JV, Cadaoas J, Castillo PR, Saini V, Slobedman B: Stimulation of B lymphocytes by cmvIL-10 but not LAcmvIL-10. Virology 2008, 374: 164-169.
Yamamoto-Tabata T, McDonagh S, Chang HT, Fisher S, Pereira L: Human cytomegalovirus interleukin-10 downregulates metalloproteinase activity and impairs endothelial cell migration and placental cytotrophoblast invasiveness in vitro. J Virol 2004, 78: 2831-2840.
Jenkins C, Abendroth A, Slobedman B: A novel viral transcript with homology to human interleukin-10 is expressed during latent human cytomegalovirus infection. J Virol 2004, 78: 1440-1447.
Jenkins C, Garcia W, Abendroth A, Slobedman B: Expression of a human cytomegalovirus latency-associated homolog of interleukin-10 during the productive phase of infection. Virology 2008, 370: 285-294.
Jenkins C, Garcia W, Godwin MJ, Spencer JV, Stern JL, Abendroth A, Slobedman B: Immunomodulatory properties of a viral homolog of human interleukin-10 expressed by human cytomegalovirus during the latent phase of infection. J Virol 2008, 82: 3736-3750.
Lockridge KM, Zhou SS, Kravitz RH, Johnson JL, Sawai ET, Blewett EL, Barry PA: Primate cytomegaloviruses encode and express an IL-10-like protein. Virology 2000, 268: 272-280.
Redpath S, Angulo A, Gascoigne NR, Ghazal P: Murine cytomegalovirus infection down-regulates MHC class II expression on macrophages by induction of IL-10. J Immunol 1999, 162: 6701-6707.
Cavanaugh VJ, Deng Y, Birkenbach MP, Slater JS, Campbell AE: Vigorous innate and virus-specific cytotoxic T-lymphocyte responses to murine cytomegalovirus in the submaxillary salivary gland. J Virol 2003, 77: 1703-1717.
Benedict CA, Butrovich KD, Lurain NS, Corbeil J, Rooney I, Schneider P, Tschopp J, Ware CF: Cutting edge: a novel viral TNF receptor superfamily member in virulent strains of human cytomegalovirus. J Immunol 1999, 162: 6967-6970.
Lurain NS, Kapell KS, Huang DD, Short JA, Paintsil J, Winkfield E, Benedict CA, Ware CF, Bremer JW: Human cytomegalovirus UL144 open reading frame: sequence hypervariability in low-passage clinical isolates. J Virol 1999, 73: 10040-10050.
Poole E, King CA, Sinclair JH, Alcami A: The UL144 gene product of human cytomegalovirus activates NFkappaB via a TRAF6-dependent mechanism. EMBO J 2006, 25: 4390-4399.
Cheung TC, Humphreys IR, Potter KG, Norris PS, Shumway HM, Tran BR, Patterson G, Jean-Jacques R, Yoon M, Spear PG, Murphy KM, Lurain NS, Benedict CA, Ware CF: Evolutionarily divergent herpesviruses modulate T cell activation by targeting the herpesvirus entry mediator cosignaling pathway. Proc Natl Acad Sci USA 2005, 102: 13218-13223.
Sedy JR, Gavrieli M, Potter KG, Hurchla MA, Lindsley RC, Hildner K, Scheu S, Pfeffer K, Ware CF, Murphy TL, Murphy KM: B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol 2005, 6: 90-98.
Poole E, Atkins E, Nakayama T, Yoshie O, Groves I, Alcami A, Sinclair J: NF-kappaB-mediated activation of the chemokine CCL22 by the product of the human cytomegalovirus gene UL144 escapes regulation by viral IE86. J Virol 2008, 82: 4250-4256.
Arav-Boger R, Battaglia CA, Lazzarotto T, Gabrielli L, Zong JC, Hayward GS, Diener-West M, Landini MP: Cytomegalovirus (CMV)-encoded UL144 (truncated tumor necrosis factor receptor) and outcome of congenital CMV infection. J Infect Dis 2006, 194: 464-473.
Arav-Boger R, Willoughby RE, Pass RF, Zong JC, Jang WJ, Alcendor D, Hayward GS: Polymorphisms of the cytomegalovirus (CMV)-encoded tumor necrosis factor-alpha and beta-chemokine receptors in congenital CMV disease. J Infect Dis 2002, 186: 1057-1064.
Bale JF Jr, Petheram SJ, Robertson M, Murph JR, Demmler G: Human cytomegalovirus a sequence and UL144 variability in strains from infected children. J Med Virol 2001, 65: 90-96.
Heo J, Petheram S, Demmler G, Murph JR, Adler SP, Bale J, Sparer TE: Polymorphisms within human cytomegalovirus chemokine (UL146/UL147) and cytokine receptor genes (UL144) are not predictive of sequelae in congenitally infected children. Virology 2008.
Mao ZQ, Huang Y, Sun M, Ruan Q, Qi Y, He R, Huang YJ, Ma YP, Ji YH, Sun ZR, Gao H: Genetic polymorphism of UL144 open reading frame of human cytomegalovirus DNA detected in colon samples from infants with Hirschsprung's disease. World J Gastroenterol 2007, 13: 4350-4354.
Picone O, Costa JM, Chaix ML, Ville Y, Rouzioux C, Leruez-Ville M: Human cytomegalovirus UL144 gene polymorphisms in congenital infections. J Clin Microbiol 2005, 43: 25-29.
Yan H, Koyano S, Inami Y, Yamamoto Y, Suzutani T, Mizuguchi M, Ushijima H, Kurane I, Inoue N: Genetic variations in the gB, UL144 and UL149 genes of human cytomegalovirus strains collected from congenitally and postnatally infected Japanese children. Arch Virol 2008, 153: 667-674.
MacDonald MR, Li XY, Virgin HWt: Late expression of a beta chemokine homolog by murine cytomegalovirus. J Virol 1997, 71: 1671-1678.
MacDonald MR, Burney MW, Resnick SB, Virgin HI: Spliced mRNA encoding the murine cytomegalovirus chemokine homolog predicts a beta chemokine of novel structure. J Virol 1999, 73: 3682-3691.
