
Abstract
Background
A characteristic difference between highly and non-highly pathogenic avian influenza strains is the presence of an extended, often multibasic, cleavage motif insertion in the hemagglutinin protein. Such motif is found in H7N3 strains from chicken farm outbreaks in 2012 in Mexico.
Methods
Through phylogenetic, sequence and structural analysis, we try to shed light on the role, prevalence, likelihood of appearance and origin of the inserted cleavage motifs in these H7N3 avian influenza strains.
Results
The H7N3 avian influenza strain which caused outbreaks in chicken farms in June/July 2012 in Mexico has a new extended cleavage site which is the likely reason for its high pathogenicity in these birds. This cleavage site appears to have been naturally acquired and was not present in the closest low pathogenic precursors. Structural modeling shows that insertion of a productive cleavage site is quite flexible to accept insertions of different length and with sequences from different possible origins. Different from recent cleavage site insertions, the origin of the insert here is not from the viral genome but from host 28S ribosomal RNA (rRNA) instead. This is a novelty for a natural acquisition as a similar insertion has so far only been observed in a laboratory strain before. Given the abundance of viral and host RNA in infected cells, the acquisition of a pathogenicity-enhancing extended cleavage site through a similar route by other low-pathogenic avian strains in future does not seem unlikely. Important for surveillance of these H7N3 strains, the structural sites known to enhance mammalian airborne transmission are dominated by the characteristic avian residues and the risk of human to human transmission should currently be low but should be monitored for future changes accordingly.
Conclusions
This highly pathogenic H7N3 avian influenza strain acquired a novel extended cleavage site which likely originated from recombination with 28S rRNA from the avian host. Notably, this new virus can infect humans but currently lacks critical host receptor adaptations that would facilitate human to human transmission.
Similar content being viewed by others

Background
Influenza viruses are classified into 3 different types (A,B,C) and influenza A is further divided into specific subtypes named after the respective combination of surface protein variants pairing 1 of 17 hemagglutinins (the “H” in HxNx) with 1 of 10 neuraminidases (the “N” in HxNx). These subtypes are known to circulate preferably in specific bird species which possess sialic acid linked to oligosaccharides via alpha (2,3) linkages, such as chickens, turkeys, and ducks.[1, 2]. There has been a recent outbreak of a new H7N3 strain in chicken farms in Mexico in June/July 2012, characterized as a highly pathogenic avian influenza (HPAI) strain[3]. While the epidemiological and initial genetic characterization of this outbreak strain has been described elsewhere[4, 5], we would like to add information on the detailed origin of the extended cleavage site possibly responsible for making the strain highly pathogenic. The hemagglutinin cleavage site in the influenza A HA0 precursor protein typically contains a monobasic cleavage site with the consensus motif Q/E-x-R, allowing for cleavage of the HA after the “R”, usually by trypsin, into the HA1 and HA2 proteins[6]. In highly pathogenic avian influenza (HPAI) viruses, the HA0 cleavage site usually contains a multibasic cleavage site (MBCS) corresponding to a canonical R-x-K/R-R motif, suggesting that this motif is at least partially involved in the increased pathogenicity of the given HPAI strain. However, in some HPAI strains, in place of an MBCS, observations have been made of an extended cleavage site with multiple basic residues at positions other than the canonical site, which usually conform to the minimal R-x-x-R cleavage motif. Such motif differences can still result in functional cleavage sites, possibly changing the range of proteases or the same protease with different efficiencies. Gain of function of cleavability by ubiquitously expressed proteases opens the door for systemic replication of the virus and consequently increased pathogenicity[7]. Of particular interest is the situation of the inserted extended cleavage site (PENPK-DRKSRHRR TR/GLF, insertion in bold) in HA of A/chicken/Jalisco/CPA1/2012(H7N3). Firstly, it turns the classical monobasic cleavage motif into an extended RxxR cleavage site which could be targeted by an increased range of proteases, including matriptase among others. Secondly, with a register shift of two positions in N-terminal direction, there is also a canonical multibasic cleavage motif (RHRR = R-x-K/R-R) which could be hypothesized to be cleavable by furin or other subtilisin-like proteases. Multibasic cleavage sites (MBCS) in the influenza A hemagglutinin protein have been studied extensively in the context of pathogenicity in different viruses[6–8]. However, only H5 and H7 subtypes have been known to naturally acquire MBCSs, and this acquisition has been attributed to 2 distinct mechanisms, either by the random insertion or gradual accumulation of basic amino acids through mutations[9, 10], or by recombination either with viral or host RNA[11, 12], a phenomenon which has only been observed in H7 strains[11]. While the insertion of an MBCS is sufficient to turn low pathogenicity strains (LPAI) into high pathogenicity strains (HPAI) in chickens[13, 14], this pathogenicity increase is not consistently observed in other poultry species such as ducks. This suggests that the acquisition of an MBCS is not the only pathogenicity determinant in these species – indeed, there are physiological differences between ducks and chickens, such as the lack of RIG-I in chickens, as well as differences in the upregulation of pro-inflammatory cytokines and interferons in response to HPAI infection[15]. Moreover, the increase of pathogenicity does not seem to translate directly to mammalian systems[16, 17]. However, MBCS acquisition has been seen to alter the route for systemic infections[18]. Here, we used phylogenetic, sequence and structural analysis to shed light on the origin of the extended cleavage motif in this H7N3 avian influenza strain from recombination with host 28S rRNA as well as evaluate its potential for human-to-human transmission.
