
Abstract
Background
In Europe, the north-south downhill cline frequency of the chemokine receptor CCR5 allele with a 32-bp deletion (CCR5-Δ32) raises interesting questions for evolutionary biologists. We had suggested first that, in the past, the European colonizers, principally Romans, might have been instrumental of a progressively decrease of the frequencies southwards. Indeed, statistical analyses suggested strong negative correlations between the allele frequency and historical parameters including the colonization dates by Mediterranean civilisations. The gene flows from colonizers to native populations were extremely low but colonizers are responsible of the spread of several diseases suggesting that the dissemination of parasites in naive populations could have induced a breakdown rupture of the fragile pathocenosis changing the balance among diseases. The new equilibrium state has been reached through a negative selection of the null allele.
Results
Most of the human diseases are zoonoses and cat might have been instrumental in the decrease of the allele frequency, because its diffusion through Europe was a gradual process, due principally to Romans; and that several cat zoonoses could be transmitted to man. The possible implication of a feline lentivirus (FIV) which does not use CCR5 as co-receptor is discussed. This virus can infect primate cells in vitro and induces clinical signs in macaque. Moreover, most of the historical regions with null or low frequency of CCR5-Δ32 allele coincide with historical range of the wild felid species which harbor species-specific FIVs.
Conclusion
We proposed the hypothesis that the actual European CCR5 allelic frequencies are the result of a negative selection due to a disease spreading. A cat zoonosis, could be the most plausible hypothesis. Future studies could provide if CCR5 can play an antimicrobial role in FIV pathogenesis. Moreover, studies of ancient DNA could provide more evidences regarding the implications of zoonoses in the actual CCR5-Δ32 distribution.
Similar content being viewed by others

Background
As infection is the greatest killer in human history [1], the strongest evidence for selection in the human genome has been obtained for genes involved in immune defense, including those which encode receptors. One of the most-celebrated examples of adaptive selection is the 32-bp coding sequence deletion, CCR5-Δ32, of the chemokine receptor CCR5. This is probably the more recent and complete example of a gene studied from clinical, epidemiological and evolutionary genetics. CCR5 function as co-receptors for the cell entry of HIV-1 and the deletion which leads to a frame shift and generates an inactive CCR5 receptor. Homozygosity for the CCR5-Δ32 allele confers almost complete, mendelian resistance to R5-tropic HIV-1 while HIV-infected individuals heterozygous for this allele were delayed in progression to AIDS [2, 3].
The CCR5-Δ32 allele is mainly present in Europeans (10% on average) and the allele frequency exhibits a north-south cline with frequencies ranging from 16% in Northern Europe to 4% or less in Greece and in most of the Mediterranean islands (Figure 1A and [4, 5]). The broadest area of high frequency is located in North-Eastern Europe, particularly in the Baltic and White Sea regions. From these maximum, the frequency gradually decreases in all directions across Europe [4]; however, some additional peaks of frequency are found in France or Russian areas [4, 6–8]. Moreover, Ashkenazi Jews have high frequencies of CCR5-Δ32, but this is likely due to founder effects unique to their history rather than the general process of dispersal that spread the allele in other populations [9]. Outside Europe, the mutation can be found at low frequencies in neighbouring regions (North Africa, Middle East, Central Asia); it is absent in Sub-Saharan Africa, East and South-East Asia and in indigenous populations of America and Oceania (Figure 1A).

Geographic distribution of the CCR5-Δ32 allele (A) compared with historical range of felids carrying species-specific FIVs (B). In (A), only the frequencies of Native populations have been evidenced in America, Asia, Africa and Oceania. Map redrawn and modified from [4, 5]. In (B), the black areas correspond to the range of wild individuals bearing species-specific FIVs in a given continent, America: bobcat, jaguarundi, ocelot and puma; Asia: Pallas cat; Africa: cheetah, leopard and lion. The pale grey areas correspond to the range where individuals of these species have been found seronegative or when their serological status is unknown in a given continent (Asia: cheetah, leopard and lion; Europe: leopard and lion). Areas where these last three species lived in sympatry with Pallas cat are in dark grey. The historical ranges are approximate by 500 BC for Europe, North Africa and Western Asia; since the European settlement in America, and during the 1500's to the beginning of the 1900's in the remainder of Africa, Asia and Oceania. These data were principally inferred from [65–71].
Because the AIDS pandemic is too recent to change allele frequencies, other infectious diseases have been suggested as the agent causing the selection of the null allele increase, such as resistance to plague and smallpox infections [10]. However, analyses of Scandinavian Mesolithic DNA which have pushed the date of the first occurrence back to around 5000 BC [11] and genomic analyses [12] have weakened the evidence for recent selection of the null allele. Due to the north-south spatial gradient, it has been proposed that the actual allele distribution could be explained by migrations of Northern populations. As suggested by Lucotte [13] in its seminal article in the field and by Balanovsky et al. [4] Vikings and Uralic speaking people, respectively, could have brought the deletion in some Southern populations. Moreover, these migrations and/or gene flow cannot explain, according to us, the whole of the European allele frequency distribution. Also, we have proposed an alternative hypothesis in which the actual allele frequency distribution might not be due to the genes spreading, but to a negative selection resulting in the spread of pathogens principally during principally Roman expansion [5]. This hypothesis is supported by several facts.
The idea that bottlenecks and founder effects could lead to an increase in damaging alleles in human populations was historically reserved for isolated populations that experienced severe founder effects (for example, Ashkenazi Jews [14] and Finns [15]). However, recently signs of a population bottleneck in variability data obtained for a number of genomic loci in European populations were described and also led to the conclusion that a severe bottleneck occurred after the appearance of the anatomically modern human in Africa, and thus presumably during, or after, the emigration out of Africa [16–18] and references therein). Moreover, the earlier European population of hunter-gatherers could suffer severe bottlenecks during the latest ice age (Pleistocene) [19]. As there is strong evidence for the unitary origin of the CCR5-Δ32 mutation [20, 21], the null allele could have been already present in the ancestors of the European populations (in spite of their present language differences) at a relatively high frequency, probably >10% as suggested by analysis of ancient DNA from Bronze age [22] and Neolithic [11], similarly to many other polymorphisms found in Europeans but not in the other populations [23].
Previous statistical analyses showed strong negative correlations in Europe between the allele frequency and two historical parameters, i.e. the first colonization dates by the great ancient Mediterranean civilisations, and the distances from the frontiers of the Roman Empire in its greatest expansion [5]. However, the possible decrease of the ancestral CCR5-Δ32 allele frequency was not due directly to the colonizers, as the gene flows to European native populations were extremely low [19]. This suggests that the role of colonizers were indirect. As evolutionary biologists have shown several evidences that infectious diseases, as a leading cause of human morbidity and mortality, have exerted important selective forces on our genomes [24, 25], the cause of the decrease of the CCR5-Δ32 allele frequency in Southern European populations is probably due to infectious agent(s). It has been suggested that the most important infectious diseases of modern food-producing human populations also include diseases that could have emerged only within the past 11,000 years, following the rise of agriculture [1, 25, 26]. The second great historical transitions occurred when great ancient conquering Eurasian civilizations (such as the Greek and Roman empires) came into military and commercial contact, ca. 3000–2000 years ago, swapping their dominant infections [27]. It is either a human disease or a zoonosis transmittable to humans. Moreover, studies on the West Nile virus have shown that host genetic factors are highly pathogen-specific and can therefore be beneficial in one context and harmful in another [28]. Which agree that the possible decrease of the CCR5-Δ32 allele frequency in the South of the Europe could be due to parasites. The introduction of parasites in naive colonized populations could have induced a breakdown of the pathocenosis and a new equilibrium has been reached through a decrease of the CCR5-Δ32 allele frequency. The theoretical framework of pathocenosis, first coined by Grmek [29, 30]) and developed by Biraben [31], offers a synthetic approach to the history of disease. Drawing on the concept of biocenosis, Grmek defines pathocenosis as "the ensemble of pathological states present in a specific population at a given moment in time" and suggests that "the frequency and overall distribution of each disease depends on the frequency and distribution of all the other diseases within a given population". The concept of pathocenosis attempts to offer a synthetic view of disease ecology, which, in our context is defined as all interdependences within pathogens, their hosts (including their genetic responses) and their environment.
The aim of this article is to critically discuss the possible nature of this (or these) parasite(s) responsible of the decrease of the CCR5-Δ32 allele frequency in the Southern European populations.
Results
Putative role of cats as host-parasite
Previous, statistical analyses suggested a decrease of the ancestral CCR5-Δ32 allele frequency in European population due principally to Roman expansion [5]. However, this negative selection was not directly due to the military or colonisation spreads, as the gene flows from colonisators to European native populations were too low [19]. Moreover, statistical analyses suggested that factor(s) responsible for the decrease of null allele frequency had partly diffused beyond the borders of the Roman Empire [5]. The diffusion of one or more factor(s) excludes the role of climatical changes, the change in allele frequency could be due to the spread of human or animal parasites that affect human populations.
More than any other civilizations, the Romans have created links between Mediterranean basin and Western and Central Europe and the great routes of infectious diseases went straight through it [32]. Not only did the first great historical pestilences pass through the Empire, but also the slow insidious penetration of endemic disease (like tuberculosis, leprosy and malaria) has invaded Europe [30]. Moreover, conquerors and invading armies brought also with them insect and rodent vectors that could introduce or sustain infectious diseases in nonendemic European areas. As, to our knowledge, no known human diseases could explain the decrease of the null allele in Europe, zoonoses might be implicated. Indeed, most of the infectious diseases affecting human populations are considered zoonotic in origin [33–35]. Close contact with animals is a risk for humans to acquire infectious diseases and it is well known that the domestication of animals has facilitated the passage of animals parasites to human [36, 37]. Many of the major human infectious diseases, including some now confined to humans and absent from animals, have arisen only after the origins of agriculture (11,000 years BP) [1, 25, 26]. The five animal species (cow, sheep, goat, pig and dog) which have had probably the most epidemic impact on the human populations are explicitly named the Pandora's pentad [38]. Moreover, few tropical but many temperate diseases arose from domestic animals, because these live mainly in the temperate zones, and their concentration there was formerly even more lopsided [35]. In Europe, the Romans were the cause of some permanent changes in the distribution of birds and beasts [39]; several animals, such as cat, donkey, mule and pheasant have been voluntarily introduced throughout Europe [40] and others involuntarily, such as malaria vector mosquito species [30]. If we consider that the most impact on the decrease of the CCR5-Δ32 allele frequency could be principally due to Roman expansion, according to us, among all the domestic animals, cat could be the best candidate. Indeed, once the cat had arrived in Rome; this animal would have spread throughout Europe, quite likely as a camp follower and companion to the constantly travelling Roman armies. Moreover, several parasitic, bacterial and viral zoonoses diagnosed in cats could be transmitted to man [41, 42]. To support this view, before investigate the type of disease which could be transmitted, the major steps of the spread of the domestic cat in Europe are summarized.
Cat's origin is yet little uncertain; however, several analysis revealed that cats were domesticated in the Near East; wildcats of Near East (F. s. lybica) are the closest group to all domestic cats [43, 44], and likely coincide with agricultural village development in the Fertile Crescent. This is congruent with archaeological studies, the earliest evidence of cat-human association involves their co-occurrence in Cyprus deposits aged at 9,500 years ago [45]. Similarly, in all the other islands of the Mediterranean Basin far beyond continent (Sardinia, Corsica and Crete), felids originated from African or Middle East wildcats which were voluntarily introduced by Neolithic navigators about 6,000–8,000 years ago [46–50]. Interestingly, the populations of these areas have the lower level of CCR5-Δ32 allele frequency (references therein [5]. The earliest records of probably tamed or domestic cats in continental Europe would be in Greece by 1000 BC; however, at that time, cats were very extremely rare until 6th–5th centuries BC [51–53]. In the Italian Peninsula, first historical evidence of tamed or early-domesticated cats was found on archaeological sites from the beginning of the 5th–4th centuries BC [50, 54]. Interestingly, in numerous parts of the Roman Empire, generally the oldest remains of the domestic cat (for example in Belgium, Netherlands, Hungary and Switzerland) dated to the Roman period [55–59]; moreover, remains of cats have been found in many of the Roman settlements excavated extensively suggesting that the spread of domesticated cats throughout continental Europe and Great Britain is principally due to Romans [40, 60]. Moreover, as contrarily to Asia, Africa and America, there was no tameable felid in the Northern Mediterranean countries, therefore numerous substitutes have been found by the European populations, principally Mustelidae species [48, 56, 61, 62].