Smith LM, Shellam GR, Redwood AJ: Genes of murine cytomegalovirus exist as a number of distinct genotypes. Virology 2006, 352: 450-465.
Saederup N, Lin YC, Dairaghi DJ, Schall TJ, Mocarski ES: Cytomegalovirus-encoded beta chemokine promotes monocyte-associated viremia in the host. Proc Natl Acad Sci USA 1999, 96: 10881-10886.
Saederup N, Aguirre SA, Sparer TE, Bouley DM, Mocarski ES: Murine cytomegalovirus CC chemokine homolog MCK-2 (m131-129) is a determinant of dissemination that increases inflammation at initial sites of infection. J Virol 2001, 75: 9966-9976.
Fleming P, Davis-Poynter N, Degli-Esposti M, Densley E, Papadimitriou J, Shellam G, Farrell H: The murine cytomegalovirus chemokine homolog, m131/129, is a determinant of viral pathogenicity. J Virol 1999, 73: 6800-6809.
Noda S, Aguirre SA, Bitmansour A, Brown JM, Sparer TE, Huang J, Mocarski ES: Cytomegalovirus MCK-2 controls mobilization and recruitment of myeloid progenitor cells to facilitate dissemination. Blood 2006, 107: 30-38.
Vink C, Beuken E, Bruggeman CA: Complete DNA sequence of the rat cytomegalovirus genome. J Virol 2000, 74: 7656-7665.
Kaptein SJ, van Cleef KW, Gruijthuijsen YK, Beuken EV, van Buggenhout L, Beisser PS, Stassen FR, Bruggeman CA, Vink C: The r131 gene of rat cytomegalovirus encodes a proinflammatory CC chemokine homolog which is essential for the production of infectious virus in the salivary glands. Virus Genes 2004, 29: 43-61.
Penfold M, Miao Z, Wang Y, Haggerty S, Schleiss MR: A macrophage inflammatory protein homolog encoded by guinea pig cytomegalovirus signals via CC chemokine receptor 1. Virology 2003, 316: 202-212.
Haggerty SM, Schleiss MR: A novel CC-chemokine homolog encoded by guinea pig cytomegalovirus. Virus Genes 2002, 25: 271-279.
Cha TA, Tom E, Kemble GW, Duke GM, Mocarski ES, Spaete RR: Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J Virol 1996, 70: 78-83.
Penfold ME, Dairaghi DJ, Duke GM, Saederup N, Mocarski ES, Kemble GW, Schall TJ: Cytomegalovirus encodes a potent alpha chemokine. Proc Natl Acad Sci USA 1999, 96: 9839-9844.
Hahn G, Revello MG, Patrone M, Percivalle E, Campanini G, Sarasini A, Wagner M, Gallina A, Milanesi G, Koszinowski U, Baldanti F, Gerna G: Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J Virol 2004, 78: 10023-10033.
Miller-Kittrell M, Sai J, Penfold M, Richmond A, Sparer TE: Functional characterization of chimpanzee cytomegalovirus chemokine, vCXCL-1(CCMV). Virology 2007, 364: 454-465.
Arav-Boger R, Zong JC, Foster CB: Loss of linkage disequilibrium and accelerated protein divergence in duplicated cytomegalovirus chemokine genes. Virus Genes 2005, 31: 65-72.
Hassan-Walker AF, Okwuadi S, Lee L, Griffiths PD, Emery VC: Sequence variability of the alpha-chemokine UL146 from clinical strains of human cytomegalovirus. J Med Virol 2004, 74: 573-579.
Bradley AJ, Kovacs IJ, Gatherer D, Dargan DJ, Alkharsah KR, Chan PK, Carman WF, Dedicoat M, Emery VC, Geddes CC, Gerna G, Ben-Ismaeil B, Kaye S, McGregor A, Moss PA, Pusztai R, Rawlinson WD, Scott GM, Wilkinson GW, Schulz TF, Davison AJ: Genotypic analysis of two hypervariable human cytomegalovirus genes. J Med Virol 2008, 80: 1615-1623.
Akter P, Cunningham C, McSharry BP, Dolan A, Addison C, Dargan DJ, Hassan-Walker AF, Emery VC, Griffiths PD, Wilkinson GW, Davison AJ: Two novel spliced genes in human cytomegalovirus. J Gen Virol 2003, 84: 1117-1122.
Wang D, Shenk T: Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc Natl Acad Sci USA 2005, 102: 18153-18158.
Wang D, Shenk T: Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J Virol 2005, 79: 10330-10338.
Chee MS, Satchwell SC, Preddie E, Weston KM, Barrell BG: Human cytomegalovirus encodes three G protein-coupled receptor homologues. Nature 1990, 344: 774-777.
Beisser PS, Lavreysen H, Bruggeman CA, Vink C: Chemokines and chemokine receptors encoded by cytomegaloviruses. Curr Top Microbiol Immunol 2008, 325: 221-242.
Murphy PM: The molecular biology of leukocyte chemoattractant receptors. Annu Rev Immunol 1994, 12: 593-633.
van Cleef KW, Smit MJ, Bruggeman CA, Vink C: Cytomegalovirus-encoded homologs of G protein-coupled receptors and chemokines. J Clin Virol 2006, 35: 343-348.
Gao JL, Murphy PM: Human cytomegalovirus open reading frame US28 encodes a functional beta chemokine receptor. J Biol Chem 1994, 269: 28539-28542.
Kuhn DE, Beall CJ, Kolattukudy PE: The cytomegalovirus US28 protein binds multiple CC chemokines with high affinity. Biochem Biophys Res Commun 1995, 211: 325-330.
Kledal TN, Rosenkilde MM, Schwartz TW: Selective recognition of the membrane-bound CX3C chemokine, fractalkine, by the human cytomegalovirus-encoded broad-spectrum receptor US28. FEBS Lett 1998, 441: 209-214.
Michelson S, Dal Monte P, Zipeto D, Bodaghi B, Laurent L, Oberlin E, Arenzana-Seisdedos F, Virelizier JL, Landini MP: Modulation of RANTES production by human cytomegalovirus infection of fibroblasts. J Virol 1997, 71: 6495-6500.