Results and discussion
First, we investigated the frequency of extended cleavage site motifs of H7N3 strains in the EpiFlu database of the Global Initiative on Sharing All Influenza Data (GISAID) since the year 2000 (Figure 1). It can be seen that most H7N3 strains collected from 2006 to 2011 lack extended cleavage motifs with the consensus R-x-x-R, while in 2012 in the Mexican chicken farm outbreak sequences, we do see a prominent reappearance of such a motif.

Frequency of occurrence of H7N3 strains with and without extended cleavage site. 257 HAs from H7N3 strains since the year 2000 were downloaded from GISAID and their absolute numbers per year shown as bar plot (values on left axis). The orange curve shows the percentage of sequences with extended cleavage site as defined by a consensus motif of R-x-x-R (where the final R is the normal cleavage site without insertion).
Since the extended and multibasic cleavage sites are of importance for pathogenicity potential of influenza strains, we tried to shed further light on its possible origins. In the case of the previous occurrence of a highly pathogenic H7N3 in an outbreak in British Columbia in February 2004, it was found that the insertion resulting in the extended HA cleavage site was derived from intersegmental recombination from the matrix gene of the same virus[11]. Similarily, H7N3 strains from an outbreak in Chile in 2002 also had an extended cleavage site inserted through intersegmental recombination but from the viral NP gene instead[12]. Consequently, we tried to find if the extended cleavage site from the current Mexican H7N3 strains would have a similar intersegmental origin by comparing it to all segments of the virus using BLAST[19] with settings for small query sequences. However, as there was no significant hit in the genome of the virus itself we extended the search to all other known influenza viruses (potential for co-infection) and again did not find a hit. Next, we searched the nr/nt database[20] with restriction to chicken sequences and found a perfect match to a chicken 28S ribosomal RNA (rRNA) covering all 24 inserted nucleotides at 100% identity (E-value 4e-05). While the acquisition of an extended cleavage site by recombination has been rarely reported in comparison to random insertions of basic amino acids[11], having 28S rRNA as source for recombination is not surprising as it is an important molecule with high copy number in eukaryotes, including chicken[21]. Indeed, a similar insertion through recombination with host 28S rRNA has already been described previously[22] in an H7N3 lab strain from 1971 (…PENPKT-SLSPLYPGRTTDLQVPTA-R/GLF…, insertion bold between hyphens, classical cleavage site indicated by “/”). Although both insertions come from the 28S rRNA host gene, they are derived from different regions of the gene as can be seen by the different insertion sequence in the 2012 H7N3 strains (…PENPK-DRKSRHRR-TR/GLF…) and are certainly independent recombination events. While the 1971 insertion was only observed in a lab strain, it is noteworthy that the 2012 Mexican H7N3 acquisition of the extended cleavage site from host 28S rRNA appeared to have been the first observation of natural recombination of this kind.
Although the 28S rRNA origin of this 24 nucleotide sequence is unambiguous as the same sequence is not found in any other gene with the described searches, the exact genome mapping of 28S rRNA genes is tricky as they are encoded in repeated blocks in different copy numbers on different chromosomes with variation among individuals and, therefore, often omitted from reference genome assemblies. It has to be noted that due to the high conservation of the 28S rRNA, there are in principle also several other organisms including other birds, horses, pigs and even humans that share the same 100% identical fragment. Therefore, while it can be deduced that the insertion most likely originated from a eukaryotic host 28S rRNA, one cannot unambiguously identify this host, although an avian host appears the most likely scenario. Also, it is not possible to distinguish if the insertion first happened in the chicken farm or already before in wildbirds. Nevertheless, in this context it is interesting to note that some but not all chicken lines selected for enhanced growth or egg quality, size and number appear to have an increased rRNA gene copy number[23].
