
Abstract
The complement system, a major component of the innate immune system, is becoming increasingly recognised as a key participant in physiology and disease. The awareness that immunological mediators support various aspects of both normal central nervous system (CNS) function and pathology has led to a renaissance of complement research in neuroscience. Various studies have revealed particularly novel findings on the wide-ranging involvement of complement in neural development, synapse elimination and maturation of neural networks, as well as the progression of pathology in a range of chronic neurodegenerative disorders, and more recently, neurotraumatic events, where rapid disruption of neuronal homeostasis potently triggers complement activation. The purpose of this review is to summarise recent findings on complement activation and acquired brain or spinal cord injury, i.e. ischaemic-reperfusion injury or stroke, traumatic brain injury (TBI) and spinal cord injury (SCI), highlighting the potential for complement-targeted therapeutics to alleviate the devastating consequences of these neurological conditions.
Similar content being viewed by others

Introduction
Injury to the central nervous system (CNS) elicits a complex series of pathophysiological events, including ischaemia, excitotoxicity and inflammation. All of these factors adversely affect the integrity of spared neurons and thus accentuate tissue damage beyond the initial site of trauma. The cellular immune response in particular has received much attention as a key mediator of secondary injury, and strategies to manipulate the activation and recruitment of neutrophils [1–5], monocytes and macrophages [6–9], and lymphocytes [10–12] after trauma have all been investigated, with the ultimate goal being to improve functional outcomes (reviewed in [13]).
Several recent studies have, however, put activation of the innate immune complement system into the spotlight as a perhaps sometimes-overlooked but potent mediator of secondary pathology [14–16]. The particular aim of this review is to summarise current knowledge and understanding of complement activation in the injured CNS, specifically in relation to post-traumatic neuroinflammatory events and associated secondary damage. Several other recent reviews have already provided a comprehensive overview of the role of complement in CNS development and chronic neurodegenerative disorders [17–19].
The complement system: an introduction and effector mechanisms
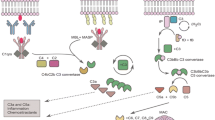
The predominant site of peripheral complement protein synthesis is the liver, where hepatocytes constantly produce and replenish circulating complement factors [20]. Activation of these circulating complement proteins in response to an injurious or infectious challenge results in a self-amplifying cascade of proteolytic reactions through any one of four major identified pathways (Figure 1).

Common pathways for complement activation. Recognition of antigen-antibody complexes by C1q initiates the ‘classical pathway’. Binding of carbohydrate antigens by mannose-binding lectin (MBL) or MBL-associated serine proteases (MASPs) initiates the ‘lectin pathway’. Both pathways lead to the formation of the C3 convertase, C4b2a. Complement activation through the ‘alternative pathway’ involves the spontaneous hydrolysis of plasma C3, generating a second C3 convertase, C3(H2O)Bb. Proteolysis of C3 then leads to production of the C3b fragment, which binds to C3 convertases to generate C5 convertases. After the cleavage of C5, the C5b fragment binds C6-C9 to generate the membrane attack complex (MAC). The coagulation cascade leads to complement activation via the ‘extrinsic pathway’; this route does not depend on the presence of C3 convertases. Anaphylatoxins C5a, C3a and C4a are generated through cleavage of C5, C3 and C4, respectively. Soluble and membrane-bound negative regulators of complement and their site of action are indicated in green. The functional significance of certain activation steps is shown in red.
The classical pathway for complement activation is initiated by the binding of the recognition molecule C1q to pathogen antigens, C-reactive protein bound to bacterial polysaccharides or antigen-antibody complexes [21]. It is of interest to note in this context that pathogen opsonisation and antibody ligation by C1q also provide a bridge to activation of the adaptive immune system, which includes an enhancement of antigen retention in lymphoid tissues, a decrease in the B cell activation threshold and increased memory B cell survival [22–24]. T cell proliferation, differentiation, activation and antigen-presenting cell (APC) function can also be significantly influenced by complement [25, 26]. The lectin pathway for complement activation involves the recognition of pathogen carbohydrate antigens by mannose-binding lectin-associated serine proteins (MASP-1 and MASP-2) [27] and the ficolins [28]. The alternative pathway of complement activation is initiated by spontaneous hydrolysis of complement component C3 in plasma, and the binding of factor B and D to C3(H2O) [29]. All of the three aforementioned activation routes lead to the formation of C3 convertases and thus converge at this level.
C3 convertases cleave the parental C3 molecule into two fragments, the larger C3b molecule and the smaller anaphylatoxin C3a. The C3b fragment opsonises pathogen-associated molecular patterns (PAMPs), which are small, conserved molecular motifs that are shared by classes of microbes and recognised by host cell pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) [30]. C3b opsonises altered-self ligands, immune complexes and/or dead cells as well, which ultimately enhances their recognition and rapid phagocytosis by scavenging leukocytes that bear C3b receptors. The C3b fragment can also bind the C3 convertase, which leads to the formation of a C5 convertase and the subsequent cleavage of the parental C5 protein into C5b and the anaphylatoxin C5a. The amplification cascade then culminates in the association of C5b with C6, C7 and C8, which induces the polymerisation of 10–16 C9 molecules in order to assemble a transmembrane pore called the terminal ‘membrane attack complex’ (MAC), with subsequent lysis of the targeted pathogens or abnormal host cells as a result [31]. Importantly, components of the blood clotting and fibrinolysis pathways, as well as other cell-derived serine proteases, can also directly cleave and activate C3 and C5 proteins, and thus initiate the formation of complement end products, independent of the C3 and C5 convertases, a process that is now referred to as the extrinsic pathway[32–34].
