
Abstract
Background
Impaired HIV-1 Gag, Pol, and Env function has been described in elite controllers (EC) who spontaneously suppress plasma viremia to < 50 RNA copies/mL; however, activity of the accessory protein Nef remains incompletely characterized. We examined the ability of 91 Nef clones, isolated from plasma of 45 EC and 46 chronic progressors (CP), to down-regulate HLA class I and CD4, up-regulate HLA class II invariant chain (CD74), enhance viral infectivity, and stimulate viral replication in PBMC.
Results
In general, EC Nef clones were functional; however, all five activities were significantly lower in EC compared to CP. Nef clones from HLA-B*57-expressing EC exhibited poorer CD4 down-regulation function compared to those from non-B*57 EC, and the number of EC-specific B*57-associated Nef polymorphisms correlated inversely with 4 of 5 Nef functions in these individuals.
Conclusion
Results indicate that decreased HIV-1 Nef function, due in part to host immune selection pressures, may be a hallmark of the EC phenotype.
Similar content being viewed by others


Background
Elite controllers (EC) are rare (<1%) HIV-1 infected individuals who spontaneously suppress plasma viral loads to undetectable levels in the absence of antiviral therapy. Several factors likely contribute to this phenotype, including host genetics [1], characteristics of HLA-restricted T-cell responses [2], immune-mediated reductions in viral protein function and/or replication [3, 4], and acquisition of attenuated viruses [5, 6]. Recombinant viruses expressing gag and pol sequences from EC exhibit reduced in vitro replication capacity, due in part to cytotoxic T lymphocyte (CTL) escape mutations selected by certain HLA class I (HLA-I) alleles [3, 4], while EC-derived viral envelopes exhibit impaired entry [7]. The in vitro function of other viral proteins in EC remains incompletely characterized.
HIV-1 Nef is an accessory protein required for maintenance of high viral loads and progression to AIDS [8], as demonstrated by slow or non-progressive disease in hosts infected with nef-deleted or otherwise nef-defective strains [5, 6, 9, 10]. Nef exhibits a variety of in vitro functions that may modulate pathogenesis, including CD4 down-regulation [11], HLA-I down-regulation [12], HLA class II invariant chain (CD74) up-regulation [13], enhancement of virion infectivity [14], and stimulation of viral replication in PBMC [15] (for reviews see [16–18]). Multiple Nef activities may act together to facilitate immune evasion and enhancement of viral spread in vivo[19]; however, multi-functional assessments of patient-derived Nef clones from HIV elite controllers are lacking. Although Nef sequence diversity is highly influenced by host HLA-I selection pressures [20], the relationship between HLA-associated polymorphisms and Nef function is largely unknown. Assessing multiple in vitro Nef functions in EC, a population that is highly enriched for protective HLA-I alleles such as B*57 [1], provides an opportunity to investigate these questions.
Previous analysis of plasma HIV RNA Nef sequences in our cohort of EC revealed no evidence of gross mutational defects [21], suggesting that any impairment in Nef protein function would have a more complex etiology. For this study, we generated recombinant viruses encoding a single representative HIV RNA Nef clone from 45 EC to assess Nef-mediated down-regulation of HLA class I, up-regulation of HLA class II invariant chain (CD74), viral infectivity, and viral replication in PBMC. The same Nef clone was engineered into a GFP-expression vector to assess its ability to down-regulate CD4. Results were compared to the activities of HIV RNA-derived Nef clones from 46 chronic progressors (CP). Finally, we assessed the role of host immune selection pressures, most notably novel polymorphisms associated with HLA-B*57 in EC, on Nef function in these individuals.
Results
Nef protein expression and viral production
For each of 45 EC and 46 CP, a single representative plasma HIV RNA-derived Nef sequence with an intact open reading frame (ORF) was cloned into a recombinant NL4.3 virus construct. Consistent with previous analyses of bulk plasma HIV RNA sequences from our EC cohort [21], clonal Nef sequences from EC showed no evidence of gross defects or recent shared ancestry (Figure 1, Additional file 1: Table S1). Western blots revealed comparable band intensities between EC and CP, indicating that EC Nefs were not markedly diminished in steady-state protein expression levels (Figure 2A, B). Similarly, p24Gag levels in culture supernatants were comparable between groups, indicating that EC Nefs were not significantly impaired in virion production (Figure 2C).

Maximum-likelihood phylogenetic tree of plasma HIV RNA-derived Nef clonal sequences. EC-derived Nefs are red, CP-derived Nefs are blue, and control strain SF2 is black.

Nef expression and progeny virus production. Panel A: Western blot detection of control strains NL4.3-NefSF2 and NL4.3-ΔNef and two representative EC and CP-derived Nefs using two different polyclonal anti-Nef sera. Panel B: Relative band intensity of EC (red) and CP (blue)-derived Nefs as detected by Western blot. Open circles identify the representative data shown in panel A. Bars depict median and interquartile ranges. Statistical significance assessed using Mann–Whitney U-Test. Panel C: Virus production (measured as p24Gag in culture supernatant) of EC (red) and CP (blue) Nef recombinant viruses.
Nef-mediated enhancement of viral infectivity and replication is impaired in EC
All viruses harboring EC Nef displayed infectivity greater than the negative control NL4.3ΔNef, which had 7.3% infectivity relative to NL4.3-NefSF2. Compared to control strain NL4.3-NefSF2, median EC Nef infectivity was 55% (IQR 38-76%), values that were significantly lower than CP-derived Nef (median 116, IQR 88-160%) (p < 0.001; Figure 3A). Similarly, all viruses harboring EC Nef displayed higher replication capacities than the negative control NL4.3ΔNef in PBMCs from four HIV-negative donors. Consistent with previous reports [22, 23], replication of patient-derived Nef recombinant viruses in PBMC differed to some extent among donors; however, viruses encoding EC Nef displayed consistently poorer ability to replicate in PBMC relative to those harboring CP Nef in all donors (p ≤ 0.01; Figure 3B, C). Averaged over all four donors, median [IQR] replication capacities were 34 [23–52]% of NL4.3-NefSF2 for EC-derived viruses and 76 [57–98]% for CP-derived viruses, respectively (p < 0.001, not shown).