World repartition of FIV-infected felids and their relationship with humans
If we hypothesize that a cat zoonosis might be transmitted to human, the corresponding infectious agent could also affect other felid species. Among, all the cat zoonoses, according us, only one parasite distribution could be correlated to those of the CCR5-Δ32 allele frequency. The corresponding infectious agent is the Feline immunodeficiency virus (FIV) which can also infect primate cells in vitro and induce clinical signs in a primate [63, 64] and references therein). Indeed, historical regions with null or low frequency of CCR5-Δ32 allele coincide with historical range of the wild felid species [65–71] which harbor species-specific FIVs (Figure 1). The two maps do not correspond perfectly, and we can only conclude that these patterns are not inconsistent with the hypothesis that allele frequency and the old presence FIV-infected felids are causally related. However, as developed below, bibliographical analyses provide several arguments in favour of this hypothesis.
FIV, as Human immunodeficiency virus (HIV) and Simian immunodeficiency virus (SIV) belong to the Lentivirus genus of the Retroviridae (reviewed in [72]). In domestic cat, FIV infection results in disease progression and outcome similar to that of HIV in humans, and offers a natural model to AIDS [73, 74]. Other felid species which are infected with FIV seem not to develop AIDS-like disease [75, 76]. However, both captive and/or wild FIV-infected lions (Panthera leo) and pumas (Puma concolor) exhibited mild to severe CD4+ T-cell depletion and some other clinical health consequences [77–80]. These findings raise the prospect that FIV is not completely benign in these species, but rather suppress host immune response and may increase the incidence of opportunistic infections or even spontaneous cancers as AIDS does in humans.
The extant felids have arisen from a common ancestor in Asia 10.8 MYA during the Miocene. The 37 felid species form eight distinct evolutionary lineages that have successfully inhabited all continents except Oceania and Antarctica through a series of migrations likely facilitated by sea-level oscillations [81]. Among the Felidae, at least 11 free ranging Felidae species harbor FIV antibodies and FIV viral genomes (Table 1). Moreover, nine of these species (lion, cheetah, leopard, Pallas cat, jaguarundi, ocelot, domestic cat, puma, and bobcat) have been shown to harbor species-specific FIVs by evaluation of complete or partial viral genomic sequences (Table 1 and [74, 82, 83]). However, the seroprevalence of FIVs varies dramatically by species and geographic areas. African lion and leopard, puma and Pallas cat populations demonstrate very high rates of seropositivity. The seroprevalence of FIV infections in natural settings is nearly 100% in Serengeti African lions and pumas of Wyoming and Montana, respectively [84–86]. In contrast, significant numbers of free-ranging lions in Namibia or from Asia were all seronegative [86, 87]. The absence of FIV-Ple in Namibia is puzzling, but may be explained by the low density of lions in this African area [88]. Moreover, several Asian lions held in captivity were noted to be 75% FIV seropositive, demonstrating that lions of Asian origin are not intrinsically resistant to infection [89]. Interestingly, a similar geographic dispersal of seropositivity was noted for Asian versus African leopards; i.e., free-ranging African populations demonstrate seropositivity of >25%, whereas Asian-born animals are seronegative [90, 91]. More than 50% of Pallas cats (Manul) tested harboured anti-FIV antibodies [91]. Other species, including the domestic cat, cheetah, and South American Neotropical free-ranging felid populations, tend to demonstrate seroprevalence rates of 10% or less. Asian species other than the Pallas cat are apparently not infected with an endemic FIV, although when during captivity, Asian felid individuals are exposed to other species harbouring FIVs, these animals may become infected (Table 1 and [74, 91]). It must be noted that a species-specific FIV-related virus has also been found in Hyaenidae, which belong to the Feloidea superfamily [91, 92].
As already shown by several authors [74, 91, 93], the FIV phylogenies does not exactly mirror that of its feline host species (Figure 2). However, the relative differences in genetic diversity among FIV strains be interpreted in the context of the evolutionary and phylogeographical history of each host species. Indeed, in spite of that free-ranging individuals of many species harbor monophyletic, species-specific strain(s) of FIV, viruses isolated from different species seem to group more by geographic region of the host than in groupings concordant with the phylogenetic relationships of host species. Moreover, molecular analyses failed to resolve the origin domestic cat FIV strains as has been already shown by other studies [74, 83]. The pattern of the strains infecting domesticated cat (FIV-Fca) which exhibit three monophyletic clades may due rapid viral diversification within the domestic cat world-wide due to the great number of individuals (some estimates put the domestic house cat population at 60 million and the feral cat population at the same number, that's 120 million animals) and to the trans-continental travels and traffics. The extreme divergence between the two highly FIV-Pco clades and the six FIV-Ple clades suggest an ancient origin of FIV infection of respectively, puma and lion [88]. Concerning FIV-Pco, this could be a consequence of two separate introductions of FIV within puma populations [83], whereas for African lion virus, each clades correspond with distinct geographic areas of endemicity [88]. The strains infecting cheetah (FIV-Aju) and leopard (FIV-Ppa) are closely related, in spite the fact that their hosts have evolved in distinctly different felid lineages, puma and cheetah are closely related, belonging to the puma linage, while lions and leopards are members of the Panthera lineage [81]. Moreover, cheetah and leopard could be sympatric; all these data suggest recent inter-species transmission. Due to the date of expansion of cheetah throughout Africa, the FIV-Aju emergence may have occurred within the last 10,000 years, perhaps acquired from leopards [93]. FIV-Oma is found in wild populations of the Eurasian Pallas cat [91], a species that arose during the late Pleistocene [81]. The monophyletic lineage of Pallas cat FIV-Oma and African lion FIV-Ple observed here could suggest more ancient inter-species transmission as the last time lions and Pallas cats were in geographic contact was during the Pleistocene when lion ranges spread throughout Asia, providing a possible opportunity for FIV transmission between these species [93]. In addition, FIV-Ccr occurs in spotted hyena, a species from the Hyaenidae family within carnivores that co-exist in the same habitats as most African felid species, affording opportunities for cross-species transmission. Interestingly, as already shown by Pecon Slattery et al. [93], all the FIV strains which infected Afro-Asian Feloidea constitute a monophyletic group. This grouping could suggest a common origin or/and old cross transmissions, in spite that in Asia, no wild seropositive individuals have been found in cheetahs, lions, leopards and hyenas which have a large Afro-Asian repartition [94]. Moreover, the geographic partitioning reflected in the amino acid phylogeny suggests evidence for an Old world/New world split (Figure 2 and [74, 91]). Lastly, similarly to the cheetah/leopard case, two American species that evolved in distinctly different felid lineages (ocelot and jaguarondi), which have almost identical distribution, are infected with closely related viruses (FIV-Lpa and FIV-Hya, respectively) suggesting recent inter-species transmission. However, with few exceptions, the strong monophyletic origin of each species-specific strain suggests that FIV has rarely undergone effective transmission between species. In addition, the monophyly of FIV sequences within each species suggests that, in most cases, FIV has been successfully introduced once and adapted, expanded, and evolved within the host.

Viral-host co-evolution. The tree on the left shows observed viral sequence relationships [82, 91] and references therein) and the tree on the right represents host species relationships [81]. FIV polymerase sequences (158 amino acids included in analysis) were analyzed phylogenetically from nine feline species representing six out of the eight feline lineages [81]. Asterisks indicate significant bootstrap values (≥ 70%). The branch lengths are not in scale. Numbers next to node define estimated time of divergence for each the felid lineages and for the Felidae/Hyaenidae split in million years.
The precise origin of FIV emergence into Felidae is not easily discerned by viral phylogenetic analyses due to its recent and rapid evolution, and to cross-transmissions. According to Pecon-Slattery et al. [93], the widespread occurrence of FIV combined with large interspecies divergence in Africa would suggest that FIV arose in Africa rather than Asia. Moreover, an African origin of all lentiviruses may be posited, indeed, Simian lentiviruses are endemic in Africa infecting over 36 species of primates [95], and caprine arthritis-encephalitis virus (CAEV), bovine immune deficiency virus (BIV) and visna are present in Africa ungulate species [96] and references therein]). Moreover, the substantial genetic difference observed among FIV lineages in Africa is consistent with a long residence time within these species, and suggest global dissemination of FIV from Africa during felid transcontinental migrations into Eurasia and the Americas [81]. Moreover, the near absence of FIV in Asian species (except for the Mongolian Pallas cats) suggests that the virus did not originate along with ancestral felids of Asia which exclude that the FIV might have arisen in Asia along with the progenitor of modern felids 10.8 MYA. In addition, FIV related strains infect African feline species and the spotted hyena; however, FIV phylogenetic analyses do not support an ancient introduction of this virus to the Felidae and Hyaenidae (i.e., prior to the Felidae/Hyaenidae, using fossil, split is dated at about 47 million years ago) [97] but more probably, a recent African crossspecies transmissions. Lastly, the presence of FIV in both old and new world felids suggests that the current viruses may have descended from transmission events that occurred the last time felid species crossed the Bering Straits in the late Pleistocene (>12,000 years ago [81]), or earlier. By contrast, like the recent emergence of HIV in humans, domestic cat lentiviral infections are relatively new diseases, with more limited distribution and lower seroprevalence than infections noted in lions and pumas [74]. The domestic cat evolved as a unique felid lineage only around 10,000 year ago [45] from subspecies of wildcat Felis silvestris inhabiting Near East Asia [43]. Seroprevalence studies, suggest that FIV is present in nearly all of the close relatives of domestic cat (Felis genus [81]) including European wildcat F. s. silvestris [91, 98, 99]. However, concerning European wildcat, it is due to recent cross transmissions from feral or domestic infected cats. In Europe, hybridization between domestic cats and wildcats are well known [50, 100, 101], showing evidence that contacts between wild and domestic cats are not rare.
As FIV-infected wild felids are present in most of the world countries since at least the end of the last glaciation, it could be interesting to analyse the historical relationship between human and felids in relation with their serological status (Table 1). The exact history of human interaction with felids is still somewhat vague; however, as wild felid species are found in all parts of the world, except Greenland, Australia and Antarctica, suggesting that contacts between men and felids were probably very numerous during the last millennia. In spite that archaeological and historical records are sketchy, there are several evidence that throughout history people have had close relationships with felids. Moreover, given that, the single domestication event within the Felidae, apart from these modern hybrids, might suggest that this group is behaviourally poorly preadapted for domestication; it is all the more surprising that in a wide variety of cultures, over many centuries, particular felid species have been "tamed" as domestic pets. In addition, tamed felids have possibly lived in association with humans far earlier than archaeological and historical records imply. A comportemental study has evidenced that numerous species of small cats have an important preadaptation to domestication [102].
As summarized in the Table 1, in Afro-Asia, numerous felid species can be tamed including the four species with specific FIV. Cheetahs, which have been considered the easiest of the exotic cats to tame, have been tamed by several ancient Afro-Asian civilizations since 2500 to 5000 years ago [40, 65, 68, 103, 104]. Lions and leopards have been tamed since the beginning of Egyptian history (2800–2650 BC) [105, 106]. Tigers were a popular animal in aristocratic collections in Asia for centuries [65],). Servals and caracals have been tamed in Egypt since at least at the 15th century AD [106] and several centuries later, caracal have been trained for hunting in Asia [105, 107–109]. The earliest remains of cats in domestic or tamed contexts from Egypt date from about 4000 to 3000 BC; moreover, archaeological remains of F. chaus and F. s. lybica have been found [52, 110]. Pallas Cats (F. manul) have been reports of this cat being kept in a semi-domestic state in Central Asia [111]. More surprisingly, concerning an Afro-Asian non felid feliformia, there is evidence from paintings and bas-reliefs in tombs that in ancient Egypt striped hyenas were tamed and kept as pets, as well as being artificially fattened as food or for medical use [112, 113].
In pre-Columbian times, relatively few animals were domesticated, and almost none of them extended beyond the geographic limits of their wild ancestors. However, jaguarondi and Geoffroy's cats have been partially domesticated as a rodents-catcher [114], and other American felids which are relatively easily tamed, like ocelot, margay, and puma have interacted with humans [65, 115, 116]. In summary, if except bobcats (however, young bobcats can be somewhat tamed), all the other American species bearing specific FIV have had closed relationships with natives [91, 117].
This bibliographic analysis suggests that both in Afro-Asia and in America, numerous people could have been in contact with FIV. However, the principal criticism could be that most of the contacts with felids have restraints to wealthy people. If it is partially true for big cats as lion, leopard, puma and cheetah, but this is not the case for Pallas's cat, Geoffrey's cat and jaguarondi. In addition, four species (cheetah, leopard, lion and spotty hyena) with specific FIV were formerly widespread throughout western Asia and Africa. To date, none wild individuals of these species have been seropositive in Asia; however, at least four empires (Egyptian, Hyksosian, Achaemenian and Greek) have been on two continents, facilitating animal trade across the Sinai Peninsula and importation of African felids in Asian countries and vice-versa.