As indicated above, the cleavage of C3 and C5 also leads to the generation of two smaller activation fragments that do not directly contribute to MAC formation, the so-called anaphylatoxins. The small cleavage product of the parental C3 protein, the anaphylatoxin C3a, is a local mediator of inflammation that signals through its G-protein coupled receptor, C3aR [35]. The anaphylatoxin C5a, generated from C5, is one of the most potent pro-inflammatory peptides known. It can act as a phagocyte chemoattractant, and promote vascular permeability, platelet and leukocyte activation through, for example, upregulation of the leukocyte adhesion molecules necessary for transendothelial extravasation to sites of infection or injury. It also induces production of proinflammatory cytokines, chemokines, leukotrienes, prostaglandins, oxidative burst and degranulation (reviewed in [31, 36]). Similar to C3a, the C5a molecule signals through a G-protein coupled receptor, which is known as C5aR or CD88 [37]. A second receptor for C5a, known as C5L2, has been reported [38], but its functional significance still remains controversial; indeed, it could have multiple roles in different species, organs and pathophysiological states [39–42].
Physiological functions for complement in the CNS
Although circulating complement proteins in blood plasma do not normally have access to the CNS because of the blood–brain and blood-spinal cord barriers (BBB and BSB, respectively), several studies have demonstrated that virtually all of the components of complement can be synthesized within the CNS [18, 19, 43]. Accordingly, various non-immune physiological roles for complement have been identified, including synaptic remodelling during development, cell survival and neurogenesis.
A seminal study by Stevens and colleagues [44] showed that components of the classical complement pathway are key mediators of synapse elimination in the developing retinogeniculate pathway in mice. Early in development, retinal ganglion cells (RGCs) from both eyes extend excessive, overlapping projections into the dorsal lateral geniculate nucleus (dLGN) of the thalamus. Weaker synaptic arborisations are then eliminated and more active connections strengthened, resulting in the adult pattern of segregated eye layers by postnatal day 20 [45]. This process is coordinated by astrocyte-driven deposition of C1q and C3 on immature or weaker RGC synapses, which then tags them for removal, most likely by activated microglia. RGC axons in C1q−/− and C3−/− mice have indeed a higher degree of overlap during and after the remodelling process, resulting in the persistent retention of excessive retinal innervations of lateral geniculate neurons [44]. The supernumerary inputs retained in C1q−/− and C3−/− neurons were shown to be immature or dysfunctional as judged by the weak magnitude of the glutamate-mediated currents carried by their AMPA receptors [44]. Interestingly, the failure to prune excessive excitatory synapses during development has been positively correlated to enhanced synaptic connectivity and epileptogenesis in C1q−/− mice compared to wild-type (WT) controls [46]. Our groups have also previously shown localized expression of CD88, the C5a anaphylatoxin receptor, on presynaptic terminals of mossy fibres within the CA3 region of the adult rat hippocampus [47]; it remains to be determined whether physiological C5a signalling here could also be involved in synaptic plasticity or whether it serves different and yet unknown functions in this structure.
In addition to a role in synaptic plasticity, complement proteins may play an important role in neuroprotection in the CNS. In the absence of other complement components, C1q has been shown to increase neuronal survival and arborisation [48]. These effects are mediated via the upregulation of genes associated with cytoskeleton function (syntaxin-3), cholesterol/lipid metabolism (CH25H, INSIG2) and neurotrophic factors (NGF, NT-3, NTN1) [49].
A functional role for the MAC has also been demonstrated. At sublytic concentrations, the MAC can be endocytosed by oligodendrocytes and cause them to re-enter the cell cycle [50]. Sublytic MAC can also reduce apoptotic cell death by increasing synthesis of Bcl-2 and inhibition of caspase-3 activation and caspase-8 processing and upregulating FLIP [51, 52].
Lastly, a physiological role for the anaphylatoxins C3a and C5a within the adult murine CNS has emerged, specifically an involvement in cell survival and neurogenesis. In mixed cultures of neurons and astrocytes, C3a protected neurons against NMDA-induced excitotoxicity in a dose- and astrocyte-dependent manner [53]. The neuroprotective action of C3a signalling could be mediated through the induction of nerve growth factor (NGF) expression in microglia [54, 55] as well as astrocytes [55]. C5a exposure also causes an upregulation of NGF mRNA expression in astrocytes [55], with similar neuroprotective effects against glutamate-mediated neuronal excitotoxicity. The neuroprotective effects of anaphylatoxins against glutamate-induced excitotoxicity were shown to be mediated via MAPK-dependent inhibition of caspase-3 [56, 57], regulation of glutamate receptor subunit 2 (GluR2) expression [58], and increased glial expression of the glutamate transporter GLT-1, which enhances the removal of extracellular glutamate [59]. Administration of C5a in vivo was also reported to protect against kainic acid-induced neuronal apoptosis [56]. With regards to a role for anaphylatoxins in neurogenesis, the respective receptors for C3a and C5a, C3aR and CD88 were shown to be expressed on neural progenitor cells as well as immature neurons [60]. Mice treated with a non-specific C3aR antagonist (SB290157) displayed decreased formation of new neurons in areas of adult neurogenesis [60]. The cellular or humoral source of C3a and the mechanism via which C3aR activation influences the creation and/or survival of new neurons remain to be elucidated. However, in the post-natal developing cerebellum, C3aR and CD88 expressions are known to increase during granular cell maturation [61]. Subdural injection of a non-specific CD88 agonist (MAP-C5a) into the cerebellum of young rats increased the proliferation of immature granule neurons, resulting in an enlarged external granule cell layer [62, 63]. This effect could be reversed through concurrent administration of PMX53, a specific CD88 antagonist [63, 64]. A C3a agonist (MAP-C3a), on the other hand, decreased the thickness of the EGL whilst increasing the thickness of the internal granule cell layer. Video microscopy revealed that C3a accelerated the migration process of granule cells from the EGL to the internal granule cell layer [63].