Considering the mechanism of recombination, previous studies[11, 12, 24] tried to find palindromic sequences at the junction of the insert to strengthen the possibility of RNA recombination but only for the 1971 insertion such motif was reported[22]. We reanalyzed previous and the current instances of cleavage site insertions for H7 strains and found previously undetected palindromic sequences at regions surrounding the respective inserts. For most of the palindromic sequences in Table 1, the midpoint of the pair of palindromic sequences occurs either exactly or in the vicinity from the start of the insert, thereby providing a plausible explanation as to how insertion might occur at the given site. While we also find a candidate palindromic recombination motif for the 2012 Mexican sequence, we acknowledge that this motif is short which increases the chance of random occurrence and not ideally positioned relative to the insertion site. Consequently, the exact mechanism of recombination for this particular insert remains to be elucidated.
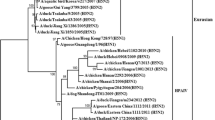
In order to investigate if the extended cleavage site was also present in possible phylogenetic precursor strains, we analyzed the relation of H7N3 hemagglutinin sequences from 2000 until 2012. The phylogenetic tree (Figure 2) shows a scattered pattern of strains with and without the generic R-x-x-R cleavage site motifs and appearance of the current 2012 Mexican motif cannot be explained by descendance from an old strain that already had the insertion but, instead, it is missing in the genetically most closely related strains from the preceding years. Therefore, this extended cleavage site appears to be a recent acquisition which is consistent with earlier analysis of H7 evolution and lineages[1, 25]. Looking at the hemagglutinin (HA) nucleotide sequences, the closest phylogenetic relatives with sequences in GISAID match the geotemporal context of occurrence in wild birds in southern states of the US in the preceding years, e.g. A/mallard/Missouri/220/2009(H7N3). Interestingly, when including all H7 subtypes into the analysis (Additional file1: Figure S1), reassortment history of the ancestral H7 of the current H7N3 strains also includes recent combinations with N5 and N7, e.g. A/mallard/California/1390/2010(H7N5) and A/northernshoveler/Mississippi/09OS643/2009(H7N7). Therefore, a future detailed analysis of the reassortment history of these strains including all segments would be of interest. It should be noted that also the other close relatives from different H7Nx subtypes did not have the additional cleavage motif insertion.

Phylogenetic relationship between 205 H7N3 full-length HA nucleotide sequences collected from 2000 till 2012. Strains with extended cleavage sites representing high pathogenic avian influenza (HPAI) strains as reported in the literature were separated by geographical regions and different years of outbreak and were highlighted with 5 different colors indicated in the legend. The recent 2012 Mexican chicken farm outbreak arose from a cluster of originally low pathogenic strains and therefore independently acquired the extended cleavage site which likely switched the strain to highly pathogenic.
The structural position of the inserted cleavage site is in the HA stem and away from the head region where functionally important host receptor and antibody binding sites are located (Figure 3). On the other hand, cleavage at this HA stem site is required for conformational changes allowing entry of the virus[6]. The assumed biomolecular mechanism of the increased pathogenicity in chickens through an extended cleavage motif is a gain of a trypsin-independent cleavage site which increases cleavage efficiency through utilizing ubiquitous proteases such as furin and other subtilisin-like proteases allowing infection of more cell types and tissues[7, 26, 27]. We show in a representative structural model of the hemagglutinin from this H7N3 strain using Yasara Structure[28] that the newly inserted cleavage site is, as expected, at the protein surface and accessible for protease cleavage (Figure 3). As seen in the crystal structure of furin with a substrate analog[29], the substrate cleavage motif structure appears linear and this is in agreement with linearly extended conformations accessible to the loop region as exemplified in the model. Furthermore, the dynamic structure of the insertion loop also suggests that mainly the relative but not absolute position of the positive charges for the new cleavage motif seems restricted and that it could flexibly accommodate different arrangements of related cleavage motifs of different length and sequence origin which increases the likelihood of insertion of a productive cleavage site and, hence, facilitates extended cleavage site occurrence. In addition to this, examination of the entire insert in the modified cleavage site using the ProP 1.0 furin cleavage predictor[30] has identified both the original RRTR motif at the P4-P1 positions, which conforms to the minimal requirement (R-x-x-R) for furin cleavage[31] as well as RHRR at the P5-P2 positions, which conforms to the canonical R-x-K/R-R furin cleavage site, as predicted furin substrates. Given the presence of these 2 motifs in such close proximity, and previous estimations of a log-difference in affinity between the canonical and minimal furin cleavage motifs[31], it is quite likely that the RHRR in the extended cleavage site would be preferentially bound in comparison to the existing monobasic RRTR cleavage site, allowing for non-specific cleavage by ubiquitous proteases.