Moreover, concerning early European contacts with FIV-infected felids, the Romans displayed lions, tigers, leopards, cheetahs and other felids in menageries, pageants and arena combats [118], most of them having been caught in Africa and southwest Asia [53], but they were rarely tamed [106]. In the Roman Empire there were many amphitheatres, e.g. in the second century AD there were more than a hundred amphitheatres in Italy and a similar number in the rest of Europe [119]. In addition, there were similar numbers of circuses. The Romans systematically collected animals for display, entertainment and slaughter in arenas, theatres and amphitheatres throughout the Empire [120]. Even if the spectacles staged in Rome did not have an equivalent importance elsewhere in the Empire, in the arenas of this large city a great number of felids were massacred. For example, the dictator Sulla (93 B.C.) exhibited lions in the Rome's arena; in 55 B.C. under Pompey's reign on two occasions 500 and 410 leopards fought against Gaetulians armed with darts; in 46 B.C. Julius Caesar had 400 lions imported primarily from North Africa; and after Trajan's victory over the Dacians the games continued for 123 successive days when 11,000 animals were killed in the arena [120–124]. Caretakers could be bitten by these felids; moreover, captive felids could infect domestic cats and vice-versa, cross-species FIV transmission involving captive felids are well documented [74]. In addition, similarly to Simian retrovirus infections [125–128], human could be infected during hunting or cutting up, most of the felid species having always been very exploited for their pelts.
In summary, with exception of Oceania, historical regions with low or null frequency of CCR5-Δ32 allele coincide with historical range of the wild felid species which harbor species-specific FIVs (Figure 1B). Among these nine felid species, four of them have the largest distributions of the members of this family. Leopards have the largest distribution of any felid and were found from South Africa across that continent to the Middle East, Java, and northward to Siberia. According to historical records, lion populations have been distributed in Middle East to India and in Africa except in desert and rainforest habitats. The distribution of cheetahs was almost identical to that of lions, except that they have not been found in Europe, but that they were distributed in semi-deserts. Historically, pumas were found from the boreal forests of northern Canada to the tip of South America. Among the four other felid species, the Pallas' cats inhabited from the Caspian Sea area to parts of Western China through Southern Asia. In nearly half of its distribution range, they were sympatric with lions, cheetahs and/or leopards. The bobcat formerly ranged from southern Canada throughout most of the United States, south to central Mexico. The distribution of the ocelot was almost identical to that of jaguarondi; they were found from Arizona and south west Texas through Central America to South America except in high mountains or plateaux and in the extreme southern cone beyond approximately 45° latitude. In the past, lions and leopards lived in Balkans, but they were not numerous in the historical time and the last specimens became extinct about 2500–2000 years ago [129, 130]. In Europe, only two species (Eurasian and Iberian lynx) and one subspecies (Eurasian wildcat) of wild felids live since historical times, and their seropositive level is null or very low and probably due to recent contamination by domestic cat [86, 98, 99].
Discussion
Previous analyses suggested that in Europe the CCR5-Δ32 allele frequency is negatively correlated with colonization by ancient Mediterranean civilizations principally Romans [5]. We have the hypothesis that a zoonosis could have played a role in the decrease of the mutation frequency or in the absence of maintenance of the null allele if it would have appeared. As the cat spread throughout Europe is principally due to Romans, a cat zoonosis could be involved. Interestingly, to the exclusion of Oceania, in the countries in which FIV infected felids are found, the lower CCR5-Δ32 allele frequency is found in native human populations. Further bibliographic analyses are needed in order to know if FIV could infect human and also if the CCR5-Δ32 mutation can be unfavourable.
Could FIVs infect humans?
More than half of the 1407 human pathogens are zoonotic [131] and recent epidemics such as HIV and severe acute respiratory syndrome (SARS) have changed the view we had about emerging infectious diseases; these epidemics showed evidences that animal reservoirs are important sources of new infectious threats to humans. Contacts between humans and animals are a crucial rate-limiting step in this process, although data describing the variables that influence animal-to-human transmission are relatively scarce. Therefore, a brief analysis of the data supporting cross-species transmissions of Simian retrovirus to humans can be instructive. Data on SIV/HIV dramatize this point; scientists now theorize that SIVs were transmitted from primates to humans on several occasions [132–138]. Although HIV causes AIDS in humans, SIV does not cause any disease in its natural hosts. However, it is not known exactly how HIV first entered the human population [139], eating raw monkey meat, drinking monkey blood, or perhaps through another method of direct exposure to monkey bodily fluids have been suggested as a possible source and remains the best candidate so far [125, 134]. These hypotheses are supported by the fact that primate handlers and those who hunt and butcher "bushmeat" (the meat of wild animals that includes chimpanzees, gorilla and other monkeys) have detectable humoral and cell-mediated immunity to SIV. There are at least eight documented incidents of zoonotic transfer of SIV to humans [137] and two laboratory workers have been accidentally infected by SIV, one infection was cleared and the second (a human infection with SIVsmB670), caused a persistent asymptomatic infection [140–142].
In addition, the family of SIV is 1 out of 5 primate borne retroviruses known to infect humans. Simian (spumaretro-) foamy viral (SFV) infection, probably acquired through bites, has also been reported in 1 to 5% of persons occupationally exposed to non-human primates in zoos, primate centers and laboratories, mainly in Northern America but also in Europe (reviewed in [143]). Recently, naturally acquired SFV infections have been described in 1% of hunters living in Cameroon, Central Africa [125] and in one person with frequent contacts with Macaca fascicularis in a Indonesian temple [144]. In Cameroon, more than 60% of the population is directly exposed to fresh nonhuman primate blood and bodily fluids from hunting, butchering or petting [125, 126]. Moreover, it has been recently demonstrated efficient transmission of SFVs to humans in natural settings in Central Africa, specifically following ape bites, and viral persistence in the human host [145]. There is currently no evidence of human-to-human transmission of SFV; however, only a few cases (n = 6) have had a short clinical follow-up [146–149]. Simian T-cell lymphotropic viruses (STLVs), enzootic in both Asian and African Old World monkeys and apes, may have repeatedly crossed the species barrier, the close relation between human and great ape primate T lymphotropic virus type 1 (PTLV-1) strains in Africa is suggestive of zoonosis [126, 127], which might result from hunting and slaughter activities. In addition, serologic studies have demonstrated evidence of primate-to-human transmission of simian type D retrovirus (SRV), a retrovirus enzootic among Old World monkeys, in laboratory workers exposed to captive primates [150]. To date, no disease has been linked to human infection with this retrovirus.
To date, concerning the FIV, for which the host is phylogenetically more distant to human than monkeys, there is no evidence that it can infect or cause disease in humans. Researchers and veterinarians who have been bitten by FIV positive cats have been consistently tested negative for FIV [151]. However, FIV infection was assessed solely by serological tests, confirmation of direct exposure to the virus was limited, and prolonged periods between potential exposure and assessment of infection existed. FIV-specific antibodies were not detected in the cynomolgus macaques (Macaca fascicularis), in which FIV infection cause clinical signs, including depletion of CD4+ cells and weight loss, which are consistent with FIV infection; moreover, FIV genes expression has been found in necropsied tissues [63]. As the most obvious effects of FIV infection in macaques were observed early after exposure, the lack of serum detection suggests that seroconversion is not indicative of prior exposure to the virus. In addition, even if FIV is antigenically distinct from the primate lentiviruses, it shares many biological properties that manifest in its ability to infect productively both primary and immortalized primate cell lines in vitro [64, 152–161]. In addition, a FIV strain which cannot naturally infect primate cells, when forced, preferred human cells to monkey cells [161] and the restricting effect of the host factor TRIM5α is fairly substantial in macaque cells, but is rather mild in human cells [162, 163]. However, the ability of FIV to express its LTR in primate cells seems to vary depending upon the viral strain, the experimental protocol, and the cell line used. Most of the restriction to expression seems to be due to limitations imposed by promoter sequences residing within the U3 region of FIV LTR [158, 159]. Once this restriction is overcome, FIV is able to express in a wide variety of cell types [64]. Moreover, it is likely that the determinants of feline cell tropism, such as envelope-mediated entry of target cells may also influence infection of primate cells by FIV, which must find cells that express the right combination of receptors and co-receptors [161]. While the chemokine receptor CXCR4 as an entry receptor and the tumor necrosis factor receptor CD134 have been well established as essential for FIVfca receptor-mediated cell entry, the receptor interactions of puma and lion FIVs are not identified, but in some cases appear to involve other cell surface determinants [73, 164–170]. Moreover, a puma FIV isolate targeted gastrointestinal peripheral lymphoid tissues or other sites in a domestic cat infection model [171].
The use of CXCR4 and CD134 as receptors is compatible with our hypothesis, as well as, analogous to primate lentivirus receptor usage, the predominant FIVfca quasispecies changes during the course of FIV infection, in that isolates from terminally infected animals have been reported to be CD134 independent [168]. However, to date, there are no firm data to support a role for CCR5 in infections of feline cells [63, 160, 172], but a FIV strain could use human CCR5 to infect some human cells [161], nevertheless, this could be the result of a recent shift in coreceptor usage. In another hand, it has been reported that env deletion mutants of FIV have adapted to replicate in human cells [159]. Moreover, the increased cell death that preceded a loss of infectious FIV in infected human peripheral blood mononuclear cells supports previous findings that infection of human cells by FIV is cytopathic, which is probably due to the expression of FIV envelope glycoproteins [158]. FIV infection of relatively few cells in culture has been associated with increased cytotoxicity in feline cultures due to the release of cytotoxic molecules [173, 174], which is similar to reports of other lentiviruses. Hence, it is conceivable that FIV-mediated cytotoxicity may limit the number of infected and potentially infectable cells leading to the loss of detectable FIV DNA in infected human cultures. So, even if there were not a true infection, a high rate of cellular death and/or an immunological depletion could be deleterious although the infection appeared to be clinically silent. Wolfe et al. [35] have delineated five stages in the transformation of an animal pathogen into a specialized pathogen of humans. According to these last authors, the present hypothesis of human infection by FIV would correspond to the stage 2: a pathogen of animals that, under natural conditions, has been transmitted from animals to humans ("primary infection") but has not been transmitted between humans ("secondary infection").
If the cause of the change of CCR5-Δ32 allele frequency was FIV infection, the characteristics of the virus that was present 2000–3000 years ago are unknown, especially since recombinations and cross-species transmissions have been shown for this virus. Discordant env phylogeny between FIV Ple subtypes reveals ancestral FIV recombination events in the wild [88]. It is probable, as with primate lentiviruses [74, 175], that recombination plays a significant role in the evolution of FIV and that different evolutionary patterns would be seen within different viral regions. Although cross-species transmissions have been rare, they likely did occur in the past to produce a pattern of viral evolution in felids that does not completely match the evolution of the Felidae. One of the best examples is the position of hyena FIV-Ccr within felid FIV suggests increased opportunities for inter-species transmission due to a greater elapsed time since the virus entered and disseminated in African felids. Finally, there are now several examples of modern inter-species transmissions (Figure 1 and [82, 91, 176, 177]). However, while there is one case of a free-ranging leopard cat that acquired FIVfca from a domestic cat [177], most cross-species transmissions of FIV have been documented in captive settings.
In natural settings there are substantial behavioural and ecological barriers to cross-species transmission of FIV, a pathogen requiring direct contact for infection to occur. The major mode of transmission for FIV in domestic cats is believed to be biting, although vertical transmission can also occur [178]. If we hypothesize that the FIV infected cats before their domestication, this suggests, after this last event, frequent transmissions of FIV by biting from cat to human. Even if it is speculative, several forms of infections could occur and it is important to underline that during Antiquity, the bodies of colonized people faced greater danger from infections new to their immune systems and that numerous infectious diseases have profoundly affected human populations. Infections could induce fever, this might pass unnoticed and moreover, several prolonged or not fevers occurred relatively frequently during Antiquity [179] and still today, fevers of unknown origin are numerous and several of them are probably zoonoses [131]. Moreover, even if the virus cannot infect productively human cells, it could induce cell death. The in vitro lytic properties of this virus in monkey and human cells suggest possible biological abnormalities associated with human FIV infection. Moreover, infections usually benign alone could have more severe effects on people which were co-infected by several epidemic or endemic pathogenous agents.
Moreover, cat zoonoses can be transmitted to man [41, 42] and the hypothesis of the role of FIV remains putative. However, the implication of a feline retrovirus could be plausible; indeed, three other species of feline retroviruses, feline foamy virus (FeFV), feline sarcoma virus (FeSV) and Feline leukemia virus (FeLV) can replicate in some human cell cultures with generally production of infectious virus and could sometimes produce morphological cell change [151, 180–192]. Moreover, cat horizontal transmission of FeLV by cat fleas has also been demonstrated [193] and FeSV can also induce malignant tumours in non felid mammalian including monkeys [194]. To date, there has been no evidence of infection of feline retrovirus in humans so far. However, all these reports suggest that numerous cat pathogen agents could have played a role in the putative decrease of the null allele frequency.