Complement and CNS disorders
Various regulators normally finely tune the complement activation repertoire so that healthy host tissue is discriminated and self-harm is avoided. However, a disturbed balance between activation and regulation can induce self-attack, and excessive or inappropriate complement activation has been implicated in the pathogenesis of numerous autoimmune, ischaemic and vascular diseases [31, 65]. Complement deregulation has also been proposed in a myriad of CNS inflammatory pathologies and, as a result, complement-targeted therapeutics are increasingly emerging into the spotlight of drug discovery endeavours for various chronic neurodegenerative diseases, including multiple sclerosis [66–74], Alzheimer’s disease [75–78], Huntington's disease [79, 80], Parkinson’s disease [81, 82] and motor neuron disease [83–86]. Although breakdown of the BBB and BSB does not occur until very late in most neurodegenerative pathologies, a prominent role for complement is perhaps not surprising when one considers again that the CNS can endogenously synthesise virtually all components of the complement system under appropriate stimuli [87]. Furthermore, neurons and oligodendrocytes express only low levels of the surface complement regulatory protein decay activating factor (DAF/CD55) and membrane cofactor protein (MCP/CD46), which renders them particularly vulnerable to complement-associated death [88, 89].
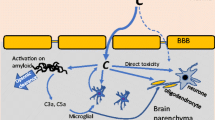
In contrast to the aforementioned chronic neurological disorders, CNS trauma is unique in that it involves a rapid and dramatic breakdown of the BBB/BSB. As a result, the immune-privileged CNS parenchyma, with relatively low endogenous expression of complement and associated negative regulators, is exposed to the full force of both innate and adaptive components of the immune system, which includes a massive influx of serum complement as well as the invasion of circulating and activated leukocytes. Epitopes exposed by cellular injury, including phosphatidylserine, DNA and myelin, are highly vulnerable to complement recognition, opsonisation and MAC deposition [90] (Figure 2). Although this process is important for the clearance of cellular and myelin debris as well as other molecules that may be inhibitory to wound healing and repair, over-activation of complement can compromise the integrity of neurons and oligodendrocytes in neighbouring tissue that was originally spared at the time of impact, thus exacerbating and widening neuropathology [91, 92]. Lastly, since micro-haemorrhaging is a hallmark of most neurotraumatic events, prominent complement activation through the extrinsic pathway can also be expected as a result of protease activity in the blood clotting and fibrinolysis pathways [32, 33]. The following sections of this review will now examine the multifarious roles that complement plays in stroke (i.e. ischaemia-reperfusion injuries), traumatic brain injury (TBI) and spinal cord injury (SCI).

Schematic diagram showing the current understanding of the role of complement activation in the pathophysiology associated with traumatic spinal cord injury (SCI). Mechanical damage to the spinal cord causes neuronal cell death and disruption of the blood-spinal cord barrier (BSB). This primary damage triggers a potent inflammatory response and initiates complement activation. Although complement activation may aid the clearance of cellular debris through opsonisation, it is also known to potentiate injury beyond the site of trauma through e.g. the opsonins C1q, C3b and MAC, which can promote clearance of only mildly compromised cells and thus contribute to secondary demyelination and apoptosis. Known functions of complement in the pathology of SCI are shown in italics; a green font colour indicates a putative reparative role, whereas a red font points towards an injurious role, 1[93], 2[12], 3[14], 4[91], 5[16], 6[94], 7[4, 94].
Complement and cerebral ischaemic-reperfusion (IR) injury
Macroscopically, cerebral IR injury is described as an arterial occlusion preceded by a thromboembolic event. Dynamic changes in cerebral blood flow produce a severe ischaemic core in the territory that is normally supplied by the affected artery, surrounded by a poorly perfused ‘penumbra’ region [95]. Recanalization of the occluded artery leads to reperfusion of blood and the induction of a series of excitotoxic and inflammatory events, which results in microvascular failure and neural cell death [95]. Analysis of blood from human patients with ischaemic stroke showed significant alterations in complement levels, including elevation of the anaphylatoxins C3a and C5a, and depression of the MAC [96]. Moreover, immunohistochemical staining of human brains revealed expression of C1q, C-reactive protein C3 and C4d (classical pathway), while MASP-2 and factor B (lectin pathway) and C9 (terminal pathway) were also present in ischaemic lesions [97]. Staining for CD59 and CD55 was identified in normal brains, but these complement regulators were absent from lesioned brains, supporting that deregulation of complement contributes to IR pathology [97].