Bodaghi B, Jones TR, Zipeto D, Vita C, Sun L, Laurent L, Arenzana-Seisdedos F, Virelizier JL, Michelson S: Chemokine sequestration by viral chemoreceptors as a novel viral escape strategy: withdrawal of chemokines from the environment of cytomegalovirus-infected cells. J Exp Med 1998, 188: 855-866.
Vieira J, Schall TJ, Corey L, Geballe AP: Functional analysis of the human cytomegalovirus US28 gene by insertion mutagenesis with the green fluorescent protein gene. J Virol 1998, 72: 8158-8165.
Randolph-Habecker JR, Rahill B, Torok-Storb B, Vieira J, Kolattukudy PE, Rovin BH, Sedmak DD: The expression of the cytomegalovirus chemokine receptor homolog US28 sequesters biologically active CC chemokines and alters IL-8 production. Cytokine 2002, 19: 37-46.
Fraile-Ramos A, Kledal TN, Pelchen-Matthews A, Bowers K, Schwartz TW, Marsh M: The human cytomegalovirus US28 protein is located in endocytic vesicles and undergoes constitutive endocytosis and recycling. Mol Biol Cell 2001, 12: 1737-1749.
Margulies BJ, Gibson W: The chemokine receptor homologue encoded by US27 of human cytomegalovirus is heavily glycosylated and is present in infected human foreskin fibroblasts and enveloped virus particles. Virus Res 2007, 123: 57-71.
Billstrom MA, Johnson GL, Avdi NJ, Worthen GS: Intracellular signaling by the chemokine receptor US28 during human cytomegalovirus infection. J Virol 1998, 72: 5535-5544.
Casarosa P, Bakker RA, Verzijl D, Navis M, Timmerman H, Leurs R, Smit MJ: Constitutive signaling of the human cytomegalovirus-encoded chemokine receptor US28. J Biol Chem 2001, 276: 1133-1137.
Wilkie TM, Scherle PA, Strathmann MP, Slepak VZ, Simon MI: Characterization of G-protein alpha subunits in the Gq class: expression in murine tissues and in stromal and hematopoietic cell lines. Proc Natl Acad Sci USA 1991, 88: 10049-10053.
Streblow DN, Soderberg-Naucler C, Vieira J, Smith P, Wakabayashi E, Ruchti F, Mattison K, Altschuler Y, Nelson JA: The human cytomegalovirus chemokine receptor US28 mediates vascular smooth muscle cell migration. Cell 1999, 99: 511-520.
Streblow DN, Orloff SL, Nelson JA: The HCMV chemokine receptor US28 is a potential target in vascular disease. Curr Drug Targets Infect Disord 2001, 1: 151-158.
McLean KA, Holst PJ, Martini L, Schwartz TW, Rosenkilde MM: Similar activation of signal transduction pathways by the herpesvirus-encoded chemokine receptors US28 and ORF74. Virology 2004, 325: 241-251.
Waldhoer M, Kledal TN, Farrell H, Schwartz TW: Murine cytomegalovirus (CMV) M33 and human CMV US28 receptors exhibit similar constitutive signaling activities. J Virol 2002, 76: 8161-8168.
Stropes MP, Miller WE: Functional analysis of human cytomegalovirus pUS28 mutants in infected cells. J Gen Virol 2008, 89: 97-105.
Miller WE, Houtz DA, Nelson CD, Kolattukudy PE, Lefkowitz RJ: G-protein-coupled receptor (GPCR) kinase phosphorylation and beta-arrestin recruitment regulate the constitutive signaling activity of the human cytomegalovirus US28 GPCR. J Biol Chem 2003, 278: 21663-21671.
Maussang D, Verzijl D, van Walsum M, Leurs R, Holl J, Pleskoff O, Michel D, van Dongen GA, Smit MJ: Human cytomegalovirus-encoded chemokine receptor US28 promotes tumorigenesis. Proc Natl Acad Sci USA 2006, 103: 13068-13073.
Zhou YF, Leon MB, Waclawiw MA, Popma JJ, Yu ZX, Finkel T, Epstein SE: Association between prior cytomegalovirus infection and the risk of restenosis after coronary atherectomy. N Engl J Med 1996, 335: 624-630.
Melnick JL, Adam E, Debakey ME: Cytomegalovirus and atherosclerosis. Eur Heart J 1993,14(Suppl K):30-38.
Harkins L, Volk AL, Samanta M, Mikolaenko I, Britt WJ, Bland KI, Cobbs CS: Specific localisation of human cytomegalovirus nucleic acids and proteins in human colorectal cancer. Lancet 2002, 360: 1557-1563.
Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, Nabors LB, Cobbs CG, Britt WJ: Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res 2002, 62: 3347-3350.
Melnychuk RM, Smith P, Kreklywich CN, Ruchti F, Vomaske J, Hall L, Loh L, Nelson JA, Orloff SL, Streblow DN: Mouse cytomegalovirus M33 is necessary and sufficient in virus-induced vascular smooth muscle cell migration. J Virol 2005, 79: 10788-10795.
Gruijthuijsen YK, Casarosa P, Kaptein SJ, Broers JL, Leurs R, Bruggeman CA, Smit MJ, Vink C: The rat cytomegalovirus R33-encoded G protein-coupled receptor signals in a constitutive fashion. J Virol 2002, 76: 1328-1338.
Casarosa P, Gruijthuijsen YK, Michel D, Beisser PS, Holl J, Fitzsimons CP, Verzijl D, Bruggeman CA, Mertens T, Leurs R, Vink C, Smit MJ: Constitutive signaling of the human cytomegalovirus-encoded receptor UL33 differs from that of its rat cytomegalovirus homolog R33 by promiscuous activation of G proteins of the Gq, Gi, and Gs classes. J Biol Chem 2003, 278: 50010-50023.
Sherrill JD, Miller WE: G protein-coupled receptor (GPCR) kinase 2 regulates agonist-independent Gq/11 signaling from the mouse cytomegalovirus GPCR M33. J Biol Chem 2006, 281: 39796-39805.