Could the null allele be unfavourable?
As other receptors for inflammatory chemokines, CCR5 contribute to leukocyte recruitment in a number of inflammatory diseases (reviewed in [195]). However, owing to the redundancy of the chemokine system, CCR5 could only play a modest role, and blocking CCR5 was predicted to be safe because individuals lacking CCR5 develop normally and seem healthy. Nevertheless, over the years, the CCR5-Δ32 allele has been linked, using epidemiologic studies, with several non infectious human diseases, including multiple sclerosis and schizophrenia [196–198] but the associations have generally been weak or inconsistent between these studies. In another hand, in mouse models of infection, CCR5 has been implicated in host defense against Influenza A virus, Listeria, Trypanosoma cruzi, Toxoplasma gondii, Cryptococcus neoformans and Chlamydia trachomatis [199–205]. In humans, similarly to HIV-1, CCR5-Δ32 carriers also have a decreased likelihood of contracting hepatitis B virus [206], but these carriers improved outcomes during hepatitis C virus infection [207] and tick-borne encephalitis virus infections (TBEV) [208]. Moreover, it has been reported that CCR5-Δ32 homozygosis was strongly associated with symptomatic West Nile virus (WNV) infection [28, 209], consistent with a previous finding that CCR5 was a crucial antiviral and survival factor in WNV infection in mice [210]. WNV and TBEV are members of the same family (Flaviviridae) and share certain similarities between them. Interestingly, like most of the infectious agents, flavivirus and influenza viruses are endemic in several tropical and subtropical regions and probably CCR5 is implicated in the defense against several other tropical viruses; this could perhaps explain why the CCR5-Δ32 allele frequency is relatively weak in these areas, even if this or another null mutation has arisen, they could be rapidly under selected. If our hypothesis is correct, this could explain the quasi-null allele frequency in Australia [211], in spite that the Aborigines have not been in contact with felids during approximately 50,000 years [212]. In the context of infectious diseases, CCR5 comprises positive and negative elements that ultimately contribute to the evolution of the gene over time. In flavivirus infections and putatively ancient cat zoonosis pathogenesis, CCR5 is antimicrobial, whereas in HIV pathogenesis, CCR5 is promicrobial.
Can archaeologists excavate evidence of cats' role in the human CCR5-Δ32 allele frequency?
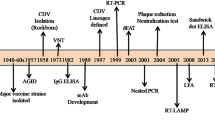
Future studies on ancient DNA will confirm or reject our hypothesis which include a great CCR5-Δ32 allele frequency in the ancient European population, followed by a progressively decrease of the frequency southwards due indirectly to Romans and other colonizers which have helped spread a possible cat zoonosis to native populations. These future analyses could also give data for characterisation of ancient European pathocenosis compositions including the genetic responses and changes to epidemic and endemic diseases. Indeed, whereas evolutionary information derived from present-day DNA sequences is, by necessity, indirect, ancient DNA sequences provide a direct view of past genetic variants and infectious agents. Moreover, technical advances in DNA extraction, multiplex DNA amplification and high-throughput sequencing have recently opened new horizons in ancient genomics (references there in [213]), and studies to elucidate the genetic basis of the environmental adaptations of the human ancestors, compared to humans today is now possible. The presence and frequencies of the CCR5-Δ32 variant in past human populations has been studied by several authors. The results of these studies have argue against the possibility that plague was a major selective force that caused a rapid increase in CCR5-Δ32 gene frequencies within European populations [22, 214] and have pushed the dating of the CCR5-Δ32 allele back to around 5000 BC [11].
Moreover, sequencing of complete genome of Homo sapiens neanderthalensis is underway [215, 216] and could give interesting data concerning the origin of the null allele. Indeed, as Neanderthals are the extinct hominid species most closely related to contemporary humans, the continuation of the Neanderthal genome project provides a unique opportunity to identify genetic changes that are specific to modern humans [215]. Dating such genomic events would help to interpret these changes mechanistically. In addition to the different methods of age estimation based on allele frequencies and sequence comparison between species, conclusive data from the analysis of prehistoric remains of members of the genus Homo (e.g. from humans and Neanderthals) would help to date such events by determining the presence and frequency of genomic variants. Moreover, Currat and Excoffier [217] using a method, which assumed environmental homogeneity, have simulated the range expansion of modern humans into Europe under realistic demographic scenarios to investigate potential admixture between colonizing humans and resident Neanderthals. Their simulations indicated that even with only a few admixture events, the contribution of Neanderthal genes to the current human gene pool should be large because new genes (which have a Neanderthal origin) have a high probability of persistence when entering a progressively expanding (modern human) population compared with those entering a stationary population. In a recent review, Hodgson and Disotell [218] have concluded that "it seems unlikely that Neanderthals contributed any substantial fraction of modern variation and it remains to be seen whether any adaptive alleles crossed the human-Neanderthal species boundary". Moreover, more recent major events in human evolution, such as the re-colonization of northern latitudes after the Ice Ages, could also be taken into account.
In addition, the analysis of the DNA of ancient micro-organisms in archaeological and palaeontological human and animals remains can contribute to the understanding of issues as different as the spreading of a new disease. The molecular resolution of extinct species' genomes raises the hope of discovering infectious agents and pathogens that might have played a regulatory role in historic ecosystems. Potentials, and sometimes pitfalls, of this research field are illustrated by the results of the various research works performed on ancient DNA. For example, DNA of bacteria of the genus Bartonella responsible of chronic bacteremia and which have mammalian reservoirs including cats has been detected in a human and a cat who lived respectively 4000 and 800 years ago [219, 220]. Moreover, the finding of ancient human T cell leukemia virus type I (HTLV-1) long terminal repeat (LTR) DNA sequences in association with a 1500-year-old Chilean mummy [221, 222], even if it has stirred vigorous debate shows that ancient provirus sequences will become available in the future. Cumulative research on felid natural history, evolution, phylogeography and ancient DNA analyses will provide important context for FIV emergence. Ancient DNAs from felids are useful not only to phylogenetic analysis but also to population genetic approaches that may increase our understanding of the incipient extinction of modern species [71, 223, 224]. Moreover, the potential role of extinct felids, such as the saber-tooth species, which co-existed with modern felids until around the end of the Pleistocene [69] in FIV origin and its dissemination could be known.
Conclusion
In this study, we have proposed the hypothesis that in Europe, the actual European CCR5 allelic frequencies are the result of a negative selection due to a disease spreading (ostensibly by the Roman Empire or some other colonizers). A cat zoonosis could be the most plausible hypothesis and even if it is speculative, the implication of FIV added to possible deleterious effects of the null allele mutation has been suggested. Future studies will prove or dismiss if in FIV pathogenesis, CCR5 can play an antimicrobial role. Moreover, this study shows that in the future all pieces of the puzzle could be put together to see the whole picture of the CCR5-Δ32 allele evolution.
Bibliographical analysis shows evidence that species-specificity of FIV might be less stringent than previously considered. The abundance of studies demonstrating the capacity of viruses, including retrovirus, to cross species raises questions about ongoing transmissions and renders the study of the adaptations required for viruses to be transmitted from one host species to another increasingly relevant. In addition, although bibliographical analysis shows that the FIV has the ability to infect primate cells in vivo, it is not our intent to suggest that FIV represents a health hazard. However, the apparent lack of pathogenicity of FIV infection in humans, which is still based on a limited number of cases, contrasts strongly with the in vitro lytic properties of these viruses in primate cells. Moreover, as the analyses concern only healthy persons this induces an important bias. Although the risks for human are considered extremely small, from a public health perspective it is often recommended that immunosuppressed people should have limited contact with infected cats. FIV infection in immunocompromised persons, especially those with HIV infection, could also heighten public health concerns because such coinfection is probable during cohabitation with infected pets.
In addition, scientific evidence for the ancient spread of a resistance allele or a pathogenic agent could become available through research on ancient DNA and this research field could be determinant in the comprehension of the interrelations with human genome, pathogenic agents and their hosts in the last millennia. Recent advances in ancient-DNA extraction have made it possible to retrieve substantial amounts of ancient DNA sequences from at least Pleistocene remains in order to analyse the pathocenoses and the corresponding genetic responses. As most of the human diseases are zoonoses, analyses of human and animal remains must be made in conjunction.
This study shows also evidence that only an integrated multidisciplinary approach has enabled us to understand the evolutionary history of the CCR5-Δ32 allele.
Methods
Data sources
We have compiled bibliographical data concerning the past distribution of felids which are now infected by species-specific pathogenic agents. Species descriptions and all references are in Table 1.
Sequence analyses
All the FIV Pol protein sequences have been extracted from GenBank. These sequences have been aligned with the BioEdit software [225]. Phylogenetic analyses were performed using the Neighbor-Joining (NJ) method [226] in PHYLIP version 3.6 alpha 3 [227] accessed at http://bioinfo.hku.hk/services/menuserv.html. Robustness of nodes was estimated by running a bootstrap test with 100 replicates.
References
Diamond J: Guns, Germs, and Steel: the Fates of Human Societies. New York: Norton; 1997.
Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, Saragosti S, Lapoumeroulie C, Cognaux J, Forceille C, Muyldermans G, Verhofstede C, Burtonboy G, Georges M, Imai T, Rana S, Yi Y, Smyth RJ, Collman RG, Doms RW, Vassart G, Parmentier M: Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996, 382: 722-725. 10.1038/382722a0
Arenzana-Seisdedos F, Parmentier M: Genetics of resistance to HIV infection: role of co-receptors and co-receptor ligands. Semin Immunol 2006, 18: 387-403. 10.1016/j.smim.2006.07.007
Balanovsky O, Pocheshkhova E, Pshenichnov A, Solovieva D, Kuznetsova M, Voronko O, Churnosov M, Tegako O, Atramentova L, Lavryashina M, Evseeva I, Borinska S, Boldyreva M, Dubova N, Balanovska E: Is spatial distribution of the HIV-1-resistant CCR5Delta32 allele formed by ecological factors? J Physiol Anthropol Appl Human Sci 2005, 24: 375-382. 10.2114/jpa.24.375
Faure E, Royer-Caranzy M: Is the European spatial distribution of the HIV-1-resistant CCR5-Delta32 allele formed by a breakdown of the pathocenosis due to the historical Roman expansion? Infect Genet Evol, in press.
Lucotte G, Mercier G: Frequency of the coreceptor CCR5 gene delta 32 mutation in different French regions. C R Acad Sci III 1998, 321: 409-413.
Lucotte G, Mercier G: Distribution of the CCR5 gene 32-bp deletion in Europe. J Acquir Immune Defic Syndr Hum Retrovirol 1998, 19: 174-177.
Lucotte G, Dieterlen F: More about the Viking hypothesis of origin of the delta32 mutation in the CCR5 gene conferring resistance to HIV-1 infection. Infect Genet Evol 2003, 3: 293-295. 10.1016/j.meegid.2003.07.001
Lucotte G, Smets P: CCR5-Delta32 allele frequencies in Ashkenazi Jews. Genet Test 2003, 7: 333-337. 10.1089/109065703322783716
Galvani AP, Slatkin M: Evaluating plague and smallpox as historical selective pressures for the CCR5-delta 32 HIV-resistance allele. Proc Natl Acad Sci USA 2003, 100: 15276-15279. 10.1073/pnas.2435085100
Lidén K, Linderholm A, Götherström A: Pushing it back. Dating the CCR5-Δ32 bp deletion. to the Mesolithic in Sweden and its implications for the Meso/Neo transition. Documenta Praehistorica 2006, 33: 29-37.