Ability of EC Nef to enhance viral infectivity and stimulate viral replication in PBMC. Panel A: Scatterplots depict the ability of EC (red) and CP (blue) Nef recombinant viruses to enhance viral infectivity. Values are normalized to that of NL4.3-NefSF2 such that values of 100% indicate infectivity equal to that of NL4.3-NefSF2, while values <100% and >100% indicate infectivity lower than or higher than that of NL4.3-NefSF2, respectively. Bars represent median and interquartile ranges. The p-value was calculated using Mann–Whitney U-test. Panel B: Growth curves are shown for EC (red) and CP (blue) Nef recombinant viruses, plus control viruses NL4.3-NefSF2 and NL4.3-ΔNef, in PBMCs from donor #1. The mean value from quadruplicate samples is shown at each time point. The means ± SEM values for control viruses NL4.3-NefSF2 and NL4.3-ΔNef at day 9 and 12 are 115 ± 27 and 1.3 ± 0.02, and 285 ± 18.8 and 4.3 ± 0.4 respectively. Panel C: Scatterplots depict the ability of recombinant EC (red) and CP (blue) Nef recombinant viruses to stimulate viral replication in PBMC from four HIV-negative donors. Values are normalized to that of control NL4.3-NefSF2. Bars represent median and interquartile ranges. P-values were calculated using Mann–Whitney U-test.
Modulation of surface HLA-I, CD74, and CD4 by EC Nef
All EC Nef clones displayed greater ability to modulate cell-surface receptors than ΔNef negative controls. Relative to control strain NL4.3-NefSF2, EC-derived Nef recombinant viruses maintained considerable HLA-I down-regulation activity (median 95 [IQR 79–106]%) that was nevertheless significantly lower compared to CP Nef viruses (median 106 [IQR 96–111]%) (p<0.001; Figure 4A, B). The ability of EC Nef viruses to up-regulate CD74 was markedly lower (median 49 [IQR 35–76]%) compared to CP Nef viruses (median 111 [IQR 68–150]%) (p < 0.001; Figure 4C, D). HIV-1 Vpu and Env proteins contribute to surface CD4 modulation [24]; therefore, Nef-mediated CD4 down-regulation activity was assessed using DNA expression plasmids. Relative to control NefSF2, most EC Nef clones maintained substantial CD4 down-regulation activity (median 91 [IQR 76–95]%) that was nevertheless significantly lower compared to CP Nef clones (median 99 [IQR 89–101]%) (p = 0.002) (Figure 4E, F). All EC Nef sequences and functional data are provided in Additional file 1: Table S1. Of interest, 32 of 45 (71%) EC Nef viruses displayed replication activity less than 50% of that of control strain NefSF2, while only one EC Nef virus showed HLA-I down-regulation activity less than 50%. Three EC Nef demonstrated activity less than 50% of NefSF2 for all functions tested, except HLA-I down-regulation (Additional file 1: Table S1).

Ability of EC Nef to modulate cell surface receptor levels. Panel A: Flow cytometry plots depicting representative staining of cell-surface HLA-I (HLA-A*2402; y-axis) vs. intracellular p24Gag (x-axis) for uninfected, NL4.3-ΔNef (negative) and NL4.3-NefSF2 (positive) controls are shown. Panel B: Scatterplots depicting the ability of recombinant EC (red) and CP (blue) Nef recombinant viruses to down-regulate HLA-I are shown. Panel C: Flow cytometry plots depicting representative staining of cell-surface CD74 (y-axis) vs. intracellular p24Gag (x-axis) using uninfected and control viruses are shown. Panel D: Scatterplots depicting the ability of recombinant EC (red) and CP (blue) Nef recombinant viruses to upregulate CD74 are shown. Panel E: Flow cytometry plots depicting representative staining of cell-surface CD4 (y-axis) vs. GFP (x-axis) after delivery of no-DNA, ΔNef and NefSF2 plasmid vectors are shown. Panel F: Scatterplots depicting the ability of EC (red) and CP (blue)-derived Nef to downregulate CD4 are shown. In these experiments, results are normalized to NL4.3-NefSF2. (positive) and NL4.3-ΔNef (negative) controls. The activity of NL4.3-ΔNef or ΔNef plasmid is inherently set to zero. Bars represent median and interquartile ranges. P-values were calculated using Mann–Whitney U-test.
Host HLA-I allele expression and Nef function in EC
Protective HLA-I alleles, most notably B*57, are over-represented in EC [1, 25]. To investigate this as a potential confounder in comparisons between EC and CP, we re-analyzed our data excluding individuals who expressed HLA-B*57 (17/45 of EC and 8/46 of CP). Measures for all five Nef functions remained significantly lower among non-B*57 EC compared to non-B*57 CP (all p < 0.01, not shown). Exclusion of individuals expressing any protective allele (defined as B*27, B*57, and B*58:01) yielded similar results (all p < 0.05, not shown).