To address whether complement activation products are simply a by-product of injury or directly contribute to pathology in human stroke patients, one study analysed the influence of genetic polymorphisms in the mannose-binding lectin (MBL) -2 and MASP-2 genes, which render the lectin pathway dysfunctional, on injury outcome. A logistic regression adjusted for age, gender and initial stroke severity determined that an unfavourable outcome at 3 months post-injury was more likely associated with a normally functioning lectin pathway [98]. This finding was further substantiated in a mouse model of middle cerebral artery occlusion (MCAO) in which MBL-deficient mice displayed smaller infarctions around the penumbra of the striatum, cortex and hippocampus, and better behavioural outcomes as well as less C3 deposition and leukocyte infiltration compared to WT mice [98]. Reconstitution of MBL−/− mice with recombinant human protein annulled the beneficial effects of MBL deficiency [98]. A detrimental role for the lectin pathway in IR injury was, however, not fully reproduced in an independent study where MBL-deficient mice subjected to cerebral IR reportedly showed no difference in systemic neutrophil activation, C3 deposits and only modest tissue sparing in a sub-cortical brain region compared to their WT counterparts [99]. It must be noted, however, that occlusion of the middle cerebral artery in the latter study lasted only for 60 min [99] as opposed to 2 h as in the earlier mentioned investigation [98]. Injury severity and the resulting degree of complement activation may thus have been an influencing factor in the outcome that could explain the seeming discrepancy between these two studies. The contribution of the lectin pathway to secondary pathology was further studied in a human recombinant C1 inhibitor (rhC1-INH), which binds MBL with high affinity [100]. RhC1-INH reduced cerebral damage when given up to 18 h after transient ischaemia and up to 6 h after permanent ischaemia, demonstrating a relatively wide therapeutic window for this treatment [100]. It must be noted, however, that this inhibitor also influences activity of the classical pathway (vide infra).
The significance of classical pathway activation has also been investigated in rodent models of IR injury. One of the subunits of the C1 protease, C1q, accumulates on neuronal cell bodies as well as necrotic cellular debris during the period of greatest infarct evolution [101, 102]. Interestingly, C1q deficiency was shown to be neuroprotective in neonatal (p7) mouse hypoxic-ischemic brain injury [103], predominantly by attenuating oxidative damage [103]. However, no beneficial effects of C1q deficiency on stroke outcomes were observed when adult C1q−/− mice were compared with their WT counterparts [104], suggesting that the presence of C1q during the acute phase does not directly mediate neuronal injury. While it could be argued that a possible detrimental role for C1q in secondary immunopathology following hypoxic-ischaemic brain injury may only become more apparent during the post-acute phase when CNS autoantibodies are likely to be present, this does not explain the discordance between acute neonatal and adult studies. Although speculative at present, an alternative explanation for this apparent discrepancy may lie in age-related differences in complement protein expression and the general maturity of the complement system in neonatal versus adult mice. Known age-related increases in the expression of factor B, C3, C4 and C5 [105] may have masked or overshadowed any neuroprotective effects of C1q deficiency in adult mice through non-classical routes of complement activation.
The therapeutic potential of a C1 inhibitor (C1-INH) that binds and inactivates C1r, C1s, MASP1 and MASP2, thus blocking both the classical and lectin pathways, has also been assessed [106]. These investigators reported a reduction in ischaemic volume (to as low as 10.8% of that of vehicle-treated mice), alongside ameliorated neurological impairments, neuronal degeneration and reduced infiltration of CD45+ leukocytes [106]. Similar effects were observed in Wistar rats following a 60-min occlusion of the middle cerebral artery, after which animals that were treated with this C1 inhibitor had smaller infarct volumes and less granulocyte accumulation [107]. This neuroprotective effect was later shown to be mediated by upregulation of the anti-inflammatory cytokine IL-10 as well as IL-6, which is known to be able to exert both pro- and anti-inflammatory effects [108, 109], in addition to a downregulation of pro-inflammatory P-selectin and ICAM-1, and known inducers of apoptosis like TNF, IL-18 and pro-caspase-3 [110]. When C1q−/− mice were treated with C1-INH, the ischaemic volume was reduced to 31.4% of that of saline-treated mice [106], indicating the protective effects of C1-INH are indeed independent of C1q. Together, these findings indicate that the lectin pathway and subcomponents of the C1 complex, but not C1q, contribute to IR pathology.
An inhibitor of both the classical and alternative pathways, soluble complement receptor-1 (sCR1), led to a significant reduction in neutrophil and platelet aggregation and improved neurological function in mouse MCAO [111, 112]. This finding was, however, not reproduced in a non-human primate model of stroke [113]. It would be of interest to more specifically pinpoint the contribution of the alternative pathway to cerebral IR injury as targeted inhibitors of this pathway have shown therapeutic benefits in both cardiac [114] and intestinal [112] IR injury. This could be achieved using factor B null mice or the targeted inhibitor of the alternative pathway, CR2-fH [112].
As illustrated in Figure 1, the classical, alternative and lectin activation pathways of complement converge at the level of C3 convertase, making this cascade centrepiece an ideal target to assess the overall impact of complement activation in IR injury. In line with the above-mentioned studies, which largely indicate a detrimental role for complement activation following injury, C3−/− mice have significantly smaller infarct volumes, improved neurological deficit scores, and reduced granulocyte infiltration and oxidative stress in a mouse model of transient focal cerebral ischaemia. These effects were reversed by reconstitution with C3 protein [104].
The precise role of C5, the next downstream intersection in the cascade, is still uncertain. C5 deficiency reduced the neurological deficit and lesion size in the MCAO model in mice [115], while C5 inhibition with a monoclonal antibody yielded similarly positive results in rats [116]. A more recent in vitro study showed that oxygen-glucose deprivation can induce the proteolytic cleavage of neuronally expressed C5, which in turn increased apoptotic cell death through a C5a-dependent mechanism [117]. The benefits of C5 deficiency on stroke outcome in mice were, however, not observed in an independent study by Mocco et al. [104]. Although we cannot fully reconcile these differences, it was noted that a later study from the same laboratory did report improved recovery when blocking C5a signalling (vide infra). Further research is therefore warranted to better characterise the role of C5 and its activated cleavage products, C5a and C5b, in the context of IR injury.