Sabeti PC, Walsh E, Schaffner SF, Varilly P, Fry B, Hutcheson HB, Cullen M, Mikkelsen TS, Roy J, Patterson N, Cooper R, Reich D, Altshuler D, O'Brien S, Lander ES: The Case for Selection at CCR5-Δ32. PLoS Biol 2005, 3: e378. 10.1371/journal.pbio.0030378
Lucotte G: Distribution of the CCR5 gene 32-basepair deletion in West Europe. A hypothesis about the possible dispersion of the mutation by the Vikings in historical times. Hum Immunol 2001, 62: 933-936. 10.1016/S0198-8859(01)00292-0
Slatkin M: A population-genetic test of founder effects and implications for Ashkenazi Jewish diseases. Am J Hum Genet 2004, 75: 282-293. 10.1086/423146
Kere J: Human population genetics: Lessons from Finland. Annu Rev Genomics Hum Genet 2001, 2: 103-128. 10.1146/annurev.genom.2.1.103
Schmegner C, Hoegel J, Vogel W, Assum G: Genetic variability in a genomic region with long-range linkage disequilibrium reveals traces of a bottleneck in the history of the European population. Hum Genet 2005, 118: 276-286. 10.1007/s00439-005-0056-2
Behar DM, Villems R, Soodyall H, Blue-Smith J, Pereira L, Metspalu E, Scozzari R, Makkan H, Tzur S, Comas D, Bertranpetit J, Quintana-Murci L, Tyler-Smith C, Wells RS, Rosset S: The Genographic Consortium. The dawn of human matrilineal diversity. Am J Hum Genet 2008, 82: 1-11. 10.1016/j.ajhg.2008.04.002
Lohmueller KE, Indap AR, Schmidt S, Boyko AR, Hernandez RD, Hubisz MJ, Sninsky JJ, White TJ, Sunyaev SR, Nielsen R, Clark AG, Bustamante CD: Proportionally more deleterious genetic variation in European than in African populations. Nature 2008, 451: 994-997. 10.1038/nature06611
Cavalli-Sforza LL, Menozzi P, Piazza A: The History and Geography of Human Genes. Princeton, NJ: Princeton University Press; 1994.
Libert F, Cochaux P, Beckman G, Samson M, Aksenova M, Cao A, Czeizel A, Claustres M, Rua C, Ferrari M, Ferrec C, Glover G, Grinde B, Guran S, Kucinskas V, Lavinha J, Mercier B, Ogur G, Peltonen L, Rosatelli C, Schwartz M, Spitsyn V, Timar L, Beckman L, Parmentier M, Vassart G: The ΔCCR5 mutation conferring protection against HIV-1 in Caucasian populations has a single and recent origin in Northeastern Europe. Hum Mol Genet 1998, 7: 399-406. 10.1093/hmg/7.3.399
Klitz W, Brautbar C, Schito AM, Barcellos LF, Oksenberg JR: Evolution of the CCR5 delta 32 mutation based on haplotype variation in Jewish and Northern European population samples. Hum Immunol 2001, 62: 530-538. 10.1016/S0198-8859(01)00239-7
Hummel S, Schmidt D, Kremeyer B, Herrmann B, Oppermann M: Detection of the CCR5-Δ32 HIV resistance gene in Bronze Age skeletons. Genes Immun 2005, 6: 371-374. 10.1038/sj.gene.6364172
Gross L: A Well-Studied Disease-Resistance Gene Shows No Signs of Selection. PLoS Biol 2005, 3: e400. 10.1371/journal.pbio.0030400
Anderson RM, May RM: Infectious Diseases of Humans: Dynamics and Control. Oxford: Oxford University Press; 1991.
Dobson AP, Carper ER: Infectious diseases and human population history. Bioscience 1996, 46: 115-126. 10.2307/1312814
Diamond J: The worst mistake in the history of the human race. Discover Mag 1987, 5: 64-66.
McMichael AJ: Environmental and social influences on emerging infectious diseases: past, present and future. Philos Trans R Soc Lond B Biol Sci 2004, 359: 1049-1058. 10.1098/rstb.2004.1480
Glass WG, McDermott DH, Lim JK, Lekhong S, Yu SF, Frank WA, Pape J, Cheshier RC, Murphy PM: CCR5 deficiency increases risk of symptomatic West Nile virus infection. J Exp Med 2006, 203: 35-40. 10.1084/jem.20051970
Grmek MD: Préliminaires d'une étude historique des maladies. Annales ESC 1969, 6: 1473-1483.
Grmek MD: Diseases in the Ancient Greek World. Baltimore and London: Johns Hopkins University Press; 1991.
Biraben JN: Disease in Europe: Equilibrium and breakdown of the pathocenosis. In Western Medical Thought from Antiquity to the Middle Ages. Edited by: Grmek M. Cambridge MA: Harvard University Press; 1998:319-353.
Retief FP, Cilliers L: Epidemics of the Roman Empire, 27 BC-AD 476. S Afr Med J 2000, 90: 267-272.
Chomel BB, Belotto A, Meslin FX: Wildlife, exotic pets, and emerging zoonoses. Emerg Infect Dis 2007, 13: 6-11.
Greger M: The human/animal interface: emergence and resurgence of zoonotic infectious diseases. Crit Rev Microbiol 2007, 33: 243-299. 10.1080/10408410701647594
Wolfe ND, Dunavan CP, Diamond J: Origins of major human infectious diseases. Nature 2007, 447: 279-283. 10.1038/nature05775
Horwitz LK, Smith P: The contribution of animal domestication to the spread of zoonoses: A case study from the southern levant. Anthropozoologica 2000, 31: 77-84.
Pearce-Duvet JM: The origin of human pathogens: evaluating the role of agriculture and domestic animals in the evolution of human disease. Biol Rev Camb Philos Soc 2006, 81: 369-382. 10.1017/S1464793106007020
Zammit J: Les nouvelles maladies du Néolithique. Dossier Pour la Science 2006, 50: 35-37.
Jennison G: Animals for show and pleasure in ancient Rome. Manchester: Manchester University Press; 1937.
Clutton-Brock J: A natural history of domesticated mammals. London and Cambridge: British Museum (Natural History) and Cambridge University Press; 1999.
Kravetz JD, Federman DG: Cat-associated zoonoses. Arch Itern Med 2002, 162: 1945-1952. 10.1001/archinte.162.17.1945
Brown RR, Elston TH, Evans L, Glaser C, Gulledge ML, Jarboe L, Lappin MR, Marcus LC: Feline zoonoses guidelines from the American Association of Feline Practitioners. J Feline Med Surg 2005, 7: 243-274. 10.1016/S1098-612X(05)00168-3
Driscoll CA, Menotti-Raymond M, Roca AL, Hupe K, Johnson WE, Geffen E, Harley EH, Delibes M, Pontier D, Kitchener AC, Yamaguchi N, O'Brien SJ, Macdonald DW: The Near Eastern origin of cat domestication. Science 2007, 317: 519-523. 10.1126/science.1139518
Lipinski MJ, Froenicke L, Baysac KC, Billings NC, Leutenegger CM, Levy AM, Longeri M, Niini T, Ozpinar H, Slater MR, Pedersen NC, Lyons LA: The ascent of cat breeds: genetic evaluations of breeds and worldwide random-bred populations. Genomics 2008, 91: 12-21. 10.1016/j.ygeno.2007.10.009
Vigne JD, Guilaine J, Debue K, Haye L, Gerard P: Early taming of the cat in Cyprus. Science 2004, 304: 259-259. 10.1126/science.1095335
Davis SJM: The Archaeology of Animals. London: Batsford BT; 1987.
Ragni B: Gatto selvatico, Felis silvestris Schreber, 1777. In Distribuzione e biologia di 22 specie di mammiferi in Italia. Edited by: Pavan M. Rome: Consiglio Nazionale delle Ricerche; 1981:105-113.
Vigne JD: Zooarchaeological and biogeographical history of the mammals of Corsica and Sardinia since the last ice age. Mammal Rev 1992, 22: 87-96. 10.1111/j.1365-2907.1992.tb00124.x
Amori G, Angelici FM, Boitani L: Mammals of Italy: a revised checklist of species and subspecies (Mammalia). Senckenb Biol 1999, 79: 271-286.
Randi E, Pierpaoli M, Beaumont M, Ragni B, Sforzi A: Genetic identification of wild and domestic cats ( Felis silvestris ) and their hybrids using Bayesian clustering methods. Mol Biol Evol 2001, 18: 1679-1693.
Bury JB: History of Greece. New York: The Modern Library; 1913.
Sunquist MN, Sunquist F: Wild Cats of the World. Chicago: University of Chicago Press; 2002.
Hughes D: Europe as consumer of exotic biodiversity: Greek and Roman times. Landscape Res 2003, 28: 21-31. 10.1080/01426390306535
Keller O: Zur Geschichte der Katze im Altertum. Mitt deutsch Arch Inst Rom Abth 1908, 23: 40-70.
Bökönyi S: History of domestic animals mammals in Central and Eastern Europe. Budapest: Akadémiai Kiado; 1974.
Kirk M: The everlasting cat. New York: Galahad Books; 1977.
Clason AT: Jager, visser, veehouder, vogellijmer. In Voltooid verleden tijd? Een hedendaagse kijk op de prehistorie. Edited by: Chamalaun M, Waterbolk HT. Amsterdam: Intermediair; 1980:131-146.
Van Neer W: De archeozoologische resten. In De Romeinse villa's van Piringen ("Mulkenveld") en Vechmaal ("Walenveld") (= Publicaties van het Provinciaal Gallo-Romeins Museum te Tongeren 42). Edited by: Vanvinckenroye W. Tongeren, Belgium: Gallo-Romeins Museum; 1990:31-33.
Canters KJ, Thissen JBM, Van Diepenbeek MAJ, Jansman HAH, Goutbeek K: The wildcat ( Felis silvestris ) finally recorded in the Netherlands. Lutra 2005, 48: 67-90.
Daniels MJ, Balharry D, Hirst D, Kitchener AC, Aspinall RJ: Morphological and pelage characteristics of wild living cats in Scotland: implications for defining the 'wildcat'. J Zool London 1998, 244: 231-247.
Masseti M: Quaternary biogeography of the mustelidae family on the Mediterranean islands. Hystrix 1995, 7: 17-34.
Delattre P, Chaste B, Silvy C: Lutte biologique et rongeurs. Dossiers de l'Environnement de l'INRA 2000, 19: 35-47.
Johnston JB, Olson ME, Rud EW, Power C: Xenoinfection of nonhuman primates by feline immunodeficiency virus. Curr Biol 2001, 11: 1109-1113. 10.1016/S0960-9822(01)00350-5
Mustafa F, Jayanth P, Phillip PS, Ghazawi A, Schmidt RD, Lew KA, Rizvi TA: Relative activity of the feline immunodeficiency virus promoter in feline and primate cell lines. Microbes Infect 2005, 7: 233-239. 10.1016/j.micinf.2004.10.015
Guggisberg CAW: Wild Cats of the World. New York: Taplinger Publishing Company; 1975.
Redford KH, Eisenberg JF: Mammals of the Neotropics. The Southern Cone: Chile, Argentina, Uruguay, Paraguay. Volume 2. Edited by: Eisenberg JF. Chicago, IL: University of Chicago Press; 1992.
Busch RH: The Cougar Almanac. New York: Lyons and Burford; 1996.
Nowell K, Jackson : Status Survey and Conservation Action Plan.Gland, Switzerland: IUCN/SSC; 1996. [http://www.carnivoreconservation.org/files/actionplans/wildcats.pdf]
Anton M, Turner A: The Big Cats and Their Fossil Relatives. New York: Columbia University Press; 2000.
Eisenberg JF, Redford KH: Mammals of the Neotropics. The Central Neotropics: Ecuador, Peru, Bolivia, Brazil. Volume 3. Chicago, IL: University of Chicago Press; 2000.
Barnett R, Barnes I, Phillips MJ, Martin LD, Harington CR, Leonard JA, Cooper A: Evolution of the extinct Sabretooths and the American cheetah-like cat. Curr Biol 2005, 15: R589-R590. 10.1016/j.cub.2005.07.052
Kanzaki LI, Looney DJ: Feline immunodeficiency virus: a concise review. Front Biosci 2004, 9: 370-377. 10.2741/1235
Willett BJ, Hosie MJ, Neil JC, Turner JD, Hoxie JA: Common mechanism of infection by lentiviruses. Nature 1997, 385: 587. 10.1038/385587a0
VandeWoude S, Apetrei C: Going wild: lessons from naturally occurring T-lymphotropic lentiviruses. Clin Microbiol Rev 2006, 19: 728-762. 10.1128/CMR.00009-06
Carpenter MA, O'Brien SJ: Coadaptation and immunodeficiency virus: lessons from the Felidae. Curr Opin Genet Dev 1995, 5: 739-745. 10.1016/0959-437X(95)80006-Q
Packer C, Altizer S, Appel M, Brown E, Martenson J, O'Brien SJ, Roelke-Parker M, Hofmann-Lehmann R, Lutz H: Viruses of the Serengeti: patterns of infection and mortality in African lions. J Anim Ecol 1999, 68: 1161-1178. 10.1046/j.1365-2656.1999.00360.x
Bull ME, Gebhard DG, Tompkins WA, Kennedy-Stoskopf S: Polymorphic expression in the CD8alpha chain surface receptor of African lions ( Panthera leo ). Vet Immunol Immunopathol 2002, 84: 181-189. 10.1016/S0165-2427(01)00401-9
Bull ME, Kennedy-Stoskopf S, Levine JF, Loomis M, Gebhard DG, Tompkins WA: Evaluation of T lymphocytes in captive african lions ( Panthera leo ) infected with feline immunodeficiency virus. Am J Vet Res 2003, 64: 1293-1300. 10.2460/ajvr.2003.64.1293
Brennan G, Podell MD, Wack R, Kraft S, Troyer JL, Bielefeldt-Ohmann H, VandeWoude S: Neurologic disease in captive lions ( Panthera leo ) with low-titer lion lentivirus infection. J Clin Microbiol 2006, 44: 4345-4352. 10.1128/JCM.00577-06
Roelke ME, Pecon-Slattery J, Taylor S, Citino S, Brown E, Packer C, Vandewoude S, O'Brien SJ: T-lymphocyte profiles in FIV infected wild lions and pumas reveal CD4 depletion. J Wildl Dis 2006, 42: 234-248.