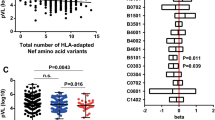
Immune selection by protective HLA-I alleles, including B*57, can modulate the in vitro function of certain HIV-1 proteins in EC [3, 4]. To examine whether this was also true for Nef, we stratified EC Nef clones by host B*57 expression and observed significantly lower CD4 down-regulation activity in B*57-derived compared to non-B*57-derived EC Nefs (median [IQR] 83 [55–94]% for B*57 vs. 92 [83–97]% for non-B*57 EC, respectively, p = 0.038). Significant differences were not seen for the other Nef activities tested (Figure 5). Of 20 HLA-I alleles expressed in a minimum of five EC, correlations with Nef function were also observed for C*06 (in linkage disequilibrium with B*57; median 74 vs 93% CD4 down-regulation activity in C*06 vs. non-C*06 EC) and A*01 (median 83 vs 97% HLA-I down-regulation activity in A*01 vs. non-A*01 EC) (both p < 0.05; q < 0.05). No HLA-I associations were observed for Nef-mediated infectivity, replication, or CD74 up-regulation activity in EC.

Relationship between host HLA-B*57 expression and Nef activities in EC. Scatterplots are shown depicting the ability of EC-derived Nefs from B*57-expressing (closed red circles) and non-B*57 expressing (open red circles) to enhance virion infectivity (panel A), enhance viral replication in PBMC (panel B; data depict means for all four PBMC donors), down-regulate HLA-I (panel C), up-regulate CD74 (panel D) and down-regulate CD4 (panel E). All results are normalized to NefSF2. Bars represent median and interquartile ranges. P-values were calculated using Mann–Whitney U-test.
Unique HLA-associated polymorphisms and Nef function in EC
Modulation of viral protein function in EC by protective HLA-I alleles may be due to the selection of unconventional HLA-associated polymorphisms in this patient group [26, 27]. To examine this, we applied phylogenetically-corrected methods [28] to identify HLA-B*57-associated Nef polymorphisms in our cohort of 45 EC. Nine associations were observed at p < 0.05 (q < 0.4) in B*57+ EC (Figure 6A). With the exception of V85L, these B*57-associated polymorphisms were distinct from those previously identified in large population-level analyses of chronically subtype B infected individuals (N >1500) [20, 29], suggesting that they may be largely unique to EC. In contrast, a search for B*57-associated polymorphisms in our cohort of 46 CP revealed several expected Nef polymorphisms at p < 0.05, including V85L and H116N [20, 29, 30] (not shown), supporting our ability to identify HLA-associated polymorphisms in cohorts of the present size. Therefore, we reasoned that the unconventional B*57-associated polymorphisms observed in EC merited further attention.

HLA-B*57-associated polymorphisms in EC, and their relationship with Nef function. Panel A: Two-dimensional map of B*57-associated polymorphisms identified in an exploratory analysis of the EC cohort using phylogenetically-corrected approaches with p<0.05 [20]. “Nonadapted” forms (those less likely to be observed in the presence of B*57 at a specific location) are shown above the Nef protein in blue; “adapted” forms (those enriched among B*57-expressing persons) are shown below the Nef protein in red. Due to limited statistical power and other reasons, both nonadapted and adapted forms are not always identified at a given position. Boxed codons indicate those where the amino acid varied in B*57 EC; the remainder were either expressed in 100% of B*57 EC (adapted forms 3G and 19F) or 0% of B*57 EC (nonadapted forms 105Q, 178G, 198M). In subsequent analyses, Nef sequences from B*57-expressing persons were counted as harboring a B*57-associated polymorphism at a given site if that site expressed anything other than the nonadapted form, or the adapted form if no nonadapted form was identified. Panel B: The locations of the nine EC-specific B*57-associated polymorphisms are indicated in green on the structure model of the Nef protein (composite crystal structure kindly provided by Art F. Y. Poon, [31]) Panels C- G: Correlations between the number of B*57-associated polymorphisms in Nef sequences from B*57-expressing EC, and five Nef functions evaluated. Statistical analyses were done using Spearman’s correlation.
Among B*57-expressing EC (N = 17), we observed significant inverse relationships between the number of EC-specific B*57-associated polymorphisms and Nef-mediated replication (Spearman R = −0.5, p = 0.04), HLA-I down-regulation (R = −0.57, p = 0.02) and CD74 up-regulation (R = −0.6, p = 0.01); and a modest, albeit not statistically significant, negative correlation with CD4 down-regulation (R = −0.37, p = 0.1) (Figure 6). No similar correlations were observed between EC-specific polymorphisms associated with other HLA-I alleles observed at comparable frequencies in our cohort (i.e. A*01, A*02, A*03, A*30, C*06, C*07) and Nef function in EC expressing these alleles (not shown).
Amino acids associated with EC Nef function
To investigate the relationship between Nef sequence and function in EC, we performed an exploratory analysis to correlate amino acids with each of the five functions tested, regardless of host HLA. A total of 23 polymorphisms occurring at 14 sites were associated with Nef-mediated modulation of HLA-I, CD74 and CD4 (p < 0.05, q < 0.4; Table 1). No Nef polymorphisms associated with infectivity or replication were observed at this threshold.
Discussion
We assessed five in vitro Nef functions using clonal plasma HIV RNA sequences from 45 EC and 46 CP. We observed that EC Nef clones were generally functional, especially for Nef’s most characteristic activities, CD4 and HLA-I down-regulation. Nevertheless, median EC Nef activities were significantly lower for all five functions when compared to those from CP. Median CP Nef activities were consistent with that of HIV-1 strain SF2 used as a normalization control for all assays, indicating that our selection of chronic Nef clones is representative of chronic Nef isolates examined previously. The range in Nef activities observed here may help to resolve discrepancies between previous studies of HIV long-term non-progressors or controllers, which have reported relative preservation of CD4 and/or HLA-down-regulation function [32, 33], inefficient Nef-mediated CD4 and/or HLA-down-regulation [34–36] and reduced infectivity [35] compared to CP. Our data suggest that there is in vivo pressure on Nef in EC to maintain CD4 and HLA-I down-regulation functions.