Interestingly, C6−/− mice have a similar degree of hypoxic-ischaemic pathology compared to WT mice [118], which appears to suggest that inhibiting MAC formation may not necessarily yield beneficial effects. However, a pathological role of excessive MAC deposition has been supported by studies of CD59a−/− mice. Deficiency in CD59a was associated with increased infarct volume, worse neurological deficits and brain swelling following a 30-min MCAO and 72-h reperfusion as compared to normal C57BL/6 mice [119]. Importantly, however, there was no difference in these outcome measures when these mice were subjected to 1 h of MCAO with 48 h reperfusion, although increased apoptosis was detected in CD59a−/− mice [119]. This highlights that the detrimental effects of excessive MAC formation relative to secondary pathology are probably concentration- and model-specific, and thus most amenable to therapeutic intervention in mild ischaemic-reperfusion injury.
Investigation of the complement system following cerebral IR would not be complete without having determined the role of the anaphylatoxins C3a and C5a. Although these proteins do not lead to MAC formation, as described earlier, they potently mediate inflammation [31]. Expression of both C3aR and CD88 is increased in mouse models of cerebral ischaemia [115, 120, 121]. Pharmacological antagonism of C3aR produced smaller stroke volumes, less ICAM-1 protein on endothelial cells and less upregulation of C3aR-positive granulocytes, but no difference in other inflammatory cell populations in mice subjected to a transient (60 min), but not permanent, MCAO [122]. It should be taken into account, however, when interpreting these findings that the compound used in this study, SB290157, reportedly has off-target effects, including neutropenia in vivo [123], and full agonist activity in a variety of cell systems in vitro[124, 125]. In comparison, mice administered a specific CD88 antagonist 30 min prior to ischaemia (PMX53, 5 mg/kg i.v.) exhibited only a moderately improved outcome when subjected to 60 min MCAO [115]. Others have reported, however, that CD88 inhibition in the same model 45 min prior to ischaemia with the same compound but at a lower dose (1 mg/kg i.v.) yielded dramatically beneficial effects, both in terms of neurological deficits and infarct volume [126]. These findings suggest that anaphylatoxin signalling may be more complex than previously thought, with concentration-specific, time-dependent and model-specific effects.
Complement activation in traumatic brain injury (TBI)
A substantial body of evidence points towards a similarly prominent role for complement activation in the secondary post-injury sequelae following brain injury. In human TBI patients that underwent frontal or temporal lobe resection for intractable intracranial hypertension (2–82 h post-injury), resected tissue was analysed for complement factors [127]. Immunoreactivity against C1q, C3, C4 C3b, C3d and C5b-9 was detected on neurons in the penumbra region of the contused brain area [127]. In the CSF of TBI patients, C3 as well as classical (C1q, C4) [128] and alternative pathway components (factor B) [129] were also elevated, and thus likely to contribute to secondary injury [129]. Furthermore, the concentration of MAC in CSF of TBI patients is up to 1,800-fold higher than in control CSF, and there is a significant correlation between intrathecal MAC levels and post-traumatic BBB dysfunction [130].
In animal models, immunoreactivity for C3 was found around the lesion but not in the uninjured contralateral hemisphere, while deposition of C9, a key component of the membrane attack complex, was also observed on damaged neurons after an experimental cerebral contusion [131]. Collectively, these findings indicate that all four complement pathways are activated in response to TBI, and it is the widely held view that complement deregulation contributes to nerve cell death.
A number of studies have attempted to address the relative contribution of specific complement activation pathways to secondary injury following TBI. Factor B null (fB −/−) mice, which lack a functional alternative pathway, show significantly attenuated complement activation and neuronal death in addition to upregulation of Fas receptor and Bcl-2 in response to TBI compared to brain homogenates of fB +/+ (i.e. WT) littermates [132]. In a follow-up study, administration of monoclonal anti-factor B antibody (mab1379), which strongly inhibits alternative pathway activation, 1 and 24 h post-injury, significantly attenuated C5a levels in serum, in addition to general inflammation and neuronal apoptosis, whilst also yielding a neuroprotective pattern of intracerebral gene expression [133]. Importantly, however, no difference was detected in neurological grade relative to controls. This was attributed to a combination of the short half-life of mab1379, compensatory inflammatory effects (i.e. release of tumour necrosis factor (TNF) and interleukins (IL) -1β, -8, -12, -18), and the need to apply more sensitive neurological testing systems [133]. Further experiments are therefore required to determine the optimal dosage, injection route and time points of mab1379 to fully determine the feasibility and therapeutic merit of targeting the alternative pathway. One option may also be to use alternative pathway-targeted therapies in combination with those targeting other complement activation routes.
Another study evaluated the combined influence of the classical and lectin pathways on the outcome from TBI by administering C1-INH to mice that underwent controlled cortical impact [134]. The investigation revealed attenuation in motor deficits and cognitive dysfunction as well as reduced contusion volumes in the group given C1-INH at 10 min post-injury. When C1-INH treatment was delayed to 60 min post-injury, improvements were only seen in recovery of motor function compared to the saline-treated group [134]. Although a more potent recombinant C1 inhibitor has been developed [100], as of yet, there are no reports detailing the efficacy of this compound in TBI or SCI models.
Unequivocal evidence that complement activation contributes to secondary damage in TBI has come from C3−/− mice, which showed less brain oedema, lower hemeoxygenase-1 levels, and reduced microglia activation and neutrophil infiltration around the clot following intracerebral haemorrhage; usage of the affected forelimb was improved as compared to WT controls [135]. In a cryoinjury model, C3−/− mice again showed fewer infiltrating immune cells, less haemorrhage and better preservation of cytoplasm compared to injured WT brains [136]. Targeted overexpression in the CNS of complement receptor type 1-related protein y (Crry), a functional homologue of the human complement-regulatory proteins CD55 and CD46 that inhibits complement activation at the C3 convertase level, also significantly improved neurological outcome for up to 4 weeks after trauma compared to WT mice [137]. Although this model is perhaps less clinically relevant, its findings led to a follow-up study that used systemic administration of the recombinant Crry molecule (Crry-Ig) in a standardised mouse model of closed head injury [138]. When given 1 and 24 h after trauma, significant neurological improvements and tissue preservation were observed, and this phenotype was associated with upregulation of neuroprotective genes (Bcl-2, C1-Inh, CD55, CD59) in the injured hemisphere compared to the vehicle-treated control group [138].