Johnson WE, Eizirik E, Pecon-Slattery J, Murphy WJ, Antunes A, Teeling E, O'Brien SJ: The late Miocene radiation of modern Felidae : a genetic assessment. Science 2006, 311: 73-77. 10.1126/science.1122277
Franklin SP, Troyer JL, Terwee JA, Lyren LM, Boyce WM, Riley SP, Roelke ME, Crooks KR, Vandewoude S: Frequent Transmission of Immunodeficiency Viruses among Bobcats and Pumas. J Virol 2007, 81: 10961-10969. 10.1128/JVI.00997-07
Troyer JL, Vandewoude S, Pecon-Slattery J, McIntosh C, Franklin S, Antunes A, Johnson W, O'Brien SJ: FIV cross-species transmission: An evolutionary prospective. Vet Immunol Immunopathol 2008, 123: 159-166. 10.1016/j.vetimm.2008.01.023
Biek R, Zarnke RL, Gillin C, Wild M, Squires JR, Poss M: Serologic survey for viral and bacterial infections in western populations of Canada lynx ( Lynx canadensis ). J Wildl Dis 2002, 38: 840-845.
Biek R, Rodrigo AG, Holley D, Drummond A, Anderson CR Jr, Ross HA, Poss M: Epidemiology, genetic diversity, and evolution of endemic feline immunodeficiency virus in a population of wild cougars. J Virol 2003, 77: 9578-9589. 10.1128/JVI.77.17.9578-9589.2003
Troyer JL, Pecon-Slattery J, Roelke ME, Black L, Packer C, O'Brien SJ: Patterns of feline immunodeficiency virus multiple infection and genome divergence in a free-ranging population of African lions. J Virol 2004, 78: 3777-3791. 10.1128/JVI.78.7.3777-3791.2004
Spencer JA, van Dijk AA, Horzinek MC, Egberink HF, Bengis RG, Keet DF, Morikawa S, Bishop DHL: Incidence of FIV reactive antibodies in free-ranging lions of the Kruger National Park and the Etosha National Park in Southern Africa detected by recombinant FIV p24 antigen. Onderstepoort J Vet Res 1992,59(4):315-322.
Pecon-Slattery J, McCracken C, Troyer JL, Roelke ME, Sondgareth K, Winterbach C, Winterbach H, VandeWoude S, Roelke M, Sondgeroth K, Winterbach C, Winterbach H, O'Brien SJ: Genomic organization, sequence divergence, and recombination of feline immunodeficiency virus from lions in the wild. BMC Genomics 2008, 9: 66. 10.1186/1471-2164-9-66
Letcher JD, O'Conner TP: Incidence of antibodies reacting to feline immunodeficiency virus in a population of Asian lions. J Zoo Wildl Med 1991, 22: 324-329.
Osofsky SA, Hirsch KJ, Zuckerman EE, Hardy WD: Feline lentivirus and feline oncovirus status of free-reanging lions ( Panthera leo ), leopards ( Panthera pardus ), and cheetahs ( Acinonyx jubatus ) in Botswana: a regional perspective. J Zoo Wildl Med 1996, 27: 453-467.
Troyer JL, Pecon-Slattery J, Roelke ME, Johnson W, VandeWoude S, Vazquez-Salat N, Brown M, Frank L, Woodroffe R, Winterbach C, Winterbach H, Hemson G, Bush M, Alexander KA, Revilla E, O'Brien SJ: Seroprevalence and genomic divergence of circulating strains of feline immunodeficiency virus among Felidae and Hyaenidae species. J Virol 2005, 79: 8282-8294. 10.1128/JVI.79.13.8282-8294.2005
Harrison TM, Mazet JK, Holekamp KE, Dubovi E, Engh AL, Nelson K, Van Horn RC, Munson L: Antibodies to canine and feline viruses in spotted hyenas ( Crocuta crocuta ) in the Masai Mara National Reserve. J Wildl Dis 2004, 40: 1-10.
Pecon-Slattery J, Troyer JL, Johnson WE, O'Brien SJ: Evolution of feline immunodeficiency virus in Felidae: Implications for human health and wildlife ecology. Vet Immunol Immunopathol 2008, 123: 32-44. 10.1016/j.vetimm.2008.01.010
Rohland N, Pollack JL, Nagel D, Beauval C, Airvaux J, Pääbo S, Hofreiter M: The population history of extant and extinct hyenas. Mol Biol Evol 2005, 22: 2435-2443. 10.1093/molbev/msi244
Sharp PM, Shaw GM, Hahn BH: Simian immunodeficiency virus infection of chimpanzees. J Virol 2005, 79: 3891-3902. 10.1128/JVI.79.7.3891-3902.2005
Woldemeskel M, Tibbo M, Potgieter LN: Ovine progressive pneumonia (Maedi-Visna): an emerging respiratory disease of sheep in Ethiopia. Dtsch Tierarztl Wochenschr 2002, 109: 486-488.
Gaubert P, Cordeiro-Estrela P: Phylogenetic systematics and tempo of evolution of the Viverrinae (Mammalia, Carnivora, Viverridae) within feliformians: Implications for faunal exchanges between Asia and Africa. Mol Phyl Evol 2006, 41: 266-278. 10.1016/j.ympev.2006.05.034
Fromont E, Sager A, Léger F, Bourguemestre F, Jouquelet E, Stahl P, Pontier D, Artois M: Prevalence and pathogenicity of retroviruses in wildcats in France. Vet Rec 2000, 146: 317-319.
Ostrowski S, Van Vuuren M, Lenain DM, Durand A: A serologic survey of wild felids from central west Saudi Arabia. J Wildl Dis 2003, 39: 696-701.
Lecis R, Pierpaoli M, Birò ZS, Szemethy L, Ragni B, Vercillo F, Randi E: Bayesian analyses of admixture in wild and domestic cats ( Felis silvestris ) using linked microsatellite loci. Mol Ecol 2006, 15: 119-131. 10.1111/j.1365-294X.2005.02812.x
Oliveira R, Godinho R, Randi E, Ferrand N, Alves PC: Molecular analysis of hybridisation between wild and domestic cats ( Felis silvestris ) in Portugal: implications for conservation. Conserv Genet 2008, 9: 1-11. 10.1007/s10592-007-9297-z
Cameron-Beaumont C, Lowe SE, Bradshaw JWS: Evidence suggesting preadaptation to domestication throughout the small Felidae. Biol J Linn Soc 2002, 75: 361-366. 10.1111/j.1095-8312.2002.tb02077.x
Burton M: Encyclopédie du monde animal. Les singes et animaux domestiques. Volume 7. Paris: Marabout Université; 1965.
Vereshchagin NK: The Mammals of the Caucasus: A History of the Evolution of the Fauna. Jerusalem: Israeli Program for Scientific Translations; 1967.
Graven J: L'homme et l'animal. Paris: Encyclopédie Planète, Éditions Planète; 1964.
Malek J: The cat in ancient Egypt. Philadelphia, PA: University of Pennsylvania Press; 1997.
Allen GO: Caracal ( Felis caracal ) and hunting leopard ( Cynailurus jubatus ) in Mirzapur, Uttar Pradesh. J Bomb Nat Hist Soc 1919, 26: 1041-1042.
Sharma VD, Sankhala K: Vanishing cats of Rajasthan. In Proceedings meeting and workshop of the IUCN/SSC Cat Specialist Group: 9–12 April 1984; Kanha, India. Edited by: Jackson P. Bougy-Villars, Switzerland: IUCN/SSC Cat Specialist Group; 1984:117-135.
Kitchener A: The natural history of the wild cats. New York: Comstock Publishing Associates; 1991.
Morrison-Scott TCS: The mummified cats of ancient Egypt. Proc Zool Soc Lond 1952, 121: 861-867.
Heptner VH, Sludskii AA, Komarov A, Komorov N: Mammals of the Soviet Union: Carnivora, Part 2 (Hyaenas and Cats). Edited by: Hoffmann RS. Washington DC: Smithsonian Inst. and the Natl. Science Fndn; 1992.
Zeuner FE: A History of Domesticated Animals. London: Hutchinson; 1963.
Lauer JP, Shoucair A: Saqqarah, la nécropole royale de Memphis, quarante siècle d'histoire, cent ving-cinq ans de recherches. Paris: Tallandier; 1976.
Rengger JR: Naturgeschichte der Säugetiere von Paraguay. Edited by: Sanderson IT. Basel, Swizzerland: Schweighausersche; 1830.
Shuffeldt RW: The mountain lion, ocelots, lynxes and their kin. Amer Forestry 1921, 27: 629-636.
Davis WB: Ocelot.denizen of the brush country. Texas Game Fish 1958, 16: 16-17.
Leal ES, Ravazzolo AP: Detecção do vírus da imunodeficiência felina (FIV) em felídeos selvagens pertencentes à Região Neotropical, através da técnica de reaão em cadeia da polimerase (PCR). Hora Veterinária 1998, 17: 57-60.
Toynbee JMC: Animals in Roman Life and Art. Baltimore: The Johns Hopkins University Press; 1973.
Scarre C: Atlas de la Rome antique, 800 av. J.-C. – 540 ap. J.-C. De la naissance de la République à la chute de l'Empire. Paris: Éditions Autrement; 1995.
Hughes JD: Ecology in ancient civilizations. Albuquerque: University of New Mexico Press; 1975.
Dio Cassius:Roman History, LXVIII.15. [http://penelope.uchicago.edu/Thayer/E/Roman/Texts/Cassius_Dio/68*.html]
Pliny the Elder:The Natural History, VIII.20, X.73. [http://www.perseus.tufts.edu/cgi-bin/ptext?lookup=Plin.+Nat.+toc]
Plutarch:Pompey, 52. [http://penelope.uchicago.edu/Thayer/E/Roman/Texts/Plutarch/Lives/Pompey*.html]
Titus Livius:The history of Rome, XXXIX.22.1. [http://bcs.fltr.ucl.ac.be/LIV/XXXIX.html]
Wolfe ND, Switzer WM, Carr JK, Bhullar VB, Shanmugam V, Tamoufe U, Prosser AT, Torimiro JN, Wright A, Mpoudi-Ngole E, McCutchan FE, Birx DL, Folks TM, Burke DS, Heneine W: Naturally acquired simian retrovirus infections in central African hunters. Lancet 2004, 363: 932-937. 10.1016/S0140-6736(04)15787-5
Wolfe ND, Heneine W, Carr JK, Garcia AD, Shanmugam V, Tamoufe U, Torimiro JN, Prosser AT, Lebreton M, Mpoudi-Ngole E, McCutchan FE, Birx DL, Folks TM, Burke DS, Switzer WM: Emergence of unique primate T-lymphotropic viruses among central African bushmeat hunters. Proc. Natl. Acad. Sci. USA 2005, 102: 7994-7999. 10.1073/pnas.0501734102
Van Dooren S, Meertens L, Lemey P, Gessain A, Vandamme AM: Full-genome analysis of a highly divergent simian T-cell lymphotropic virus type 1 strain in Macaca arctoides . J Gen Virol 2005, 86: 1953-1959. 10.1099/vir.0.80520-0
Van Dooren S, Verschoor EJ, Fagrouch Z, Vandamme AM: Phylogeny of primate T lymphotropic virus type 1 (PTLV-1) including various new Asian and African non-human primate strains. Infect Genet Evol 2007, 7: 374-381. 10.1016/j.meegid.2006.06.003
Aristotle:History of animals, VI.31, VIII.28. [http://ebooks.adelaide.edu.au/a/aristotle/history/]
Pausanias:Description of Greece, VI.5.4. [http://www.theoi.com/Text/Pausanias1A.html]
Cleri DJ, Ricketti AJ, Vernaleo JR: Fever of unknown origin due to zoonoses. Infect Dis Clin North Am 2007, 21: 963-996. 10.1016/j.idc.2007.08.009
Myers G, MacInnes K, Korber B: The emergence of simian/human immunodeficiency viruses. AIDS Res hum Retroviruses 1992, 8: 373-386.