Structure of the Influenza A hemagglutinin monomer showing a representative extended cleavage site. A) The crystal structure representation of an uncleaved hemagglutinin HA0 precursor from subtype H3 (PDBID: 1HA0 [6], cleavage region green) overlaid with a structure of a cleaved HA1/HA2 heterodimer from a highly pathogenic H7 virus (PDBID: 4DJ6 [40], cleavage region blue) and an energy-minimized average representation of the cleavage loop for the highly pathogenic H7 with the new insertion modelled in YASARA Structure (yellow). Cleavage site indicated with dashed box at the stem region. B) Detailed structure of hemagglutinin cleavage site. Uncleaved loop (PDBID: 1HA0, green) shown with cleaved ends (PDBID: 4DJ6, blue) Glu317 of HA1 and Gly1 of HA2 connected with pink dots representative of the cleaved loop fragment. Basic arginine residues at positions 323, 326, 328 and 329 in the loop model are indicated.
The 2012 motif’s uniform presence in the outbreak sequences indicates that it must have quickly replaced any precursor without motif indicating a possible advantage for the new virus through the additional cleavage site, although this will have to be confirmed experimentally. In the case of the related H5N1 avian influenza viruses, there has been some increase in measures of severity in mice and ferrets through addition of an MBCS motif while there has been a lack of increased pathogenicity in non-human primate hosts[17]. In principle, H7 viruses have the potential to infect humans[2] and there have indeed been two human cases linked to the recent Mexican H7N3 outbreak[5]. Both cases recovered fully and only had mild symptoms, such as conjunctivitis. In this context it is important to note that animal to human influenza transmissions are rare and most often limited to close contact with the respective animals[32] as was also the case for the human H7N3 infections in Mexico[5].
Recently, 7 candidate positions have been identified where sets of 4 to 5 mutations allowed airborne transmission between ferrets of influenza viruses with an avian-derived hemagglutinin[33–35]. As the ferret setup serves as model for human to human transmission, we investigated the status of these key structural positions for transmission in the current H7N3 virus (including the human infection case with available HA virus sequence: A/Mexico/InDRE7218/2012(H7N3)). The human and chicken derived strains are identical at these positions and only one out of 7 candidate positions has the mammal-adapted residue while the others show the typically avian-cell preferring residues (Table 2). Since a critical number of 4 or more such adaptive mutations would be necessary to facilitate mammalian transmission[34], the risk for human-to-human transmissions of the current strain should be low.
Conclusions
This H7N3 outbreak strain is of special interest as an extended cleavage site including a shifted multibasic cleavage site has been newly acquired by the virus with likely origin from host 28S rRNA. We discuss that structural insertion of a productive cleavage site is quite flexible to accept insertions of different length and with sequences from different possible origins. Given the abundance of viral and certain host RNA in infected cells, the acquisition of a pathogenicity-enhancing extended cleavage site through a similar route by other low-pathogenic avian strains is possible, although other mechanisms of basic residue introduction through mutation proximal to the hemagglutinin cleavage site may be more common. Importantly, although this virus may be highly pathogenic in chickens, the few reported cases of human infections seemed to have had only mild symptoms and the structural sites known to enhance mammalian airborne transmission currently are dominated by the characteristic avian residues, so the risk for human-to-human transmission is low. Nevertheless, these positions should continue to be monitored if this strain continues to cause outbreaks in birds or even further human infections.