The role of the MAC in focal closed head injury was investigated using CD59−/− mice, which have over-exuberant MAC deposition because of a lack of this negative complement regulator [139]. As anticipated, CD59−/− mice had significantly exacerbated ‘neurological severity scores’ and displayed increased neuronal cell death. Interestingly, there was no difference in Fas, FasL, Bax or Bcl-2 expression between CD59−/− mice and WT littermates [139].
The role of the anaphylatoxins in the pathophysiology of TBI has also been explored in recent years. A reduction in secondary damage after traumatic brain cyroinjury was observed in C5−/− mice or in mice treated with a CD88 antagonist [136]. Similarly, treatment with a C5aR antagonist [hexapeptide-derived macrocycle AcF(OPdChaWR)] in a mouse model of intracerebral haemorrhage significantly improved neurological function as assessed by spatial memory retention in the Morris water-maze test, corner turn test and a 28-point neurological scale at 24, 48 and 72 h post-injury, and decreased oedema and granulocyte infiltration relative to vehicle-treated animals [140]. These effects were more marked when combined with a C3aR antagonist, although this result should again be interpreted with some caution as the compound SB290157 can display full agonist activity in certain cell types, as mentioned earlier [124, 125]. Thus, anaphylatoxin signalling also appears to negatively influence TBI outcomes, at least acutely. It is, however, necessary to determine whether the beneficial effects of anaphylatoxin antagonism are dependent on timing of the treatment and if they can be sustained long-term without producing deleterious side effects to the host, as observed in other models of neurotrauma (vide infra[4]).
In summary, the above-mentioned studies demonstrate that complement is potently activated following TBI and that targeted interventions can rescue neighbouring intact tissue after head injury with the potential to improve functional outcomes.
Dual roles for complement activation in spinal cord injury?
Following traumatic spinal cord injury (SCI), a robust and complex inflammatory response is initiated through the recruitment and activation of infiltrating leukocytes and resident microglia. It is the widely held view that this inflammatory cascade again exacerbates the primary injury by damaging neighbouring neurons that were originally spared [141, 142]. A prominent role for complement activation in post-SCI inflammation and associated secondary damage is becoming increasingly clear.
Early studies on complement activation in a rat weight-drop model of SCI established that the classical (C1q and C4), alternative (Factor B) and terminal (C5b-9) pathways are strongly activated within 1 day post-injury, and that activation fragments remain on neurons and oligodendrocytes for up to 6 weeks as far as 20 mm rostral to the site of injury [14]. Complement inhibitor proteins such as factor H and clusterin are also reportedly expressed at elevated levels on both neurons and oligodendrocytes after SCI in rats, perhaps in an endogenous attempt to constrain inflammation to the primary injury area [14, 93]. These findings led to several studies in which the therapeutic potential of complement pathway inhibition was tested via pharmacological agents.
The effect of inhibiting the classical/alternative pathway [143] and C3b/C4b activity [144] was investigated in Sprague–Dawley rats subjected to moderate weight-drop SCI. In these studies, treated animals had decreased complement deposition and leukocyte infiltration, which was paralleled by increased tissue sparing and improved locomotor recovery compared to vehicle-treated animals subjected to SCI.
In mouse models of contusive SCI, genetic deficiency of factor B [16], C1q [91] C3 [93, 94] resulted in improved sensory and locomotor outcomes as well as increased tissue sparing in comparison to WT mice. Blocking the alternative pathway with the CR2 inhibitor, Crry [93] or a factor B neutralising antibody [16] similarly improved histological and functional parameters as compared to untreated mice. It has also been shown that SCI potently activates B cells, resulting in the production of pathogenic autoantibodies that bind CNS antigens [12]. Such immune complexes serve as a substrate for ligation by C1q and phagocytic/cytolytic cells bearing IgG receptors (Fc receptors). Indeed, injection of purified antibodies into uninjured spinal cord produced consistent paralysis and pathology, involving the activation of C1q and cells bearing Fc receptors. Conversely, in B cell-deficient mice, which lack antibody production and thus cannot form immune complexes, minimal C1q deposition was observed at and near the lesion site, which in turn was associated with improved recovery of locomotor function [12].
Although the above-detailed studies point to a prominent role for complement-mediated pathology after SCI, particularly via the MAC, the precise role of other activation fragments such as the anaphylatoxins is yet to be conclusively determined. A recent study illustrated these complexities in a rat model of contusive SCI where antagonism of the high-affinity C5a receptor CD88 at 14 days post-SCI worsened locomotor recovery, demyelination and altered macrophage/microglia recruitment [4]. We have now independently confirmed these results in CD88−/− mice, while a similarly novel anti-inflammatory role for C3aR in SCI was also determined in our laboratory. These intriguing findings suggest that activation of these receptors, at least during certain phases post-injury, serve a regulating and perhaps positive role in repair processes, which may be by aiding in the elimination of toxic proteins and debris [4]. With other studies having demonstrated clearly detrimental roles for general complement activation following CNS injury, most likely through formation of the MAC, it is at present difficult to judge the true importance of anaphylatoxin signalling following neurotraumatic events, and whether or not the above detailed findings are specific to SCI. It would therefore be of interest to further study the role of C3a and C5a under conditions where MAC assembly is prevented but anaphylatoxins can still be generated, e.g. through the use of C6-deficient mice [145].