Relative functional impairments between EC and CP clones are not likely to be due to differences in Nef protein stability or expression levels, nor to recent descent from a defective common ancestor. Similarly, while enrichment of protective HLA alleles in EC may contribute to Nef sequence, it is not likely to be the only explanation for relative functional attenuation observed here, since differences between groups persisted after persons who expressed protective HLA alleles were excluded from analysis. Indeed, although significantly lower CD4 down-regulation activity was observed in B*57 compared to non-B*57 EC (Figure 5E), this was not true for other Nef functions, indicating that B*57 expression alone does not guarantee Nef attenuation in this group.
Rather, our results are consistent with functional variability of naturally occurring Nef sequences from EC, which may be attributable in part to non-canonical HLA-associated escape mutations selected in this rare group. Previously, in order to investigate the influence of HLA-associated viral polymorphisms on HIV-1 protein function in EC, we have made use of reference lists of common HLA-associated polymorphisms derived from population-level studies of chronically infected individuals [20, 29]. However, such lists may not capture rare escape mutations that are unique to EC [26, 27]. Therefore, we used our EC dataset to identify HLA-associated polymorphisms specific to this population, in a novel analysis of this type. It is important to note that this analysis was aimed at identifying HLA-B*57-associated polymorphisms in the EC dataset itself (and is therefore distinct from the comparative analyses described in [21] that sought to identify polymorphisms enriched among EC compared to CP). Statistical power is a major limitation of this analysis, but HLA-associated polymorphisms can be identified in modestly sized datasets (e.g. [37]), especially if analyses are limited to specific alleles. At p < 0.05, we identified nine B*57-associated polymorphic sites specific to EC (Figure 6A), most of which differed from B*57-associated polymorphisms commonly identified in population-level analyses [20, 29]. In contrast, B*57-associated polymorphisms identified in our CP dataset using the same approach were largely as expected [20, 29, 30]. ELISpot reactivity to overlapping peptides spanning codons 28, 55, 85, 178 and 198 has been documented in B*57+ EC [27]; and V85L has been described to function as an escape mutation in the B*57/58-KF9 epitope [38], further suggesting that these polymorphism may be due to CTL selection pressure in B*57 EC. In addition, paired Nef sequences from PBMC and plasma were available for two B*57 EC in the present study. The first exhibited identical amino acids in both compartments at all 7 of the residues putatively associated with HLA-B*57, while the second exhibited different amino acids at three of the seven B*57-associated sites (codons 85, 105 and 198). Although it is not possible to make conclusions based on only two patients, these data support evolution of these sites in vivo.
Notably, we observed dramatic inverse associations between the number of EC-specific B*57-associated polymorphisms and Nef-mediated replication, HLA-I down-regulation and CD74 up-regulation. CD4 down-regulation, the only function in which host expression of B*57 was in itself significantly associated with poorer function in EC, also displayed a modest, albeit not significant, negative relationship between the burden of B*57-associated escape mutations and function (Figure 6). These remarkable inverse relationships were particular to B*57 and not observed for other HLA alleles with similar frequency in our EC cohort. Taken together, results suggest that HLA-B*57-associated CTL pressures select for non-canonical polymorphisms in EC, which contribute additively to multiple functional impairments in EC Nef. Building upon previous studies of recombinant viruses encoding gag and pol sequences from the same EC cohort [3, 4], our results support a complex relationship between B*57-associated immune pressures and Nef function.
In an exploratory analysis of Nef amino acid sequences, we identified 23 polymorphisms, located at 14 residues, associated with Nef function in EC. None overlapped with mutations previously identified in HIV non-progressors [39] or with sites reported to affect HLA-I down-regulation activity in chronic infection [40], a discrepancy that might be due to non-canonical polymorphisms observed in EC. Of interest, 8S was associated with Nef-mediated modulation of cell-surface CD4, HLA-I and CD74, an observation consistent with codon 8’s involvement in myristylation [41]. Also, 28D and 105X (in this case R), associated with modulation of CD74 and CD4, respectively, are EC-specific B*57-associated polymorphisms identified in the present study.
Viral genetic studies of EC feature numerous challenges and limitations. Although care was taken to choose a Nef clone that reflected each patient’s original bulk HIV RNA sequence, and to rule out proviral DNA contamination, potential biases associated with PCR amplification from extremely low copy-number templates must be acknowledged. On the other hand, use of a single sequence per patient alleviates potential biases associated with quasispecies approaches to compare samples with low vs. high genetic diversity. As our goal was to specifically investigate the function of Nef in EC, we employed recombinant virus (and single-protein expression) approaches to eliminate potential confounding effects of other HIV-1 proteins; however, such approaches may not reflect the characteristics of infectious molecular clones or whole-virus isolates recovered from PBMC, procedures that are rarely successful in EC [21, 42]. Recombinant virus approaches are also inherently limited by potential incompatibilities between insert and backbone; our choice of a recombinant control strain (NL4.3-NefSF2) alleviates this to a minor extent. Although we assessed Nef activity using primary PBMCs and immortalized cell lines, Nef’s multiple functions [16–18, 43] may vary in different cell types [43–45]. Furthermore, the in vivo relevance of our observations - in particular, the extent to which these functional differences contribute to the viremia control in EC - remains unclear. Although our results are consistent with the transmission of partially attenuated Nef sequences in at least some EC, and/or further immune-mediated attenuation in others [3, 4, 46], it is not possible to disentangle cause and effect in cross-sectional studies. Furthermore, Nef function may change over the infection course [47], therefore longitudinal analysis of Nef function in controllers, beginning in the acute/early phase of infection, is warranted. Finally, although our results suggest that non-canonical polymorphisms in EC may contribute to attenuated Nef function, it will be important to validate these findings in larger EC cohorts. Despite these limitations, our study represents the largest linked analysis of multiple in vitro Nef functions in EC to date, and to our knowledge the only study assessing CD74 up-regulation and replication capacity in this group.