Gao F, Bailes E, Robertson DL, Chen Y, Rodenburg CM, Michael SF, Cummins LB, Arthur LO, Peeters M, Shaw GM, Sharp PM, Hahn BH: Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature 1999, 397: 436-441. 10.1038/17130
Hahn BH, Shaw GM, De Cock KM, Sharp PM: AIDS as a zoonosis: scientific and public health implications. Science 2000, 287: 607-614. 10.1126/science.287.5453.607
Korber B, Muldoon M, Theiler J, Gao F, Gupta R, Lapedes A, Hahn BH, Wolinsky S, Bhattacharya T: Timing the ancestor of the HIV-1 pandemic strains. Science 2000, 288: 1789-1796. 10.1126/science.288.5472.1789
Sharp PM, Bailes E, Chaudhuri RR, Rodenburg CM, Santiago MO, Hahn BH: The origins of acquired immune deficiency viruses: where and when? Philos Trans R Soc Lond B Biol Sci 2001, 356: 867-876. 10.1098/rstb.2001.0863
Peeters M, Courgnaud V, Abela B, Auzel P, Pourrut X, Bibollet-Ruche F, Loul S, Liegeois F, Butel C, Koulagna D, Mpoudi-Ngole E, Shaw GM, Hahn BH, Delaporte E: Risk to human health from a plethora of simian immunodeficiency viruses in primate bushmeat. Emerg Infect Dis 2002, 8: 451-457.
Van Heuverswyn F, Peeters M: The Origins of HIV and Implications for the Global Epidemic. Curr Infect Dis Rep 2007, 9: 338-346. 10.1007/s11908-007-0052-x
Apetrei C, Marx PA: Simian retroviral infections in human beings. Lancet 2004, 364: 137-138. 10.1016/S0140-6736(04)16620-8
Khabbaz RF, Rowe T, Murphey-Corb M, Heneine WM, Schable CA, George JR, Pau CP, Parekh BS, Lairmore MD, Curran JW: Simian immunodeficiency virus needlestick accident in a laboratory worker. Lancet 1992, 340: 271-273. 10.1016/0140-6736(92)92358-M
Khabbaz RF, Heneine W, George J, Parekh B, Rowe T, Woods T, Switzer WM, McClure HM, Murphey-Corb M, Folks TM: Brief report: infection of a laboratory worker with simian immunodeficiency virus. N Engl J Med 1994, 330: 172-177. 10.1056/NEJM199401203300304
Reynolds SJ, Bessong PO, Quinn TC: Human retroviral infections in the tropics. In Tropical infectious diseasesdprinciples, pathogens, practice. Volume 1. 2nd edition. Edited by: Guerrant RL, Walker DH, Weller PF. Philadelphia: Elsevier Churchill Livingston; 2006:852-883.
Gessain A, Calattini S: Emergence of simian foamy viruses in humans: facts and unanswered questions. Future Virol 2008, 3: 71-81. 10.2217/17460794.3.1.71
Jones-Engel L, Engel GA, Schillaci MA, Rompis A, Putra A, Suaryana KG, Fuentes A, Beer B, Hicks S, White R, Wilson B, Allan JS: Primate-to-human retroviral transmission in Asia. Emerg Infect Dis 2005, 11: 1028-1035.
Calattini S, Betsem EB, Froment A, Mauclère P, Tortevoye P, Schmitt C, Njouom R, Saib A, Gessain A: Simian foamy virus transmission from apes to humans, rural Cameroon. Emerg Infect Dis 2007, 13: 1314-1320.
Boneva RS, Grindon AJ, Orton SL, Switzer WM, Shanmugam V, Hussain AI, Bhullar VB, Chamberland ME, Heneine W, Folks TM, Chapman LE: Simian foamy virus infection in a blood donor. Transfusion 2002, 42: 886-891. 10.1046/j.1537-2995.2002.00134.x
Heneine W, Switzer WM, Sandstrom P, Brown J, Vedapuri S, Schable CA, Khan AS, Lerche NW, Schweizer M, Neumann-Haefelin D, Chapman LE, Folks TM: Identification of a human population infected with simian foamy viruses. Nat Med 1998, 4: 403-407. 10.1038/nm0498-403
Heneine W, Schweizer M, Sandstrom P, Folks T: Human infection with foamy viruses. Curr Top Microbiol Immunol 2003, 277: 181-196.
Switzer WM, Bhullar V, Shanmugam V, Cong ME, Parekh B, Lerche NW, Yee JL, Ely JJ, Boneva R, Chapman LE, Folks TM, Heneine W: Frequent simian foamy virus infection in persons occupationally exposed to nonhuman primates. J Virol 2004, 78: 2780-2789. 10.1128/JVI.78.6.2780-2789.2004
Lerche NW, Switzer WM, Yee JL, Shanmugam V, Rosenthal AN, Chapman LE, Folks TM, Heneine W: Evidence of infection with simian type D retrovirus in persons occupationally exposed to nonhuman primates. J Virol 2001, 75: 1783-1789. 10.1128/JVI.75.4.1783-1789.2001
Butera ST, Brown J, Callahan ME, Owen SM, Matthews AL, Weigner DD, Chapman LE, Sandstrom PA: Survey of veterinary conference attendees for evidence of zoonotic infection by feline retroviruses. J Am Vet Med Assoc 2000, 217: 1475-1479. 10.2460/javma.2000.217.1475
Dow SW, Dreitz MJ, Hoover EA: Feline immunodeficiency virus neurotropism: evidence that astrocytes and microglia are the primary target cells. Vet Immunol Immunop 1992, 35: 23-35. 10.1016/0165-2427(92)90118-A
Tochikura TS, Tanabe-Tochikura A, Hayes KA, Lazo A, Bailer RT, Blakeslee JRJr, Lafrado LJ, Roy-Burman P, Pandey R, Olsen RG, Mathes LE: Fusion activity dissociated from replication ability in feline immunodeficiency virus (FIV) in human cells. J Acquir Immune Defic Syndr 1993,6(12):1301-1310.
Ikeda Y, Tomonaga K, Kawaguchi Y, Kohmoto M, Inoshima Y, Tohya Y, Miyazawa T, Kai C, Mikami T: Feline immunodeficiency virus can infect a human cell line (MOLT-4) but establishes a state of latency in the cells. J Gen Virol 1996, 77: 1623-1630. 10.1099/0022-1317-77-8-1623
Miyazawa T, Kawaguchi Y, Kohmoto M, Sakuragi J, Adachi A, Fukasawa M, Mikami T: Production of feline immunodeficiency virus in feline and non-feline non-lymphoid cell lines by transfection of an infectious molecular clone. J Gen Virol 1992, 73: 1543-1546. 10.1099/0022-1317-73-6-1543
Miyazawa T, Kawaguchi Y, Kohmoto M, Tomonaga K, Mikami T: Comparative functional analysis of the various lentivirus long terminal repeats in human colon carcinoma cell line (SW480 cells) and feline renal cell line (CRFK cells). J Vet Med Sci 1994, 56: 895-899.
Miyazawa CK, Mikami T: Feline immunodeficiency virus can infect a human cell line (MOLT-4) but establishes a state of latency in the cells. J Gen Virol 1996, 77: 1623-1630. 10.1099/0022-1317-77-8-1623
Poeschla EM, Looney DJ: CXCR4 is required by a nonprimate lentivirus: heterologous expression of feline immunodeficiency virus in human, rodent, and feline cells. J Virol 1998, 72: 6858-6866.
Poeschla EM, Wong-Staal F, Looney DJ: Efficient transduction of nondividing human cells by feline immunodeficiency virus lentiviral vectors. Nat Med 1998, 4: 354-357. 10.1038/nm0398-354
Johnston J, Power C: Productive infection of human peripheral blood mononuclear cells by feline immunodeficiency virus: implications for vector development. J Virol 1999, 73: 2491-2498.
Johnston JB, Power C: Feline immunodeficiency virus xenoinfection: the role of chemokine receptors and envelope diversity. J Virol 2002, 76: 3626-3636. 10.1128/JVI.76.8.3626-3636.2002
Saenz DT, Teo W, Olsen JC, Poeschla E: Restriction of feline immunodeficiency virus by Ref1, LV1 and primate TRIM5α proteins. J Virol 2005, 79: 15175-15188. 10.1128/JVI.79.24.15175-15188.2005
Saenz DT, Barraza R, Loewen N, Teo W, Poeschla E: Production and use of feline immunodeficiency virus (FIV)-based lentiviral vectors. In Gene Transfer: ACold Spring Harbor Laboratory Manual. Edited by: Rossi J, Friedman T. New York: Cold Spring Harbor Laboratory Press; 2006:57-74.
Willett BJ, McMonagle EL, Ridha S, Hosie MJ: Differential utilization of CD134 as a functional receptor by diverse strains of feline immunodeficiency virus. J Virol 2006, 80: 3386-3394. 10.1128/JVI.80.7.3386-3394.2006
Egberink HF, De Clerq E, Van Vliet AL, Balzarini J, Bridger GJ, Henson G, Horzinek MC, Schols D: Bicyclams, selective antagonists of the human chemokine receptor CXCR4, potently inhibit feline immunodeficiency virus replication. J Virol 1999, 73: 6346-6352.
Richardson J, Pancino G, Merat R, Leste-Lasserre T, Moraillon A, Schneider-Mergener J, Alizon M, Sonigo P, Heveker N: Shared usage of the chemokine receptor CXCR4 by primary and laboratory-adapted strains of feline immunodeficiency virus. J Virol 1999, 73: 3661-3671.
de Parseval A, Chatterji U, Sun P, Elder JH: Feline immunodeficiency virus targets activated CD4+ T cells by using CD134 as a binding receptor. Proc Natl Acad Sci USA 2004, 101: 13044-13049. 10.1073/pnas.0404006101
de Parseval A, Grant CK, Sastry KJ, Elder JH: Sequential CD134-CXCR4 interactions in feline immunodeficiency virus (FIV): soluble CD134 activates FIV Env for CXCR4-dependent entry and reveals a cryptic neutralization epitope. J Virol 2006, 80: 3088-3091. 10.1128/JVI.80.6.3088-3091.2006
Shimojima M, Miyazawa T, Ikeda Y, McMonagle EL, Haining H, Akashi H, Takeuchi Y, Hosie MJ, Willett BJ: Use of CD134 as a primary receptor by the feline immunodeficiency virus. Science 2004, 303: 1192-1195. 10.1126/science.1092124
Smirnova N, Troyer JL, Schissler J, Terwee J, Poss M, VandeWoude S: Feline lentiviruses demonstrate differences in receptor repertoire and envelope structural elements. Virology 2005, 342: 60-76. 10.1016/j.virol.2005.07.024
Terwee JA, Yactor JK, Sondgeroth KS, Vandewoude S: Puma lentivirus is controlled in domestic cats after mucosal exposure in the absence of conventional indicators of immunity. J Virol 2005, 79: 2797-2806. 10.1128/JVI.79.5.2797-2806.2005
Lerner DL, Elder JH: Expanded host cell tropism and cytopathic properties of feline immunodeficiency virus strain PPR subsequent to passage through interleukin-2-independent T cells. J Virol 2000, 74: 1854-1863. 10.1128/JVI.74.4.1854-1863.2000
Johnson CM, Benson NA, Papadi GP: Apoptosis and CD4+ lymphocyte depletion following feline immunodeficiency virus infection of a T-lymphocyte cell line. Vet Pathol 1996, 33: 195-203.
Bishop SA, Williams NA, Gruffydd Jones TJ, Harbour DA, Stokes CR: Impaired T-cell priming and proliferation in cats infected with feline immunodeficiency virus. AIDS 1992, 6: 287-293. 10.1097/00002030-199203000-00004
Bailes E, Gao F, Bibollet-Ruche F, Courgnaud V, Peeters M, Marx PA, Hahn BH, Sharp PM: Hybrid origin of SIV in chimpanzees. Science 2003, 300: 1713. 10.1126/science.1080657
Carpenter MA, Brown EW, Culver M, Johnson WE, Pecon-Slattery J, Brousset D, O'Brien SJ: Genetic and phylogenetic divergence of feline immunodeficiency virus in the puma ( Puma concolor ). J Virol 1996, 70: 6682-6693.
Nishimura Y, Goto Y, Yoneda K, Endo Y, Mizuno T, Hamachi M, Maruyama H, Kinoshita H, Koga S, Komori M, Fushuku S, Ushinohama K, Akuzawa M, Watari T, Hasegawa A, Tsujimoto H: Interspecies transmission of feline immunodeficiency virus from the domestic cat to the Tsushima cat ( Felis bengalensis euptilura ) in the wild. J Virol 1999, 73: 7916-7921.