Margulies BJ, Browne H, Gibson W: Identification of the human cytomegalovirus G protein-coupled receptor homologue encoded by UL33 in infected cells and enveloped virus particles. Virology 1996, 225: 111-125.
Davis-Poynter NJ, Lynch DM, Vally H, Shellam GR, Rawlinson WD, Barrell BG, Farrell HE: Identification and characterization of a G protein-coupled receptor homolog encoded by murine cytomegalovirus. J Virol 1997, 71: 1521-1529.
Beisser PS, Vink C, Van Dam JG, Grauls G, Vanherle SJ, Bruggeman CA: The R33 G protein-coupled receptor gene of rat cytomegalovirus plays an essential role in the pathogenesis of viral infection. J Virol 1998, 72: 2352-2363.
Case R, Sharp E, Benned-Jensen T, Rosenkilde MM, Davis-Poynter N, Farrell HE: Functional analysis of the murine cytomegalovirus chemokine receptor homologue M33: ablation of constitutive signaling is associated with an attenuated phenotype in vivo. J Virol 2008, 82: 1884-1898.
Streblow DN, Kreklywich CN, Smith P, Soule JL, Meyer C, Yin M, Beisser P, Vink C, Nelson JA, Orloff SL: Rat cytomegalovirus-accelerated transplant vascular sclerosis is reduced with mutation of the chemokine-receptor R33. Am J Transplant 2005, 5: 436-442.
Oliveira SA, Shenk TE: Murine cytomegalovirus M78 protein, a G protein-coupled receptor homologue, is a constituent of the virion and facilitates accumulation of immediate-early viral mRNA. Proc Natl Acad Sci USA 2001, 98: 3237-3242.
Beisser PS, Grauls G, Bruggeman CA, Vink C: Deletion of the R78 G protein-coupled receptor gene from rat cytomegalovirus results in an attenuated, syncytium-inducing mutant strain. J Virol 1999, 73: 7218-7230.
Kaptein SJ, Beisser PS, Gruijthuijsen YK, Savelkouls KG, van Cleef KW, Beuken E, Grauls GE, Bruggeman CA, Vink C: The rat cytomegalovirus R78 G protein-coupled receptor gene is required for production of infectious virus in the spleen. J Gen Virol 2003, 84: 2517-2530.
Michel D, Milotic I, Wagner M, Vaida B, Holl J, Ansorge R, Mertens T: The human cytomegalovirus UL78 gene is highly conserved among clinical isolates, but is dispensable for replication in fibroblasts and a renal artery organ-culture system. J Gen Virol 2005, 86: 297-306.
Seet BT, McFadden G: Viral chemokine-binding proteins. J Leukoc Biol 2002, 72: 24-34.
Lalani AS, McFadden G: Secreted poxvirus chemokine binding proteins. J Leukoc Biol 1997, 62: 570-576.
Wang D, Bresnahan W, Shenk T: Human cytomegalovirus encodes a highly specific RANTES decoy receptor. Proc Natl Acad Sci USA 2004, 101: 16642-16647.
O'Brien V: Viruses and apoptosis. J Gen Virol 1998,79(Pt 8):1833-1845.
Vaux DL, Korsmeyer SJ: Cell death in development. Cell 1999, 96: 245-254.
Mocarski ES Jr: Immunomodulation by cytomegaloviruses: manipulative strategies beyond evasion. Trends Microbiol 2002, 10: 332-339.
McCormick AL: Control of apoptosis by human cytomegalovirus. Curr Top Microbiol Immunol 2008, 325: 281-295.
Goldmacher VS: Cell death suppression by cytomegaloviruses. Apoptosis 2005, 10: 251-265.
Goldmacher VS: vMIA, a viral inhibitor of apoptosis targeting mitochondria. Biochimie 2002, 84: 177-185.
McCormick AL, Meiering CD, Smith GB, Mocarski ES: Mitochondrial cell death suppressors carried by human and murine cytomegalovirus confer resistance to proteasome inhibitor-induced apoptosis. J Virol 2005, 79: 12205-12217.
Goldmacher VS, Bartle LM, Skaletskaya A, Dionne CA, Kedersha NL, Vater CA, Han JW, Lutz RJ, Watanabe S, Cahir McFarland ED, Kieff ED, Mocarski ES, Chittenden T: A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc Natl Acad Sci USA 1999, 96: 12536-12541.
Skaletskaya A, Bartle LM, Chittenden T, McCormick AL, Mocarski ES, Goldmacher VS: A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc Natl Acad Sci USA 2001, 98: 7829-7834.
Reboredo M, Greaves RF, Hahn G: Human cytomegalovirus proteins encoded by UL37 exon 1 protect infected fibroblasts against virus-induced apoptosis and are required for efficient virus replication. J Gen Virol 2004, 85: 3555-3567.
Mavinakere MS, Williamson CD, Goldmacher VS, Colberg-Poley AM: Processing of human cytomegalovirus UL37 mutant glycoproteins in the endoplasmic reticulum lumen prior to mitochondrial importation. J Virol 2006, 80: 6771-6783.
Mavinakere MS, Colberg-Poley AM: Dual targeting of the human cytomegalovirus UL37 exon 1 protein during permissive infection. J Gen Virol 2004, 85: 323-329.
Bozidis P, Williamson CD, Colberg-Poley AM: Mitochondrial and secretory human cytomegalovirus UL37 proteins traffic into mitochondrion-associated membranes of human cells. J Virol 2008, 82: 2715-2726.
Hayajneh WA, Colberg-Poley AM, Skaletskaya A, Bartle LM, Lesperance MM, Contopoulos-Ioannidis DG, Kedersha NL, Goldmacher VS: The sequence and antiapoptotic functional domains of the human cytomegalovirus UL37 exon 1 immediate early protein are conserved in multiple primary strains. Virology 2001, 279: 233-240.
Sharon-Friling R, Goodhouse J, Colberg-Poley AM, Shenk T: Human cytomegalovirus pUL37x1 induces the release of endoplasmic reticulum calcium stores. Proc Natl Acad Sci USA 2006, 103: 19117-19122.
Arnoult D, Bartle LM, Skaletskaya A, Poncet D, Zamzami N, Park PU, Sharpe J, Youle RJ, Goldmacher VS: Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc Natl Acad Sci USA 2004, 101: 7988-7993.