Conclusions
EC Nef clones were generally functional; however, all five activities assessed were significantly impaired compared to CP Nef clones. HLA-I-restricted immune pressure, most notably by B*57, may contribute to the differences observed. Taken together with previous studies of HIV Gag, Pol, and Env function in EC [3, 4, 7], our results support decreased viral protein function as a hallmark of the EC phenotype and underscore the potential role of immune pressures in modulating viral protein function in this rare group.
Methods
Study subjects
45 EC (median [interquartile range, IQR] pVL 2 [0.2-14] RNA copies/ml [25]; median [IQR] CD4 count 811 [612–1022] cells/mm3) and 46 CP (median [IQR] pVL 80500 [25121–221250] RNA copies/ml); median [IQR] CD4 count 292.5 [72.5-440] cells/mm3) were studied as described previously [3, 4, 21, 25]. All EC and CP were HIV-1 subtype B-infected, untreated at the time of sample collection, recruited from the Boston area, and comparable with respect to ethnicity and date of HIV diagnosis (1985–2006 for EC vs. 1981–2003 for CP). This study was approved by the institutional review board of Massachusetts General Hospital, Boston USA; all participants provided written informed consent.
Cloning and analysis of nef genes
For EC, HIV RNA was extracted from a starting volume of 4.5 to 35.0 ml of plasma and amplified using nested RT-PCR, as described [21]. Given that the median pVL in our EC cohort was 2 RNA copies/ml [IQR 0.2-14] [25], we estimate that on average, 40 viral RNA templates were extracted for each EC patient. To rule out proviral DNA contamination, all extractions included a DNAse treatment step; controls lacking RT enzyme were also performed [21]. For CP, HIV RNA was extracted from 0.5ml of plasma and amplified in the same manner. Nef amplicons were cloned into pIRES2-EGFP expression vector (Clontech). A minimum of three Nef clones were sequenced per patient, and a single clone with an intact Nef reading frame that closely resembled the original bulk plasma RNA sequence was selected. Genbank accession numbers for clonal Nef sequences are JX171199-JX171243 (EC) and JX440926-JX440971 (CP).
Recombinant virus construction and verification of Nef expression
Nef clones were transferred into a pNL4.3ΔNef plasmid as described [23] and confirmed by DNA sequencing. Recombinant viruses harboring nef from HIV strain SF2 (NL4.3-NefSF2), and lacking nef (NL4.3ΔNef) were used as positive and negative controls, respectively. Infectious viruses were generated as described [48]. Briefly, HEK-293T cells were transfected with each proviral clone. Virus-containing culture supernatants were harvested 48 hr following transfection, titered by p24Gag ELISA (ZeptoMetrix Corp.) and aliquots stored at −80°C until use.
HEK-293T cell pellets collected at this time point were used to prepare total cell lysates as described [45] that were subjected to SDS-PAGE in duplicate and transferred to nitrocellulose membranes. Nef genetic diversity poses a challenge to antibody-based detection as differences in reactivity may reflect suboptimal antibody binding rather than variation in protein levels. To ensure detection of patient-derived Nef, duplicate blots were probed using unique anti-Nef polyclonal antisera, developed from sheep (ARP 444; provided by O.T. Fackler, Heidelberg University, Germany) or rabbit (NIH AIDS Research and Reference Reagent Program). Band intensities were quantified using ImageQuant LAS 4000 (GE Healthcare Life Sciences).
Virion infectivity and replication assays
Recombinant virus infectivity was determined by exposing 104 TZM-bl cells (NIH AIDS Research and Reference Reagent Program) to 3 ng p24Gag recombinant virus followed by chemiluminescence detection 48 hr later as described [49]. Infectivity values represented the mean of triplicate experiments, normalized to control strain NL4.3-NefSF2, such that values > 100% and < 100% indicated increased or decreased infectivity, respectively. Recombinant virus replication was assessed by exposing 106 freshly isolated PBMC from four HIV-negative donors to 10 ng p24Gag recombinant virus for 8 hr, washing twice, and then resuspending cells in a culture medium (RPMI 1640, 10% FCS) as described [22, 23]. Three days later, PBMCs were stimulated with phytohemagglutinin at 5 μg/ml. Culture supernatants were collected and replaced with fresh medium supplemented with human rIL-2 every 3 days. Viral replication was monitored by measuring p24Gag in the culture supernatant using ELISA over 12 days. ELISA values during the initial burst of viral replication (on day 9) were used as our measure of replication capacity. Results were expressed as the mean of quadruplicate assessments in each donor, normalized to control strain NL4.3-NefSF2.
Analysis of receptor modulation HIV-infected cell surface
To assess Nef-mediated HLA-I down-regulation and CD74 up-regulation, 721.221 cells stably expressing CD4 and HLA-A*24:02 (provided by Masafumi Takiguchi,Kumamoto University, Japan) were exposed to 300 ng p24Gag recombinant virus for 48 hr, followed by staining with anti-HLA-A24-PE (MBL), anti-CD74-Alexa Fluor-647 (BioLegend), 7-amino-actinomycin D (BioLegend), and anti-p24 Gag-FITC (Beckman Coulter), as described [23]. Mean fluorescence intensity (MFI) of each receptor in live p24Gag positive and negative subsets was determined by flow cytometry (FACS Canto II; BD Biosciences). Results were expressed as the mean of duplicate experiments, normalized to control strain NL4.3-NefSF2.
Analysis of Nef-mediated CD4 down-regulation
To assess Nef-mediated CD4 down-regulation, 3 × 105 CEM-SS cells were transfected with 5 µg plasmid DNA encoding Nef protein and GFP by electroporation (BioRad GenePulser MX) and stained 24 hr later with anti-CD4-APC (BD Biosciences). MFI of GFP-negative and GFP-positive (Nef-expressing) subsets was determined by flow cytometry (Guava easyCyte 8HT, Millipore). Results were normalized to plasmid expressing NefSF2.