Methods
Extended cleavage site motif definition and determination of pathogenicity
In this study, the extended cleavage is defined by the consensus motif of R-x-x-R (where the final R is the normal cleavage site without insertion). The R-x-x-R sequence motif was used instead of the canonical R-x-R/K-R (commonly referred to as multibasic cleavage site) because there are examples of confirmed highly pathogenic strains that do not match the restrictive R-x-R/K-R pattern (for example A/chicken/Chile/2002 with motif R-E-T-R and A/chicken/BC/2004 with motif R-M-T-R, see Additional file2: Table S1). Hence, the more general R-x-x-R motif has been used in our analyses. It should be noted that this did not increase the number of strains classified as HPAI except for including the Chile and BC strains with the degenerate motifs. Consequently, the large majority of strains analyzed here as having an extended cleavage site also conform to the canonical multibasic cleavage site. At the same time, MBCS or extended cleavage motif presence does not guarantee increased HA cleavability. Similarly, increased cleavability can, but also not necessarily has to, result in increased pathogenicity. Due to its importance, the endpoint of low and high pathogenicity is analyzed more often compared to HA cleavability directly and the experimental test of low or high pathogenicity as reported in the literature is hence used in this work as indirect evidence for increased cleavability of the observed motifs. Therefore, all H7Nx sequences that were found to contain the extended R-x-x-R motif (Additional file2: Table S1) were traced to the source literature of their outbreak where they were distinguished as either LPAI strains or HPAI strains by the intravenous pathogenicity index in chickens as reported in the literature.
Sequence data
257 HAs from H7N3 strains since the year 2000 with protein sequence information around the cleavage site were downloaded from the EpiFlu database of the Global Initiative on Sharing All Influenza Data (GISAID) and used to count the occurrence of extended cleavage sites in recent H7N3 sequences (Figure 1). For the phylogenetic analysis (Figure 2), 205 isolates (a subset of the 257 above) with complete HA nucleotide sequences were used. Another phylogenetic analysis was conducted on 1032 H7Nx strains using complete HA nucleotide sequences since the earliest available H7 sequence in GISAID in the year 1902. Files with the complete list of isolates and acknowledgment of submitting laboratories are available in Additional files 3 and 4 from the journal website.
BLAST search: The origin of the cleavage site inserts with length of 16 bases or more were derived by searching against the chicken reference genome using the NCBI megablast[19]. Next, the inserts were searched against the NCBI non-redundant database limited to chicken taxid for the best hit. The best hit was subsequently searched against the chicken reference genome to ensure that the insert and the predicted gene (also the best blast hit) maps to the same genomic location. The inserts were also searched against the non-redundant database limited to bird taxid and mammal taxid, and against influenza viruses.
Search for palindromic sequences in vicinity of the insert
The nucleotide sequences of the predicted origin of the insert were aligned with representative H7 HA sequences. 25 bases flanking the insert were searched for palindromic sequences.
Phylogenetic analyses
To examine the relationship between recent H7N3 outbreaks, 205 H7N3 full-length HA nucleotide sequences (collection date from 2000 till 2012) were aligned with MAFFT[36]. Next, a maximum likelihood tree was constructed using PHYML[37] with bootstrap test (500 steps), the HKY85 substitution model with gamma distribution (4 categories) and shape parameter (0.372) estimated by the program. The tree is displayed and colored in MEGA[38]. In order to understand the phylogeny of the 2012 Mexican strains (Additional file1: Figure S1), 1032 H7Nx full-length HA nucleotide sequences (collection date from 1902 till 2012) were similarly aligned with MAFFT[36] and a neighbour joining tree using the Tamura-Nei model with gamma distribution (5 categories) was generated with MEGA[38].
Structural modeling
The HA structure with cleavage loop for the highly pathogenic H7N3 strain was modelled in YASARA using the homology modelling procedure used in the CASP competition which has been shown to give accurate structures in the model refinement category[28]. The hemagglutinin sequence from A/chicken/Jalisco/Jal0612/2012 was used as a target and HA monomers from 2 subtype H7 structures (PDBID: 3M5G[39] and PDBID: 4DJ6[40]) served as templates. It is important to note that the loop is flexible and can take up multiple conformations which are however constrained by the fixed endpoints and similar to each other and the minimized average conformation is shown in the Figure.
Abbreviations
- HA:
-
Hemagglutinin
- MBCS:
-
Multibasic cleavage site
- rRNA:
-
Ribosomal RNA.