Poncet D, Larochette N, Pauleau AL, Boya P, Jalil AA, Cartron PF, Vallette F, Schnebelen C, Bartle LM, Skaletskaya A, Boutolleau D, Martinou JC, Goldmacher VS, Kroemer G, Zamzami N: An anti-apoptotic viral protein that recruits Bax to mitochondria. J Biol Chem 2004, 279: 22605-22614.
Pauleau AL, Larochette N, Giordanetto F, Scholz SR, Poncet D, Zamzami N, Goldmacher VS, Kroemer G: Structure-function analysis of the interaction between Bax and the cytomegalovirus-encoded protein vMIA. Oncogene 2007, 26: 7067-7080.
Smith GB, Mocarski ES: Contribution of GADD45 family members to cell death suppression by cellular Bcl-xL and cytomegalovirus vMIA. J Virol 2005, 79: 14923-14932.
McCormick AL, Roback L, Mocarski ES: HtrA2/Omi terminates cytomegalovirus infection and is controlled by the viral mitochondrial inhibitor of apoptosis (vMIA). PLoS Pathog 2008, 4: e1000063.
McCormick AL, Skaletskaya A, Barry PA, Mocarski ES, Goldmacher VS: Differential function and expression of the viral inhibitor of caspase 8-induced apoptosis (vICA) and the viral mitochondria-localized inhibitor of apoptosis (vMIA) cell death suppressors conserved in primate and rodent cytomegaloviruses. Virology 2003, 316: 221-233.
Norris KL, Youle RJ: Cytomegalovirus proteins vMIA and m38.5 link mitochondrial morphogenesis to Bcl-2 family proteins. J Virol 2008,82(13):6232-6243.
Arnoult D: Apoptosis-associated mitochondrial outer membrane permeabilization assays. Methods 2008, 44: 229-234.
Patterson CE, Shenk T: Human cytomegalovirus UL36 protein is dispensable for viral replication in cultured cells. J Virol 1999, 73: 7126-7131.
Andoniou CE, Degli-Esposti MA: Insights into the mechanisms of CMV-mediated interference with cellular apoptosis. Immunol Cell Biol 2006, 84: 99-106.
Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schroter M, Scaffidi C, Krammer PH, Peter ME, Tschopp J: Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 1997, 386: 517-521.
Smith JA, Pari GS: Expression of human cytomegalovirus UL36 and UL37 genes is required for viral DNA replication. J Virol 1995, 69: 1925-1931.
Liu Y, Biegalke BJ: Characterization of a cluster of late genes of guinea pig cytomegalovirus. Virus Genes 2001, 23: 247-256.
Menard C, Wagner M, Ruzsics Z, Holak K, Brune W, Campbell AE, Koszinowski UH: Role of murine cytomegalovirus US22 gene family members in replication in macrophages. J Virol 2003, 77: 5557-5570.
Cicin-Sain L, Ruzsics Z, Podlech J, Bubic I, Menard C, Jonjic S, Reddehase MJ, Koszinowski UH: Dominant-negative FADD rescues the in vivo fitness of a cytomegalovirus lacking an antiapoptotic viral gene. J Virol 2008, 82: 2056-2064.
Lembo D, Gribaudo G, Hofer A, Riera L, Cornaglia M, Mondo A, Angeretti A, Gariglio M, Thelander L, Landolfo S: Expression of an altered ribonucleotide reductase activity associated with the replication of murine cytomegalovirus in quiescent fibroblasts. J Virol 2000, 74: 11557-11565.
Lembo D, Donalisio M, Hofer A, Cornaglia M, Brune W, Koszinowski U, Thelander L, Landolfo S: The ribonucleotide reductase R1 homolog of murine cytomegalovirus is not a functional enzyme subunit but is required for pathogenesis. J Virol 2004, 78: 4278-4288.
Brune W, Menard C, Heesemann J, Koszinowski UH: A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science 2001, 291: 303-305.
Upton JW, Kaiser WJ, Mocarski ES: Cytomegalovirus M45 cell death suppression requires RHIM-dependent interaction with receptor-interacting protein 1 (RIP1). J Biol Chem 2008.
Mack C, Sickmann A, Lembo D, Brune W: Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1-interacting protein. Proc Natl Acad Sci USA 2008, 105: 3094-3099.
Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J: Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 2000, 1: 489-495.
Wiertz EJ, Devlin R, Collins HL, Ressing ME: Herpesvirus interference with major histocompatibility complex class II-restricted T-cell activation. J Virol 2007, 81: 4389-4396.
Alcami A, Koszinowski UH: Viral mechanisms of immune evasion. Trends Microbiol 2000, 8: 410-418.
Hengel H, Reusch U, Gutermann A, Ziegler H, Jonjic S, Lucin P, Koszinowski UH: Cytomegaloviral control of MHC class I function in the mouse. Immunol Rev 1999, 168: 167-176.
Khan N, Hislop A, Gudgeon N, Cobbold M, Khanna R, Nayak L, Rickinson AB, Moss PA: Herpesvirus-specific CD8 T cell immunity in old age: cytomegalovirus impairs the response to a coresident EBV infection. J Immunol 2004, 173: 7481-7489.
Trzonkowski P, Mysliwska J, Szmit E, Wieckiewicz J, Lukaszuk K, Brydak LB, Machala M, Mysliwski A: Association between cytomegalovirus infection, enhanced proinflammatory response and low level of anti-hemagglutinins during the anti-influenza vaccination – an impact of immunosenescence. Vaccine 2003, 21: 3826-3836.
Jones TR, Hanson LK, Sun L, Slater JS, Stenberg RM, Campbell AE: Multiple independent loci within the human cytomegalovirus unique short region down-regulate expression of major histocompatibility complex class I heavy chains. J Virol 1995, 69: 4830-4841.
del Val M, Hengel H, Hacker H, Hartlaub U, Ruppert T, Lucin P, Koszinowski UH: Cytomegalovirus prevents antigen presentation by blocking the transport of peptide-loaded major histocompatibility complex class I molecules into the medial-Golgi compartment. J Exp Med 1992, 176: 729-738.