Statistical analyses, including identification of B*57-associated polymorphisms in patient-derived Nef sequences
Phylogenetically-informed methods were used to identify Nef amino acids significantly associated with HLA-B*57allele expression in our EC ([28], implemented at http://research.microsoft.com/en-us/um/redmond/projects/mscompbio/phylododdsratio/default.aspx). Multiplecomparisons were addressed using q-values, the p-value analogue of the false discovery rate (FDR) [50]. The FDR is the expected proportion of false positives among results deemed significant at a given p-value threshold; for example, at a q ≤ 0.2, we expect 20% of identified associations to be false positives.
References
Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, Walker BD, Ripke S, Brumme CJ, Pulit SL, Carrington M, et al: The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010, 330: 1551-1557.
Chen H, Ndhlovu ZM, Liu D, Porter LC, Fang JW, Darko S, Brockman MA, Miura T, Brumme ZL, Schneidewind A, et al: TCR clonotypes modulate the protective effect of HLA class I molecules in HIV-1 infection. Nat Immunol. 2012, 13: 691-700. 10.1038/ni.2342.
Brumme ZL, Li C, Miura T, Sela J, Rosato PC, Brumme CJ, Markle TJ, Martin E, Block BL, Trocha A, et al: Reduced replication capacity of NL4-3 recombinant viruses encoding reverse transcriptase-integrase sequences from HIV-1 elite controllers. J Acquir Immune Defic Syndr. 2011, 56: 100-108.
Miura T, Brockman MA, Brumme ZL, Brumme CJ, Pereyra F, Trocha A, Block BL, Schneidewind A, Allen TM, Heckerman D, Walker BD: HLA-associated alterations in replication capacity of chimeric NL4-3 viruses carrying gag-protease from elite controllers of human immunodeficiency virus type 1. J Virol. 2009, 83: 140-149. 10.1128/JVI.01471-08.
Deacon NJ, Tsykin A, Solomon A, Smith K, Ludford-Menting M, Hooker DJ, McPhee DA, Greenway AL, Ellett A, Chatfield C, et al: Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science. 1995, 270: 988-991. 10.1126/science.270.5238.988.
Kirchhoff F, Greenough TC, Brettler DB, Sullivan JL, Desrosiers RC: Brief report: absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N Engl J Med. 1995, 332: 228-232. 10.1056/NEJM199501263320405.
Lassen KG, Lobritz MA, Bailey JR, Johnston S, Nguyen S, Lee B, Chou T, Siliciano RF, Markowitz M, Arts EJ: Elite Suppressor–Derived HIV-1 Envelope Glycoproteins Exhibit Reduced Entry Efficiency and Kinetics. PLoS Pathog. 2009, 5: e1000377-10.1371/journal.ppat.1000377.
Kestler HW, Ringler DJ, Mori K, Panicali DL, Sehgal PK, Daniel MD, Desrosiers RC: Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell. 1991, 65: 651-662. 10.1016/0092-8674(91)90097-I.
Daniel MD, Kirchhoff F, Czajak SC, Sehgal PK, Desrosiers RC: Protective effects of a live attenuated SIV vaccine with a deletion in the nef gene. Science. 1992, 258: 1938-1941. 10.1126/science.1470917.
Zaunders JJ, Geczy AF, Dyer WB, McIntyre LB, Cooley MA, Ashton LJ, Raynes-Greenow CH, Learmont J, Cooper DA, Sullivan JS: Effect of long-term infection with nef-defective attenuated HIV type 1 on CD4+ and CD8+ T lymphocytes: increased CD45RO+CD4+ T lymphocytes and limited activation of CD8+ T lymphocytes. AIDS Res Hum Retroviruses. 1999, 15: 1519-1527. 10.1089/088922299309801.
Garcia JV, Miller AD: Serine phosphorylation-independent downregulation of cell-surface CD4 by nef. Nature. 1991, 350: 508-511. 10.1038/350508a0.
Schwartz O, Marechal V, Le Gall S, Lemonnier F, Heard JM: Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med. 1996, 2: 338-342. 10.1038/nm0396-338.
Schindler M, Wurfl S, Benaroch P, Greenough TC, Daniels R, Easterbrook P, Brenner M, Munch J, Kirchhoff F: Down-modulation of mature major histocompatibility complex class II and up-regulation of invariant chain cell surface expression are well-conserved functions of human and simian immunodeficiency virus nef alleles. J Virol. 2003, 77: 10548-10556. 10.1128/JVI.77.19.10548-10556.2003.
Münch J, Rajan D, Schindler M, Specht A, Rücker E, Novembre FJ, Nerrienet E, Müller-Trutwin MC, Peeters M, Hahn BH, Kirchhoff F: Nef-Mediated Enhancement of Virion Infectivity and Stimulation of Viral Replication Are Fundamental Properties of Primate Lentiviruses. J Virol. 2007, 81: 13852-13864. 10.1128/JVI.00904-07.
Miller MD, Warmerdam MT, Gaston I, Greene WC, Feinberg MB: The human immunodeficiency virus-1 nef gene product: a positive factor for viral infection and replication in primary lymphocytes and macrophages. J Exp Med. 1994, 179: 101-113. 10.1084/jem.179.1.101.
Arhel NJ, Kirchhoff F: Implications of Nef: host cell interactions in viral persistence and progression to AIDS. Curr Top Microbiol Immunol. 2009, 339: 147-175. 10.1007/978-3-642-02175-6_8.
Arien KK, Verhasselt B: HIV Nef: role in pathogenesis and viral fitness. Curr HIV Res. 2008, 6: 200-208. 10.2174/157016208784325001.
Foster JL, Denial SJ, Temple BR, Garcia JV: Mechanisms of HIV-1 Nef function and intracellular signaling. J Neuroimmune Pharmacol. 2011, 6: 230-246. 10.1007/s11481-011-9262-y.