References
Lebarbenchon C, Stallknecht DE: Host shifts and molecular evolution of H7 avian influenza virus hemagglutinin. Virol J 2011, 8: 328. 10.1186/1743-422X-8-328
Gambaryan AS, Matrosovich TY, Philipp J, Munster VJ, Fouchier RAM, Cattoli G, Capua I, Krauss SL, Webster RG, Banks J, Bovin NV, Klenk H-D, Matrosovich MN: Receptor-binding profiles of H7 subtype influenza viruses in different host species. J Virol 2012, 86: 4370-4379. 10.1128/JVI.06959-11
Highly pathogenic avian influenza, Mexico. Follow-up report No. 1. Information received on 26/06/2012 from Dr Hugo fragoso sánchez, director general de salud animal, SENASICA, SAGARPA, Mexico. http://www.oie.int/wahis_2/public/wahid.php/Reviewreport/Review?reportid=12074
FAO: Highly pathogenic avian influenza in Mexico (H7N3) - a significant threat to poultry production not to be underestimated. EMPRES WATCH 2012., 26: http://www.fao.org/docrep/016/an395e/an395e.pdf
Notes from the field: highly pathogenic avian influenza a (H7N3) virus infection in Two poultry workers — Jalisco, Mexico, July 2012. [http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6136a4.htm?s_cid=mm6136a4_e]
Chen J, Lee KH, Steinhauer DA, Stevens DJ, Skehel JJ, Wiley DC: Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 1998, 95: 409-417. 10.1016/S0092-8674(00)81771-7
Rott R, Klenk HD, Nagai Y, Tashiro M: Influenza viruses, cell enzymes, and pathogenicity. Am J Respir Crit Care Med 1995, 152: S16-19. 10.1164/ajrccm/152.4_Pt_2.S16
Zambon MC: The pathogenesis of influenza in humans. Rev Med Virol 2001, 11: 227-241. 10.1002/rmv.319
Horimoto T, Rivera E, Pearson J, Senne D, Krauss S, Kawaoka Y, Webster RG: Origin and molecular changes associated with emergence of a highly pathogenic H5N2 influenza virus in Mexico. Virology 1995, 213: 223-230. 10.1006/viro.1995.1562
García M, Crawford JM, Latimer JW, Rivera-Cruz E, Perdue ML: Heterogeneity in the haemagglutinin gene and emergence of the highly pathogenic phenotype among recent H5N2 avian influenza viruses from Mexico. J Gen Virol 1996,77(Pt 7):1493-1504.
Pasick J, Handel K, Robinson J, Copps J, Ridd D, Hills K, Kehler H, Cottam-Birt C, Neufeld J, Berhane Y, Czub S: Intersegmental recombination between the haemagglutinin and matrix genes was responsible for the emergence of a highly pathogenic H7N3 avian influenza virus in British Columbia. J Gen Virol 2005, 86: 727-731. 10.1099/vir.0.80478-0
Suarez DL, Senne DA, Banks J, Brown IH, Essen SC, Lee C-W, Manvell RJ, Mathieu-Benson C, Moreno V, Pedersen JC, Panigrahy B, Rojas H, Spackman E, Alexander DJ: Recombination resulting in virulence shift in avian influenza outbreak. Chile. Emerging Infect. Dis. 2004, 10: 693-699. 10.3201/eid1004.030396
Veits J, Weber S, Stech O, Breithaupt A, Gräber M, Gohrbandt S, Bogs J, Hundt J, Teifke JP, Mettenleiter TC, Stech J: Avian influenza virus hemagglutinins H2, H4, H8, and H14 support a highly pathogenic phenotype. Proc Natl Acad Sci USA 2012, 109: 2579-2584. 10.1073/pnas.1109397109
Munster VJ, Schrauwen EJA, De Wit E, Van den Brand JMA, Bestebroer TM, Herfst S, Rimmelzwaan GF, Osterhaus ADME, Fouchier RAM: Insertion of a multibasic cleavage motif into the hemagglutinin of a low-pathogenic avian influenza H6N1 virus induces a highly pathogenic phenotype. J Virol 2010, 84: 7953-7960. 10.1128/JVI.00449-10
Liang Q, Luo J, Zhou K, Dong J, He H: Immune-related gene expression in response to H5N1 avian influenza virus infection in chicken and duck embryonic fibroblasts. Mol Immunol 2011, 48: 924-930. 10.1016/j.molimm.2010.12.011