Rogers AB, Hoover EA: Maternal-fetal feline immunodeficiency virus transmission: timing and tissue tropisms. J Infect Dis 1998, 178: 960-967. 10.1086/515692
Cunha CB: Prolonged and perplexing fevers in antiquity: malaria and typhoid fever. Infect Dis Clin North Am 2007, 21: 857-866. 10.1016/j.idc.2007.08.010
Jarrett O, Laird HM, Hay D: Growth of feline leukaemia virus in human cells. Nature 1969, 224: 1208-1209. 10.1038/2241208a0
Jarrett O, Laird HM, Hay D: Growth of feline leukemia virus in human, canine and porcine cells. Bibl Haematol 1970, 36: 387-392.
Jarrett O, Laird HM, Hay D: Restricted host range of a feline leukaemia virus. Nature 1972, 238: 220-221. 10.1038/238220a0
Jarrett O, Laird HM, Hay D: Determinants of the host range of feline leukaemia viruses. J Gen Virol 1973, 20: 169-175. 10.1099/0022-1317-20-2-169
Chang RS, Golden HD, Harrold B: Propagation in human cells of a filterable agent from the ST feline sarcoma. J Virol 1970, 6: 599-603.
Sarma PS, Huebner RJ, Basker Lee Vernon JF, Gilden RV: Feline leukemia and sarcoma viruses susceptibility of human cells to infections. Science 1970, 168: 1098-1100. 10.1126/science.168.3935.1098
Bardell D, Essex M: Glycolysis during early infection of feline and human cells with feline leukaemia virus. Infect Immun 1974, 9: 824-827.
Chan EW, Schiop-Stansly PE, O'Connor TE: Mammalian sarcoma-leukemia viruses. I. Infection of feline, bovine, and human cell cultures with Snyder-Theilen feline sarcoma virus. J Natl Cancer Inst 1974, 52: 473-481.
Azocar J, Essex M: Susceptibility of human cell lines to feline leukemia virus and feline sarcoma virus. J Natl Cancer Inst 1979, 63: 1179-1184.
Morgan RA, Dornsife RE, Anderson WF, Hoover EA: In vitro infection of human bone marrow by feline leukemia viruses. Virology 1993, 193: 439-442. 10.1006/viro.1993.1141
Wesseling B, Kopelovich L: Differentiation of human fibroblasts to tissue macrophages by the Snyder-Theilen feline sarcoma virus (ST:FeSV): growth modulation of human tumor cell lines in agar. Anticancer Res 1997, 17: 2599-2602.
Nakata R, Miyazawa T, Shin YS, Watanabe R, Mikami T, Matsuura Y: Reevaluation of host ranges of feline leukemia virus subgroups. Microbes Infect 2003, 5: 947-950. 10.1016/S1286-4579(03)00180-1
Shojima T, Nakata R, Miyazawa T: Host cell range of T-lymphotropic feline leukemia virus in vitro. Biochem Biophys Res Commun 2006, 345: 1466-1470. 10.1016/j.bbrc.2006.05.039
Vobis M, D'Haese J, Mehlhorn H, Mencke N: Evidence of horizontal transmission of feline leukemia virus by the cat flea ( Ctenocephalides felis ). Parasitol Res 2003, 91: 467-470. 10.1007/s00436-003-0949-8
Deinhardt F, Wolfe LG, Theilen GH, Snyder SP: ST-Feline fibrosarcoma virus: Induction of tumors in marmoset monkeys. Science 1970, 167: 881. 10.1126/science.167.3919.881
Oppermann M: Chemokine receptor CCR5: insights into structure, function, and regulation. Cell Signal 2004, 16: 1201-1210. 10.1016/j.cellsig.2004.04.007
Gade-Andavolu R, Comings DE, MacMurray J, Rostamkhani M, Cheng LS, Tourtellotte WW, Cone LA: Association of CCR5 delta32 deletion with early death in multiple sclerosis. Genet Med 2004, 6: 126-131.
Pulkkinen K, Luomala M, Kuusisto H, Lehtimäki T, Saarela M, Jalonen TO, Elovaara I: Increase in CCR5 Delta32/Delta32 genotype in multiple sclerosis. Acta Neurol Scand 2004, 109: 342-347. 10.1046/j.1600-0404.2003.00233.x
Rasmussen HB, Timm S, Wang AG, Søeby K, Lublin H, Fenger M, Hemmingsen R, Werge T: Association between the CCR5 32-bp deletion allele and late onset of schizophrenia. Am J Psychiatry 2006, 163: 507-511. 10.1176/appi.ajp.163.3.507
Zhu T, Korber BT, Nahmias AJ, Hooper E, Sharp PM, Ho DD: An African HIV-1 sequence from 1959 and implications for the origin of the epidemic. Nature 1998, 391: 594-597. 10.1038/35400
Huffnagle GB, McNeil LK, McDonald RA, Murphy JW, Toews GB, Maeda N, Kuziel WA: Cutting edge: Role of C-C chemokine receptor 5 in organ-specific and innate immunity to Cryptococcus neoformans . J Immunol 1999, 163: 4642-4646.
Dawson TC, Beck MA, Kuziel WA, Henderson F, Maeda N: Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am J Pathol 2000, 156: 1951-1959.
Aliberti J, Sher A: Role of G-protein-coupled signaling in the induction and regulation of dendritic cell function by Toxoplasma gondii . Microbes Infect 2002, 4: 991-997. 10.1016/S1286-4579(02)01618-0
Barr EL, Ouburg S, Igietseme JU, Morré SA, Okwandu E, Eko FO, Ifere G, Belay T, He Q, Lyn D, Nwankwo G, Lillard J, Black CM, Ananaba GA: Host inflammatory response and development of complications of Chlamydia trachomatis genital infection in CCR5-deficient mice and subfertile women with the CCR5delta32 gene deletion. J Microbiol Immunol Infect 2005, 38: 244-254.
Machado FS, Koyama NS, Carregaro V, Ferreira BR, Milanezi CM, Teixeira MM, Rossi MA, Silva JS: CCR5 plays a critical role in the development of myocarditis and host protection in mice infected with Trypanosoma cruzi . J Infect Dis 2005, 191: 627-636. 10.1086/427515
Hardison JL, Wrightsman RA, Carpenter PM, Kuziel WA, Lane TE, Manning JE: The CC chemokine receptor 5 is important in control of parasite replication and acute cardiac inflammation following infection with Trypanosoma cruzi . Infect Immun 2006, 74: 135-143. 10.1128/IAI.74.1.135-143.2006
Thio CL, Astemborski J, Bashirova A, Mosbruger T, Greer S, Witt MD, Goedert JJ: Hilgartner M, Majeske A, O'Brien SJ, Thomas DL: Carrington M., Genetic protection against hepatitis B virus conferred by CCR5-Δ32 : evidence that CCR5 contributes to viral persistence. J Virol 2007, 81: 441-445. 10.1128/JVI.01897-06
Goulding C, McManus R, Murphy A, MacDonald G, Barrett S, Crowe J, Hegarty J, McKiernan S, Kelleher D: The CCR5-Δ32 mutation: impact on disease outcome in individuals with hepatitis C infection from a single source. Gut 2005, 54: 1157-1161. 10.1136/gut.2004.055699
Kindberg E, Mickiene A, Ax C, Akerlind B, Vene S, Lindquist L, Lundkvist A, Svensson L: A deletion in the chemokine receptor 5 (CCR5) gene is associated with tickborne encephalitis. J Infect Dis 2008, 197: 266-269. 10.1086/524709
Lim JK, Louie CY, Glaser C, Jean C, Johnson B, Johnson H, McDermott DH, Murphy PM: Genetic deficiency of chemokine receptor CCR5 is a strong risk factor for symptomatic West Nile virus infection: a meta-analysis of 4 cohorts in the US epidemic. J Infect Dis 2008, 197: 262-265. 10.1086/524691
Glass WG, Lim JK, Cholera R, Pletnev A, Gao JL, Murphy PM: Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J Exp Med 2005, 202: 1087-1098. 10.1084/jem.20042530
Stephens JC, Reich DE, Goldstein DB, Shin HD, Smith MW, Carrington M, Winkler C, Huttley GA, Allikmets R, Schriml L, Gerrard B, Malasky M, Ramos MD, Morlot S, Tzetis M, Oddoux C, di Giovine FS, Nasioulas G, Chandler D, Aseev M, Hanson M, Kalaydjieva L, Glavac D, Gasparini P, Kanavakis E, Claustres M, Kambouris M, Ostrer H, Duff G, Baranov V, Sibul H, Metspalu A, Goldman D, Martin N, Duffy D, Schmidtke J, Estivill X, O'Brien SJ, Dean M: Dating the origin of the CCR5-Δ32 AIDS-resistance allele by the coalescence of haplotypes. Am J Hum Genet 1998, 62: 1507-1515. 10.1086/301867
Bowler JM, Johnston H, Olley JM, Prescott JR, Roberts RG, Shawcross W, Spooner NA: New ages for human occupation and climatic change at Lake Mungo, Australia. Nature 2003, 421: 837-840. 10.1038/nature01383
Schöneberg T, Hofreiter M, Schulz A, Römpler H: Learning from the past: evolution of GPCR functions. Trends Pharmacol Sci 2007, 28: 117-121. 10.1016/j.tips.2007.01.001
Zawicki P, Witas HW: HIV-1 protecting CCR5-Delta32 allele in medieval Poland. Infect Genet Evol 2008, 8: 146-151. 10.1016/j.meegid.2007.11.003
Green RE, Krause J, Ptak SE, Briggs AW, Ronan MT, Simons J, Du L, Egholm M, Rothberg JM, Paunovic M, Pääbo S: Analysis of one million base pairs of Neanderthal DNA. Nature 2006, 444: 330-336. 10.1038/nature05336
Noonan JP, Coop G, Kudaravalli S, Smith D, Krause J, Alessi J, Chen F, Platt D, Pääbo S, Pritchard JK, Rubin EM: Sequencing and analysis of Neanderthal genomic DNA. Science 2006, 314: 1113-1118. 10.1126/science.1131412
Currat M, Excoffier L: Modern humans did not admix with Neanderthals during their range expansion into Europe. PLoS Biol 2004, 2: e421. 10.1371/journal.pbio.0020421
Hodgson JA, Disotell TR: No evidence of a Neanderthal contribution to modern human diversity. Genome Biol 2008, 9: 206. 10.1186/gb-2008-9-2-206
La VD, Clavel B, Lepetz S, Aboudharam G, Raoult D, Drancourt M: Molecular detection of Bartonella henselae DNA in the dental pulp of 800-year-old French cats. Clin Infect Dis 2004, 39: 1391-1394. 10.1086/424884
Drancourt M, Tran-Hung L, Courtin J, Lumley H, Raoult D: Bartonella quintana in a 4000-year-old human tooth. J Infect Dis 2005, 191: 607-611. 10.1086/427041
Li HC, Fujiyoshi T, Lou H, Yashiki S, Sonoda S, Cartier L, Nunez L, Munoz I, Horai S, Tajima K: The presence of ancient human T-cell lymphotropic virus type I provirus DNA in an Andean mummy. Nat Med 1999, 5: 1428-1432. 10.1038/15256
Sonoda S, Li HC, Cartier L, Nunez L, Tajima K: Ancient HTLV type 1 provirus DNA of Andean mummy. AIDS Res Hum Retroviruses 2000, 16: 1753-1756. 10.1089/08892220050193263
Janczewski DN, Yuhki N, Gilbert DA, Jefferson GT, O'Brien SJ: Molecular phylogenetic inference from saber-toothed cat fossils of Rancho La Brea. Proc Natl Acad Sci USA 1992, 89: 9769-9773. 10.1073/pnas.89.20.9769
Burger J, Rosendahl W, Loreille O, Hemmer H, Eriksson T, Götherström A, Hiller J, Collins MJ, Wess T, Alt KW: Molecular phylogeny of the extinct cave lion Panthera leo spelaea . Mol Phylogenet Evol 2004, 30: 841-849. 10.1016/j.ympev.2003.07.020
Hall TA: BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 1999, 41: 95-98.
Saitou N, Nei M: The neighbour-joining method: A new method for reconstructing phylogenetic trees. Mol Biol Evol 1987, 4: 406-425.
Felsenstein J: PHYLIP (Phylogeny Inference Package), version 3.6. Seattle, Washington: University of Washington, Department of Genetics; 2002.
Acknowledgements
We thank helpful comments on the manuscript were provided by Prof. J.P. Casanova (University of Provence, France).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author declares that he has no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Faure, E. Could FIV zoonosis responsible of the breakdown of the pathocenosis which has reduced the European CCR5-Delta32 allele frequencies?. Virol J 5, 119 (2008). https://doi.org/10.1186/1743-422X-5-119
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-5-119