Fackler OT, Alcover A, Schwartz O: Modulation of the immunological synapse: a key to HIV-1 pathogenesis?. Nat Rev Immunol. 2007, 7: 310-317. 10.1038/nri2041.
Carlson JM, Brumme CJ, Martin E, Listgarten J, Brockman MA, Le AQ, Chui C, Cotton LA, Knapp DJ, Riddler SA, et al: Correlates of protective cellular immunity revealed by analysis of population-level immune escape pathways in HIV-1. J Virol. 2012, 86: 13202-13216. 10.1128/JVI.01998-12.
Miura T, Brockman MA, Brumme CJ, Brumme ZL, Carlson JM, Pereyra F, Trocha A, Addo MM, Block BL, Rothchild AC, et al: Genetic characterization of human immunodeficiency virus type 1 in elite controllers: lack of gross genetic defects or common amino acid changes. J Virol. 2008, 82: 8422-8430. 10.1128/JVI.00535-08.
Fackler OT, Moris A, Tibroni N, Giese SI, Glass B, Schwartz O, Krausslich HG: Functional characterization of HIV-1 Nef mutants in the context of viral infection. Virology. 2006, 351: 322-339. 10.1016/j.virol.2006.03.044.
Ueno T, Motozono C, Dohki S, Mwimanzi P, Rauch S, Fackler OT, Oka S, Takiguchi M: CTL-mediated selective pressure influences dynamic evolution and pathogenic functions of HIV-1 Nef. J Immunol. 2008, 180: 1107-1116.
Neri F, Giolo G, Potesta M, Petrini S, Doria M: CD4 downregulation by the human immunodeficiency virus type 1 Nef protein is dispensable for optimal output and functionality of viral particles in primary T cells. J Gen Virol. 2011, 92: 141-150. 10.1099/vir.0.026005-0.
Pereyra F, Addo MM, Kaufmann DE, Liu Y, Miura T, Rathod A, Baker B, Trocha A, Rosenberg R, Mackey E, et al: Genetic and immunologic heterogeneity among persons who control HIV infection in the absence of therapy. J Infect Dis. 2008, 197: 563-571. 10.1086/526786.
Miura T, Brockman MA, Schneidewind A, Lobritz M, Pereyra F, Rathod A, Block BL, Brumme ZL, Brumme CJ, Baker B, et al: HLA-B57/B*5801 human immunodeficiency virus type 1 elite controllers select for rare gag variants associated with reduced viral replication capacity and strong cytotoxic T-lymphocyte [corrected] recognition. J Virol. 2009, 83: 2743-2755. 10.1128/JVI.02265-08.
Bailey JR, Brennan TP, O'Connell KA, Siliciano RF, Blankson JN: Evidence of CD8+ T-Cell-Mediated Selective Pressure on Human Immunodeficiency Virus Type 1 nef in HLA-B*57+ Elite Suppressors. J Virol. 2009, 83: 88-97. 10.1128/JVI.01958-08.
Carlson JM, Listgarten J, Pfeifer N, Tan V, Kadie C, Walker BD, Ndung'u T, Shapiro R, Frater J, Brumme ZL, et al: Widespread impact of HLA restriction on immune control and escape pathways of HIV-1. J Virol. 2012, 86: 5230-5243. 10.1128/JVI.06728-11.
Brumme ZL, John M, Carlson JM, Brumme CJ, Chan D, Brockman MA, Swenson LC, Tao I, Szeto S, Rosato P, et al: HLA-associated immune escape pathways in HIV-1 subtype B Gag, Pol and Nef proteins. PLoS One. 2009, 4: e6687-10.1371/journal.pone.0006687.
Frater AJ, Brown H, Oxenius A, Gunthard HF, Hirschel B, Robinson N, Leslie AJ, Payne R, Crawford H, Prendergast A, et al: Effective T-cell responses select human immunodeficiency virus mutants and slow disease progression. J Virol. 2007, 81: 6742-6751. 10.1128/JVI.00022-07.
Poon AF, Swenson LC, Dong WW, Deng W, Kosakovsky Pond SL, Brumme ZL, Mullins JI, Richman DD, Harrigan PR, Frost SD: Phylogenetic analysis of population-based and deep sequencing data to identify coevolving sites in the nef gene of HIV-1. Mol Biol Evol. 2010, 27: 819-832. 10.1093/molbev/msp289.
Zuo J, Suen J, Wong A, Lewis M, Ayub A, Belzer M, Church J, Yang OO, Krogstad P: Functional analysis of HIV type 1 Nef gene variants from adolescent and adult survivors of perinatal infection. AIDS Res Hum Retroviruses. 2012, 28: 486-492.
Nou E, Zhou Y, Nou DD, Blankson JN: Effective Downregulation of HLA-A*2 and HLA-B*57 by Primary Human Immunodeficiency Virus Type 1 Isolates Cultured from Elite Suppressors. J Virol. 2009, 83: 6941-6946. 10.1128/JVI.00306-09.
Casartelli N, Di Matteo G, Potestà M, Rossi P, Doria M: CD4 and Major Histocompatibility Complex Class I Downregulation by the Human Immunodeficiency Virus Type 1 Nef Protein in Pediatric AIDS Progression. J Virol. 2003, 77: 11536-11545. 10.1128/JVI.77.21.11536-11545.2003.
Tobiume M, Takahoko M, Yamada T, Tatsumi M, Iwamoto A, Matsuda M: Inefficient enhancement of viral infectivity and CD4 downregulation by human immunodeficiency virus type 1 Nef from Japanese long-term nonprogressors. J Virol. 2002, 76: 5959-5965. 10.1128/JVI.76.12.5959-5965.2002.