Schrauwen EJA, Bestebroer TM, Munster VJ, De Wit E, Herfst S, Rimmelzwaan GF, Osterhaus ADME, Fouchier RAM: Insertion of a multibasic cleavage site in the haemagglutinin of human influenza H3N2 virus does not increase pathogenicity in ferrets. J Gen Virol 2011, 92: 1410-1415. 10.1099/vir.0.030379-0
Suguitan AL Jr, Matsuoka Y, Lau Y-F, Santos CP, Vogel L, Cheng LI, Orandle M, Subbarao K: The multibasic cleavage site of the hemagglutinin of highly pathogenic A/Vietnam/1203/2004 (H5N1) avian influenza virus acts as a virulence factor in a host-specific manner in mammals. J Virol 2012, 86: 2706-2714. 10.1128/JVI.05546-11
Schrauwen EJA, Herfst S, Leijten LM, Van Run P, Bestebroer TM, Linster M, Bodewes R, Kreijtz JHCM, Rimmelzwaan GF, Osterhaus ADME, Fouchier RAM, Kuiken T, Van Riel D: The multibasic cleavage site in H5N1 virus is critical for systemic spread along the olfactory and hematogenous routes in ferrets. J Virol 2012, 86: 3975-3984. 10.1128/JVI.06828-11
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997, 25: 3389-402. 10.1093/nar/25.17.3389
Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Federhen S, Feolo M, Fingerman IM, Geer LY, Helmberg W, Kapustin Y, Landsman D, Lipman DJ, Lu Z, Madden TL, Madej T, Maglott DR, Marchler-Bauer A, Miller V, Mizrachi I, Ostell J, Panchenko A, Phan L, Pruitt KD, Schuler GD, Sequeira E, Sherry ST, Shumway M, Sirotkin K, Slotta D, Souvorov A, Starchenko G, Tatusova TA, Wagner L, Wang Y, Wilbur WJ, Yaschenko E, Ye J: Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 2011, 39: D38-51. 10.1093/nar/gkq1172
Delany ME, Muscarella DE, Bloom SE: Effects of rRNA gene copy number and nucleolar variation on early development: inhibition of gastrulation in rDNA-deficient chick embryos. J Hered 1994, 85: 211-217.
Khatchikian D, Orlich M, Rott R: Increased viral pathogenicity after insertion of a 28S ribosomal RNA sequence into the haemagglutinin gene of an influenza virus. Nature 1989, 340: 156-157. 10.1038/340156a0
Su MH, Delany ME: Ribosomal RNA gene copy number and nucleolar-size polymorphisms within and among chicken lines selected for enhanced growth. Poult Sci 1998, 77: 1748-1754.
Orlich M, Gottwald H, Rott R: Nonhomologous recombination between the hemagglutinin gene and the nucleoprotein gene of an influenza virus. Virology 1994, 204: 462-465. 10.1006/viro.1994.1555
Röhm C, Horimoto T, Kawaoka Y, Süss J, Webster RG: Do hemagglutinin genes of highly pathogenic avian influenza viruses constitute unique phylogenetic lineages? Virology 1995, 209: 664-670. 10.1006/viro.1995.1301
Bertram S, Glowacka I, Steffen I, Kühl A, Pöhlmann S: Novel insights into proteolytic cleavage of influenza virus hemagglutinin. Rev Med Virol 2010, 20: 298-310. 10.1002/rmv.657
Morsy J, Garten W, Rott R: Activation of an influenza virus A/turkey/Oregon/71 HA insertion variant by the subtilisin-like endoprotease furin. Virology 1994, 202: 988-991. 10.1006/viro.1994.1424
Krieger E, Joo K, Lee J, Lee J, Raman S, Thompson J, Tyka M, Baker D, Karplus K: Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins 2009,77(Suppl 9):114-122.
Henrich S, Cameron A, Bourenkov GP, Kiefersauer R, Huber R, Lindberg I, Bode W, Than ME: The crystal structure of the proprotein processing proteinase furin explains its stringent specificity. Nat Struct Biol 2003, 10: 520-526. 10.1038/nsb941
Duckert P, Brunak S, Blom N: Prediction of proprotein convertase cleavage sites. Protein Eng Des Sel 2004, 17: 107-112. 10.1093/protein/gzh013
Nakayama K: Furin: a mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem J 1997,327(Pt 3):625-635.