Thrower AR, Bullock GC, Bissell JE, Stinski MF: Regulation of a human cytomegalovirus immediate-early gene (US3) by a silencer-enhancer combination. J Virol 1996, 70: 91-100.
Greijer AE, Verschuuren EA, Dekkers CA, Adriaanse HM, Bij W, The TH, Middeldorp JM: Expression dynamics of human cytomegalovirus immune evasion genes US3, US6, and US11 in the blood of lung transplant recipients. J Infect Dis 2001, 184: 247-255.
Jones TR, Wiertz EJ, Sun L, Fish KN, Nelson JA, Ploegh HL: Human cytomegalovirus US3 impairs transport and maturation of major histocompatibility complex class I heavy chains. Proc Natl Acad Sci USA 1996, 93: 11327-11333.
Ahn K, Angulo A, Ghazal P, Peterson PA, Yang Y, Fruh K: Human cytomegalovirus inhibits antigen presentation by a sequential multistep process. Proc Natl Acad Sci USA 1996, 93: 10990-10995.
Jun Y, Kim E, Jin M, Sung HC, Han H, Geraghty DE, Ahn K: Human cytomegalovirus gene products US3 and US6 down-regulate trophoblast class I MHC molecules. J Immunol 2000, 164: 805-811.
Gruhler A, Peterson PA, Fruh K: Human cytomegalovirus immediate early glycoprotein US3 retains MHC class I molecules by transient association. Traffic 2000, 1: 318-325.
Hegde NR, Tomazin RA, Wisner TW, Dunn C, Boname JM, Lewinsohn DM, Johnson DC: Inhibition of HLA-DR assembly, transport, and loading by human cytomegalovirus glycoprotein US3: a novel mechanism for evading major histocompatibility complex class II antigen presentation. J Virol 2002, 76: 10929-10941.
Lee S, Yoon J, Park B, Jun Y, Jin M, Sung HC, Kim IH, Kang S, Choi EJ, Ahn BY, Ahn K: Structural and functional dissection of human cytomegalovirus US3 in binding major histocompatibility complex class I molecules. J Virol 2000, 74: 11262-11269.
Zhao Y, Biegalke BJ: Functional analysis of the human cytomegalovirus immune evasion protein, pUS3(22kDa). Virology 2003, 315: 353-361.
Lee S, Park B, Ahn K: Determinant for endoplasmic reticulum retention in the luminal domain of the human cytomegalovirus US3 glycoprotein. J Virol 2003, 77: 2147-2156.
Misaghi S, Sun ZY, Stern P, Gaudet R, Wagner G, Ploegh H: Structural and functional analysis of human cytomegalovirus US3 protein. J Virol 2004, 78: 413-423.
Park B, Kim Y, Shin J, Lee S, Cho K, Fruh K, Ahn K: Human cytomegalovirus inhibits tapasin-dependent peptide loading and optimization of the MHC class I peptide cargo for immune evasion. Immunity 2004, 20: 71-85.
Shin J, Park B, Lee S, Kim Y, Biegalke BJ, Kang S, Ahn K: A short isoform of human cytomegalovirus US3 functions as a dominant negative inhibitor of the full-length form. J Virol 2006, 80: 5397-5404.
Wiertz EJ, Tortorella D, Bogyo M, Yu J, Mothes W, Jones TR, Rapoport TA, Ploegh HL: Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 1996, 384: 432-438.
Wiertz EJ, Jones TR, Sun L, Bogyo M, Geuze HJ, Ploegh HL: The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 1996, 84: 769-779.
Jones TR, Sun L: Human cytomegalovirus US2 destabilizes major histocompatibility complex class I heavy chains. J Virol 1997, 71: 2970-2979.
Rehm A, Engelsberg A, Tortorella D, Korner IJ, Lehmann I, Ploegh HL, Hopken UE: Human cytomegalovirus gene products US2 and US11 differ in their ability to attack major histocompatibility class I heavy chains in dendritic cells. J Virol 2002, 76: 5043-5050.
Kikkert M, Hassink G, Barel M, Hirsch C, Wal FJ, Wiertz E: Ubiquitination is essential for human cytomegalovirus US11-mediated dislocation of MHC class I molecules from the endoplasmic reticulum to the cytosol. Biochem J 2001, 358: 369-377.
Hassink GC, Barel MT, Van Voorden SB, Kikkert M, Wiertz EJ: Ubiquitination of MHC class I heavy chains is essential for dislocation by human cytomegalovirus-encoded US2 but not US11. J Biol Chem 2006, 281: 30063-30071.
Shamu CE, Story CM, Rapoport TA, Ploegh HL: The pathway of US11-dependent degradation of MHC class I heavy chains involves a ubiquitin-conjugated intermediate. J Cell Biol 1999, 147: 45-58.
Flierman D, Coleman CS, Pickart CM, Rapoport TA, Chau V: E2-25K mediates US11-triggered retro-translocation of MHC class I heavy chains in a permeabilized cell system. Proc Natl Acad Sci USA 2006, 103: 11589-11594.
Shamu CE, Flierman D, Ploegh HL, Rapoport TA, Chau V: Polyubiquitination is required for US11-dependent movement of MHC class I heavy chain from endoplasmic reticulum into cytosol. Mol Biol Cell 2001, 12: 2546-2555.
Tirosh B, Iwakoshi NN, Lilley BN, Lee AH, Glimcher LH, Ploegh HL: Human cytomegalovirus protein US11 provokes an unfolded protein response that may facilitate the degradation of class I major histocompatibility complex products. J Virol 2005, 79: 2768-2779.
Chevalier MS, Daniels GM, Johnson DC: Binding of human cytomegalovirus US2 to major histocompatibility complex class I and II proteins is not sufficient for their degradation. J Virol 2002, 76: 8265-8275.
Barel MT, Hassink GC, van Voorden S, Wiertz EJ: Human cytomegalovirus-encoded US2 and US11 target unassembled MHC class I heavy chains for degradation. Mol Immunol 2006, 43: 1258-1266.