Corro G, Rocco C, De Candia C, Catano G, Turk G, Aulicino P, Bologna R, Sen L: Genetic and functional analysis of HIV-1 nef gene derived from LTNP children: association of attenuated variants with slow progression to pediatric AIDS. AIDS Res Hum Retroviruses. 2012, 28: 1617-1626. 10.1089/aid.2012.0020.
Bhattacharya T, Daniels M, Heckerman D, Foley B, Frahm N, Kadie C, Carlson J, Yusim K, McMahon B, Gaschen B, et al: Founder effects in the assessment of HIV polymorphisms and HLA allele associations. Science. 2007, 315: 1583-1586. 10.1126/science.1131528.
O'Connell KA, Xu J, Durbin AP, Apuzzo LG, Imteyaz H, Williams TM, Ray SC, Margolick JB, Siliciano RF, Blankson JN: HIV-1 evolution following transmission to an HLA-B*5801-positive patient. J Infect Dis. 2009, 200: 1820-1824. 10.1086/648377.
Kirchhoff F, Easterbrook PJ, Douglas N, Troop M, Greenough TC, Weber J, Carl S, Sullivan JL, Daniels RS: Sequence variations in human immunodeficiency virus type 1 Nef are associated with different stages of disease. J Virol. 1999, 73: 5497-5508.
Lewis MJ, Lee P, Ng HL, Yang OO: Immune selection in vitro reveals human immunodeficiency virus type 1 Nef sequence motifs important for its immune evasion function in vivo. J Virol. 2012, 86: 7126-7135. 10.1128/JVI.00878-12.
Geyer M, Fackler OT, Peterlin BM: Structure–function relationships in HIV-1 Nef. EMBO Rep. 2001, 2: 580-585. 10.1093/embo-reports/kve141.
Julg B, Pereyra F, Buzon MJ, Piechocka-Trocha A, Clark MJ, Baker BM, Lian J, Miura T, Martinez-Picado J, Addo MM, Walker BD: Infrequent recovery of HIV from but robust exogenous infection of activated CD4(+) T cells in HIV elite controllers. Clin Infect Dis. 2010, 51: 233-238. 10.1086/653677.
Kirchhoff F, Schindler M, Specht A, Arhel N, Munch J: Role of Nef in primate lentiviral immunopathogenesis. Cell Mol Life Sci. 2008, 65: 2621-2636. 10.1007/s00018-008-8094-2.
Suzu S, Harada H, Matsumoto T, Okada S: HIV-1 Nef interferes with M-CSF receptor signaling through Hck activation and inhibits M-CSF bioactivities. Blood. 2005, 105: 3230-3237. 10.1182/blood-2004-06-2084.
Mwimanzi P, Hasan Z, Hassan R, Suzu S, Takiguchi M, Ueno T: Effects of naturally-arising HIV Nef mutations on cytotoxic T lymphocyte recognition and Nef's functionality in primary macrophages. Retrovirology. 2011, 8: 50-10.1186/1742-4690-8-50.
Lobritz MA, Lassen KG, Arts EJ: HIV-1 replicative fitness in elite controllers. Curr Opin HIV AIDS. 2011, 6: 214-220. 10.1097/COH.0b013e3283454cf5.
Carl S, Greenough TC, Krumbiegel M, Greenberg M, Skowronski J, Sullivan JL, Kirchhoff F: Modulation of different human immunodeficiency virus type 1 Nef functions during progression to AIDS. J Virol. 2001, 75: 3657-3665. 10.1128/JVI.75.8.3657-3665.2001.
Mwimanzi P, Hasan Z, Tokunaga M, Gatanaga H, Oka S, Ueno T: Naturally arising HIV-1 Nef variants conferring escape from cytotoxic T lymphocytes influence viral entry co-receptor expression and susceptibility to superinfection. Biochem Biophys Res Commun. 2010, 403: 422-427. 10.1016/j.bbrc.2010.11.047.
Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC: Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother. 2002, 46: 1896-1905. 10.1128/AAC.46.6.1896-1905.2002.
Storey JD, Tibshirani R: Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003, 100: 9440-9445. 10.1073/pnas.1530509100.
Acknowledgments
We thank Dr. Jonathan Carlson for helpful discussions. This study was supported by a grant-in-aid for scientific research and a Global COE Program (Global Education and Research Center Aiming at the control of AIDS) from the Ministry of Education, Science, Sports, and Culture of Japan, a grant-in-aid for AIDS research from the Ministry of Health, Labor, and Welfare of Japan (to TU), and the Canadian Institutes for Health Research (CIHR) operating grant MOP-93536 (to ZLB and MAB). We acknowledge and thank the International HIV Controllers study, funded by Bill and Melinda Gates Foundation, the Schwartz Foundation and the Harvard University Center for AIDS Research. PM is a postdoctoral fellow who received support from the Japan AIDS Foundation and the Global Health Research Initiative (GHRI), a collaborative research funding partnership of the CIHR, the Canadian International Development Agency, and the International Development Research Centre. EM is supported by a Master’s Scholarship from the Canadian Association of HIV Research and Abbott Virology. ZLB is the recipient of a CIHR New Investigator Award and a Scholar Award from the Michael Smith Foundation for Health Research. MAB holds a Canada Research Chair, Tier 2, in Viral Pathogenesis and Immunity.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
PM, TJM, ZLB, MAB, and TU designed the study; PM, TJM, EM, YO, XTK, MT, and MM performed the experiments; FP, TM and BDW provided access to patient samples and analyzed clinical data; PM, TJM, EM, ZLB, MAB, and TU analyzed data; and PM, ZLB, MAB, and TU wrote the paper. All authors read and approved the final manuscript.
Philip Mwimanzi, Tristan J Markle contributed equally to this work.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Mwimanzi, P., Markle, T.J., Martin, E. et al. Attenuation of multiple Nef functions in HIV-1 elite controllers. Retrovirology 10, 1 (2013). https://doi.org/10.1186/1742-4690-10-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-4690-10-1