Conclusion and future outlook
Although complement activation is a necessary part of normal wound healing in the body, its deregulation or excessive activation following neurotraumatic events has emerged as a major contributor to secondary tissue damage. All available lines of evidence suggest that targeting complement may represent a novel and effective strategy for attenuating or ameliorating acute CNS trauma. However, because of the multifarious roles of complement in the normal CNS, non-selective and chronic anti-complement therapies may be a less favourable option when aiming to translate complement-directed therapeutics into the clinic. Detrimental outcomes have already been observed in SCI following long-term interference with anaphylatoxin receptor signalling and similar effects could perhaps be anticipated with other approaches, particularly when one considers that, similar to development, complement activation may play vital roles in post-injury plasticity; specifically the rewiring of local neural circuits that is thought to underpin functional recovery following SCI [146]. As such, it will be critical to elucidate the precise spatiotemporal function of specific complement activation fragments in future years. This will allow the design of novel and optimally effective therapeutic strategies that can harness ameliorative factors and neutralise or regulate toxic components in an appropriate and timely fashion.
As progress is being made towards the translation of complement-based therapeutics into the clinic, consideration should also be given to the fact that there are known functional differences in complement system activity between species that could influence the efficacy of the intervention and thus trial outcomes. Although studies in genetically modified mice have greatly advanced our understanding of complement activation in neurotrauma, unlocking a number of promising avenues for therapeutic intervention, certain strains of mice are also known to have lower complement activity compared to other mammals such as rats, rabbits, guinea pigs and humans [147–151]. These observations highlight the importance of understanding the limitations of experimental models and, where appropriate, the need to use intermediate animal models between mice and humans in translational complement research.
Abbreviations
- APC:
-
Antigen-presenting cell
- BBB:
-
Blood–brain barrier
- BSB:
-
Blood-spinal cord barrier
- C1-INH:
-
Complement component 1 inhibitor
- cAMP:
-
cyclic adenosine monophosphate
- CNS:
-
Central nervous system
- Crry:
-
Complement receptor type 1-related protein y
- DAF:
-
Decay-accelerating factor
- dLGN:
-
dorsal lateral geniculate nucleus
- GLT-1:
-
Glial glutamate transporter 1
- GluR2:
-
Glutamate receptor subunit 2
- IL:
-
Interleukin
- IR:
-
Ischemic-reperfusion
- MAC:
-
Membrane attack complex
- MAPK:
-
Mitogen-activated protein kinase
- MASP:
-
Mannose-binding lectin-associated serine protein
- MBL:
-
Mannose-binding lectin
- MCAO:
-
Middle cerebral artery occlusion
- NGF:
-
Nerve growth factor
- PAMP:
-
Pathogen-associated molecular pattern
- RGC:
-
Retinal ganglion cell
- SCI:
-
Spinal cord injury
- sCR1:
-
soluble complement receptor 1
- TBI:
-
Traumatic brain injury
- TNF:
-
Tumour necrosis factor
- WT:
-
Wild type.
References
Taoka Y, Okajima K, Uchiba M, Murakami K, Kushimoto S, Johno M, Naruo M, Okabe H, Takatsuki K: Role of neutrophils in spinal cord injury in the rat. Neuroscience 1997, 79:1177–1182.
Gris D, Marsh DR, Oatway MA, Chen Y, Hamilton EF, Dekaban GA, Weaver LC: Transient blockade of the CD11d/CD18 integrin reduces secondary damage after spinal cord injury, improving sensory, autonomic, and motor function. J Neurosci 2004, 24:4043–4051.
Stirling DP, Liu S, Kubes P, Yong VW: Depletion of Ly6G/Gr-1 leukocytes after spinal cord injury in mice alters wound healing and worsens neurological outcome. J Neurosci 2009, 29:753–764.
Beck KD, Nguyen HX, Galvan MD, Salazar DL, Woodruff TM, Anderson AJ: Quantitative analysis of cellular inflammation after traumatic spinal cord injury: evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain 2010, 133:433–447.
Geremia NM, Bao F, Rosenzweig TE, Hryciw T, Weaver L, Dekaban GA, Brown A: CD11d antibody treatment improves recovery in spinal cord-injured mice. J Neurotrauma 2012, 29:539–550.
Popovich P, Guan Z, Wei P, Huitinga I, van Rooijen N, Stokes B: Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol 1999, 158:351–365.
Mabon PJ, Weaver LC, Dekaban GA: Inhibition of monocyte/macrophage migration to a spinal cord injury site by an antibody to the integrin alphaD: a potential new anti-inflammatory treatment. Exp Neurol 2000, 166:52–64.
Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, Rolls A, Mack M, Pluchino S, Martino G, Jung S, Schwartz M: Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med 2009, 6:e1000113.
Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG: Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci 2009, 29:13435–13444.
Popovich PG, Stokes BT, Whitacre CC: Concept of autoimmunity following spinal cord injury: possible roles for T lymphocytes in the traumatized central nervous system. J Neurosci Res 1996, 45:349–363.
Schwartz M, Hauben E: T cell-based therapeutic vaccination for spinal cord injury. Prog Brain Res 2002, 137:401–406.
Ankeny DP, Guan Z, Popovich PG: B cells produce pathogenic antibodies and impair recovery after spinal cord injury in mice. J Clin Invest 2009, 119:2990–2999.
Hawthorne AL, Popovich PG: Emerging concepts in myeloid cell biology after spinal cord injury. Neurotherapeutics 2011, 8:252–261.