Oresic K, Tortorella D: Endoplasmic reticulum chaperones participate in human cytomegalovirus US2-mediated degradation of class I major histocompatibility complex molecules. J Gen Virol 2008, 89: 1122-1130.
Lilley BN, Ploegh HL: A membrane protein required for dislocation of misfolded proteins from the ER. Nature 2004, 429: 834-840.
Barel MT, Ressing M, Pizzato N, van Leeuwen D, Le Bouteiller P, Lenfant F, Wiertz EJ: Human cytomegalovirus-encoded US2 differentially affects surface expression of MHC class I locus products and targets membrane-bound, but not soluble HLA-G1 for degradation. J Immunol 2003, 171: 6757-6765.
Schust DJ, Tortorella D, Seebach J, Phan C, Ploegh HL: Trophoblast class I major histocompatibility complex (MHC) products are resistant to rapid degradation imposed by the human cytomegalovirus (HCMV) gene products US2 and US11. J Exp Med 1998, 188: 497-503.
Machold RP, Wiertz EJ, Jones TR, Ploegh HL: The HCMV gene products US11 and US2 differ in their ability to attack allelic forms of murine major histocompatibility complex (MHC) class I heavy chains. J Exp Med 1997, 185: 363-366.
Furman MH, Ploegh HL, Tortorella D: Membrane-specific, host-derived factors are required for US2- and US11-mediated degradation of major histocompatibility complex class I molecules. J Biol Chem 2002, 277: 3258-3267.
Gewurz BE, Gaudet R, Tortorella D, Wang EW, Ploegh HL, Wiley DC: Antigen presentation subverted: Structure of the human cytomegalovirus protein US2 bound to the class I molecule HLA-A2. Proc Natl Acad Sci USA 2001, 98: 6794-6799.
Gewurz BE, Wang EW, Tortorella D, Schust DJ, Ploegh HL: Human cytomegalovirus US2 endoplasmic reticulum-lumenal domain dictates association with major histocompatibility complex class I in a locus-specific manner. J Virol 2001, 75: 5197-5204.
Story CM, Furman MH, Ploegh HL: The cytosolic tail of class I MHC heavy chain is required for its dislocation by the human cytomegalovirus US2 and US11 gene products. Proc Natl Acad Sci USA 1999, 96: 8516-8521.
Thilo C, Berglund P, Applequist SE, Yewdell JW, Ljunggren HG, Achour A: Dissection of the interaction of the human cytomegalovirus-derived US2 protein with major histocompatibility complex class I molecules: prominent role of a single arginine residue in human leukocyte antigen-A2. J Biol Chem 2006, 281: 8950-8957.
Barel MT, Pizzato N, van Leeuwen D, Bouteiller PL, Wiertz EJ, Lenfant F: Amino acid composition of alpha1/alpha2 domains and cytoplasmic tail of MHC class I molecules determine their susceptibility to human cytomegalovirus US11-mediated down-regulation. Eur J Immunol 2003, 33: 1707-1716.
Lee SO, Hwang S, Park J, Park B, Jin BS, Lee S, Kim E, Cho S, Kim Y, Cho K, Shin J, Ahn K: Functional dissection of HCMV US11 in mediating the degradation of MHC class I molecules. Biochem Biophys Res Commun 2005, 330: 1262-1267.
Hengel H, Koopmann JO, Flohr T, Muranyi W, Goulmy E, Hammerling GJ, Koszinowski UH, Momburg F: A viral ER-resident glycoprotein inactivates the MHC-encoded peptide transporter. Immunity 1997, 6: 623-632.
Ahn K, Gruhler A, Galocha B, Jones TR, Wiertz EJ, Ploegh HL, Peterson PA, Yang Y, Fruh K: The ER-luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity 1997, 6: 613-621.
Ahn JH, Hayward GS: The major immediate-early proteins IE1 and IE2 of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies at very early times in infected permissive cells. J Virol 1997, 71: 4599-4613.
Halenius A, Momburg F, Reinhard H, Bauer D, Lobigs M, Hengel H: Physical and functional interactions of the cytomegalovirus US6 glycoprotein with the transporter associated with antigen processing. J Biol Chem 2006, 281: 5383-5390.
Kyritsis C, Gorbulev S, Hutschenreiter S, Pawlitschko K, Abele R, Tampe R: Molecular mechanism and structural aspects of transporter associated with antigen processing inhibition by the cytomegalovirus protein US6. J Biol Chem 2001, 276: 48031-48039.
Dugan GE, Hewitt EW: Structural and Functional Dissection of the Human Cytomegalovirus Immune Evasion Protein US6. J Virol 2008, 82: 3271-3282.
Hewitt EW, Gupta SS, Lehner PJ: The human cytomegalovirus gene product US6 inhibits ATP binding by TAP. EMBO J 2001, 20: 387-396.
Hengel H, Flohr T, Hammerling GJ, Koszinowski UH, Momburg F: Human cytomegalovirus inhibits peptide translocation into the endoplasmic reticulum for MHC class I assembly. J Gen Virol 1996,77(Pt 9):2287-2296.
Llano M, Guma M, Ortega M, Angulo A, Lopez-Botet M: Differential effects of US2, US6 and US11 human cytomegalovirus proteins on HLA class Ia and HLA-E expression: impact on target susceptibility to NK cell subsets. Eur J Immunol 2003, 33: 2744-2754.
Tirabassi RS, Ploegh HL: The human cytomegalovirus US8 glycoprotein binds to major histocompatibility complex class I products. J Virol 2002, 76: 6832-6835.
Furman MH, Dey N, Tortorella D, Ploegh HL: The human cytomegalovirus US10 gene product delays trafficking of major histocompatibility complex class I molecules. J Virol 2002, 76: 11753-11756.
Hansen SG, Strelow LI, Franchi DC, Anders DG, Wong SW: Complete sequence and genomic analysis of rhesus cytomegalovirus. J Virol 2003, 77: 6620-6636.
Pande NT, Powers C, Ahn K, Fruh K: Rhesus cytomegalovirus contains functional homologues of US2, US3, US6, and US11. J Virol 2005, 79: 5786-5798.