Maurer-Stroh S, Paing SST, Lee RTC, Eisenhaber F: Sporadic human cases of swine-origin influenza before 2009 share the Sa epitope. Cell Cycle 2010, 9: 3826-3828. 10.4161/cc.9.18.13166
Herfst S, Schrauwen EJA, Linster M, Chutinimitkul S, De Wit E, Munster VJ, Sorrell EM, Bestebroer TM, Burke DF, Smith DJ, Rimmelzwaan GF, Osterhaus ADME, Fouchier RAM: Airborne transmission of influenza A/H5N1 virus between ferrets. Science 2012, 336: 1534-1541. 10.1126/science.1213362
Russell CA, Fonville JM, Brown AEX, Burke DF, Smith DL, James SL, Herfst S, Van Boheemen S, Linster M, Schrauwen EJ, Katzelnick L, Mosterín A, Kuiken T, Maher E, Neumann G, Osterhaus ADME, Kawaoka Y, Fouchier RAM, Smith DJ: The potential for respiratory droplet-transmissible A/H5N1 influenza virus to evolve in a mammalian host. Science 2012, 336: 1541-1547. 10.1126/science.1222526
Imai M, Watanabe T, Hatta M, Das SC, Ozawa M, Shinya K, Zhong G, Hanson A, Katsura H, Watanabe S, Li C, Kawakami E, Yamada S, Kiso M, Suzuki Y, Maher EA, Neumann G, Kawaoka Y: Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature 2012, 486: 420-428.
Katoh K, Toh H: Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinformatics 2008, 9: 286-98. 10.1093/bib/bbn013
Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O: New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 2010, 59: 307-321. 10.1093/sysbio/syq010
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011, 28: 2731-2739. 10.1093/molbev/msr121
Yang H, Chen L-M, Carney PJ, Donis RO, Stevens J: Structures of receptor complexes of a North American H7N2 influenza hemagglutinin with a loop deletion in the receptor binding site. PLoS Pathog 2010, 6: e1001081. 10.1371/journal.ppat.1001081
Yang H, Carney PJ, Donis RO, Stevens J: Structure and Receptor Complexes of the Hemagglutinin from a Highly Pathogenic H7N7 Influenza Virus. J Virol 2012, 86: 8645-8652. 10.1128/JVI.00281-12
Acknowledgements
We would like to particularly acknowledge the laboratories that made the Mexican outbreak sequences publicly available (INDRE - Instituto Nacional de Diagnostico y Referencia Epidemiologicos; CENAPA - Centro Nacional de Servicios de Constatacion en Salud Animal; CPA - Mexico-United States Commission for the Prevention of the Foot and Mouth Disease and Other Exotic Diseases of Animals). We would also like to acknowledge and list all laboratories that submitted the complete set of sequences used for the phylogenetic and motif occurrence analyses to Genbank or GISAID. Due to space constraints for the publication, the respective list can be found online in Additional files 1 and 2 from the journal website.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare no competing interests.
Authors’ contributions
SMS and FE conceived of the study. RTCL and SMS analyzed the motif occurrence and insertion origin. RTCL contributed the phylogenetic analysis. VG and SMS carried out the structural modeling. RTCL, VG, SMS and FE wrote parts of the manuscript. All authors read and approved the final manuscript.
Sebastian Maurer-Stroh, Raphael TC Lee, Vithiagaran Gunalan contributed equally to this work.
Electronic supplementary material
12985_2012_2154_MOESM2_ESM.docx
Additional file 2:We acknowledge the authors, originating and submitting laboratories of the sequences from GISAID’s EpiFlu™ Database on which this research is based. The list of submitters of H7Nx isolates used for analysis in Figure S1 is detailed below. (DOCX 23 KB)
12985_2012_2154_MOESM3_ESM.xlsx
Additional file 3:Phylogenetic tree of the 2012 Mexican strains with 1032 H7Nx full-length HA nucleotide sequences.(XLSX 26 KB)
12985_2012_2154_MOESM4_ESM.xlsx
Additional file 4: Table S1: Comparison of H7 HA sequences containing inserts at the cleavage site. H7 sequences from GISAID were screened for the R-x-x-R consensus motif where the final R is the normal cleavage site without insertion. Each row in the table is a representative strain that contains a distinctive insert sorted by date. The amino acid sequences at the extended cleavage site are coloured based on their similarity and origin, with the same colouring scheme as the phylogenetic tree in supplementary Figure 1, while the inserted amino acids are in bold. The pathotype of the strains was inferred from literature reports. (XLSX 73 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Maurer-Stroh, S., Lee, R.T., Gunalan, V. et al. The highly pathogenic H7N3 avian influenza strain from July 2012 in Mexico acquired an extended cleavage site through recombination with host 28S rRNA. Virol J 10, 139 (2013). https://doi.org/10.1186/1743-422X-10-139
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-10-139