Powers CJ, Fruh K: Signal peptide-dependent inhibition of MHC class I heavy chain translation by rhesus cytomegalovirus. PLoS Pathog 2008, 4: e1000150.
Ziegler H, Thale R, Lucin P, Muranyi W, Flohr T, Hengel H, Farrell H, Rawlinson W, Koszinowski UH: A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity 1997, 6: 57-66.
Reusch U, Muranyi W, Lucin P, Burgert HG, Hengel H, Koszinowski UH: A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J 1999, 18: 1081-1091.
Krmpotic A, Messerle M, Crnkovic-Mertens I, Polic B, Jonjic S, Koszinowski UH: The immunoevasive function encoded by the mouse cytomegalovirus gene m152 protects the virus against T cell control in vivo. J Exp Med 1999, 190: 1285-1296.
Holtappels R, Gillert-Marien D, Thomas D, Podlech J, Deegen P, Herter S, Oehrlein-Karpi SA, Strand D, Wagner M, Reddehase MJ: Cytomegalovirus encodes a positive regulator of antigen presentation. J Virol 2006, 80: 7613-7624.
Kavanagh DG, Gold MC, Wagner M, Koszinowski UH, Hill AB: The multiple immune-evasion genes of murine cytomegalovirus are not redundant: m4 and m152 inhibit antigen presentation in a complementary and cooperative fashion. J Exp Med 2001, 194: 967-978.
Wagner M, Gutermann A, Podlech J, Reddehase MJ, Koszinowski UH: Major histocompatibility complex class I allele-specific cooperative and competitive interactions between immune evasion proteins of cytomegalovirus. J Exp Med 2002, 196: 805-816.
LoPiccolo DM, Gold MC, Kavanagh DG, Wagner M, Koszinowski UH, Hill AB: Effective inhibition of K(b)- and D(b)-restricted antigen presentation in primary macrophages by murine cytomegalovirus. J Virol 2003, 77: 301-308.
Hengel H, Reusch U, Geginat G, Holtappels R, Ruppert T, Hellebrand E, Koszinowski UH: Macrophages escape inhibition of major histocompatibility complex class I-dependent antigen presentation by cytomegalovirus. J Virol 2000, 74: 7861-7868.
Gold MC, Munks MW, Wagner M, Koszinowski UH, Hill AB, Fling SP: The murine cytomegalovirus immunomodulatory gene m152 prevents recognition of infected cells by M45-specific CTL but does not alter the immunodominance of the M45-specific CD8 T cell response in vivo. J Immunol 2002, 169: 359-365.
Pinto AK, Munks MW, Koszinowski UH, Hill AB: Coordinated function of murine cytomegalovirus genes completely inhibits CTL lysis. J Immunol 2006, 177: 3225-3234.
Munks MW, Pinto AK, Doom CM, Hill AB: Viral interference with antigen presentation does not alter acute or chronic CD8 T cell immunodominance in murine cytomegalovirus infection. J Immunol 2007, 178: 7235-7241.
Bohm V, Podlech J, Thomas D, Deegen P, Pahl-Seibert MF, Lemmermann NA, Grzimek NK, Oehrlein-Karpi SA, Reddehase MJ, Holtappels R: Epitope-specific in vivo protection against cytomegalovirus disease by CD8 T cells in the murine model of preemptive immunotherapy. Med Microbiol Immunol 2008, 197: 135-144.
Kleijnen MF, Huppa JB, Lucin P, Mukherjee S, Farrell H, Campbell AE, Koszinowski UH, Hill AB, Ploegh HL: A mouse cytomegalovirus glycoprotein, gp34, forms a complex with folded class I MHC molecules in the ER which is not retained but is transported to the cell surface. EMBO J 1997, 16: 685-694.
Lu X, Kavanagh DG, Hill AB: Cellular and molecular requirements for association of the murine cytomegalovirus protein m4/gp34 with major histocompatibility complex class I molecules. J Virol 2006, 80: 6048-6055.
Odeberg J, Soderberg-Naucler C: Reduced expression of HLA class II molecules and Iinterleukin-10- and transforming growth factor beta1-independent suppression of T-cell proliferation in human cytomegalovirus-infected macrophage cultures. J Virol 2001, 75: 5174-5181.
Tomazin R, Boname J, Hegde NR, Lewinsohn DM, Altschuler Y, Jones TR, Cresswell P, Nelson JA, Riddell SR, Johnson DC: Cytomegalovirus US2 destroys two components of the MHC class II pathway, preventing recognition by CD4+ T cells. Nat Med 1999, 5: 1039-1043.
Hegde NR, Johnson DC: Human cytomegalovirus US2 causes similar effects on both major histocompatibility complex class I and II proteins in epithelial and glial cells. J Virol 2003, 77: 9287-9294.
Chevalier MS, Johnson DC: Human cytomegalovirus US3 chimeras containing US2 cytosolic residues acquire major histocompatibility class I and II protein degradation properties. J Virol 2003, 77: 4731-4738.
Odeberg J, Plachter B, Branden L, Soderberg-Naucler C: Human cytomegalovirus protein pp65 mediates accumulation of HLA-DR in lysosomes and destruction of the HLA-DR alpha-chain. Blood 2003, 101: 4870-4877.
Heise MT, Connick M, Virgin HWt: Murine cytomegalovirus inhibits interferon gamma-induced antigen presentation to CD4 T cells by macrophages via regulation of expression of major histocompatibility complex class II-associated genes. J Exp Med 1998, 187: 1037-1046.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MMK carried out the literature review and wrote this manuscript. TES conceived of the review and edited the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12985_2008_518_MOESM1_ESM.doc
Additional file 1: Cytomegalovirus modulation of the innate immune response. A listing of the cytomegalovirus encoded proteins that interfere with the innate immune response. (DOC 124 KB)
12985_2008_518_MOESM2_ESM.doc
Additional file 2: CMV Modulation of the adaptive immune response. A listing of the cytomegalovirus encoded proteins that interfere with the adaptive immune response. (DOC 64 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Miller-Kittrell, M., Sparer, T.E. Feeling manipulated: cytomegalovirus immune manipulation. Virol J 6, 4 (2009). https://doi.org/10.1186/1743-422X-6-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-6-4