Anderson A, Robert S, Huang W, Young W, Cotman C: Activation of complement pathways after contusion-induced spinal cord injury. J Neurotrauma 2004, 21:1831–1846.
Nguyen H, Galvan M, Anderson A: Characterization of early and terminal complement proteins associated with polymorphonuclear leukocytes in vitro and in vivo after spinal cord injury. J Neuroinflammation 2008, 5:26.
Qiao F, Atkinson C, Kindy MS, Shunmugavel A, Morgan BP, Song H, Tomlinson S: The alternative and terminal pathways of complement mediate post-traumatic spinal cord inflammation and injury. Am J Pathol 2010, 177:3061–3070.
Rutkowski MJ, Sughrue ME, Kane AJ, Mills SA, Fang S, Parsa AT: Complement and the central nervous system: emerging roles in development, protection and regeneration. Immunol Cell Biol 2010, 88:781–786.
Woodruff TM, Ager RR, Tenner AJ, Noakes PG, Taylor SM: The role of the complement system and the activation fragment C5a in the central nervous system. Neuromolecular Med 2010, 12:179–192.
Veerhuis R, Nielsen HM, Tenner AJ: Complement in the brain. Mol Immunol 2011, 48:1592–1603.
Alper CA, Johnson AM, Birtch AG, Moore FD: Human C'3: evidence for the liver as the primary site of synthesis. Science 1969, 163:286–288.
Gaboriaud C, Thielens NM, Gregory LA, Rossi V, Fontecilla-Camps JC, Arlaud GJ: Structure and activation of the C1 complex of complement: unraveling the puzzle. Trends Immunol 2004, 25:368–373.
Carroll MC: The complement system in regulation of adaptive immunity. Nat Immunol 2004, 5:981–986.
Holers VM, Kulik L: Complement receptor 2, natural antibodies and innate immunity: Inter-relationships in B cell selection and activation. Mol Immunol 2007, 44:64–72.
Carroll MC: Complement and humoral immunity. Vaccine 2008,26(Suppl 8):I28-I33.
Dunkelberger JR, Song WC: Role and mechanism of action of complement in regulating T cell immunity. Mol Immunol 2010, 47:2176–2186.
Kwan WH, van der Touw W, Heeger PS: Complement regulation of T cell immunity. Immunol Res 2012,:. Epub ahead of print
Stahl PD, Ezekowitz RA: The mannose receptor is a pattern recognition receptor involved in host defense. Curr Opin Immunol 1998, 10:50–55.
Lu J, Le Y: Ficolins and the fibrinogen-like domain. Immunobiology 1998, 199:190–199.
Pangburn MK, Schreiber RD, Muller-Eberhard HJ: Formation of the initial C3 convertase of the alternative complement pathway. Acquisition of C3b-like activities by spontaneous hydrolysis of the putative thioester in native C3. J Exp Med 1981, 154:856–67.
Bianchi ME: DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 2007, 81:1–5.
Ricklin D, Hajishengallis G, Yang K, Lambris JD: Complement: a key system for immune surveillance and homeostasis. Nat Immunol 2010, 11:785–797.
Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA, Ward PA: Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med 2006, 12:682–687.
Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, Bruckner UB, Nilsson B, Gebhard F, Lambris JD, Huber-Lang M: Molecular intercommunication between the complement and coagulation systems. J Immunol 2010, 185:5628–5636.
Perl M, Denk S, Kalbitz M, Huber-Lang M: Granzyme B: a new crossroad of complement and apoptosis. Adv Exp Med Biol 2012, 946:135–146.
Ames RS, Li Y, Sarau HM, Nuthulaganti P, Foley JJ, Ellis C, Zeng Z, Su K, Jurewicz AJ, Hertzberg RP, Bergsma DJ, Kumar C: Molecular cloning and characterization of the human anaphylatoxin C3a receptor. J Biol Chem 1996, 271:20231–20234.
Manthey HD, Woodruff TM, Taylor SM, Monk PN: Complement component 5a (C5a). Int J Biochem Cell Biol 2009, 41:2114–2117.
Gerard NP, Gerard C: The chemotactic receptor for human C5a anaphylatoxin. Nature 1991, 349:614–617.
Cain SA, Monk PN: The orphan receptor C5L2 has high affinity binding sites for complement fragments C5a and C5a des-Arg(74). J Biol Chem 2002, 277:7165–7169.
Chen NJ, Mirtsos C, Suh D, Lu YC, Lin WJ, McKerlie C, Lee T, Baribault H, Tian H, Yeh WC: C5L2 is critical for the biological activities of the anaphylatoxins C5a and C3a. Nature 2007, 446:203–207.
Scola AM, Johswich KO, Morgan BP, Klos A, Monk PN: The human complement fragment receptor, C5L2, is a recycling decoy receptor. Mol Immunol 2009, 46:1149–1162.
Rittirsch D, Flierl MA, Nadeau BA, Day DE, Huber-Lang M, Mackay CR, Zetoune FS, Gerard NP, Cianflone K, Kohl J, Gerard C, Sarma JV, Ward PA: Functional roles for C5a receptors in sepsis. Nat Med 2008, 14:551–557.
Bamberg CE, Mackay CR, Lee H, Zahra D, Jackson J, Lim YS, Whitfeld PL, Craig S, Corsini E, Lu B, Gerard C, Gerard NP: The C5a receptor (C5aR) C5L2 is a modulator of C5aR-mediated signal transduction. J Biol Chem 2010, 285:7633–7644.
Perry VH, O'Connor V: C1q: the perfect complement for a synaptic feast? Nat Rev Neurosci 2008, 9:807–811.
Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA: The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131:1164–1178.
Jaubert-Miazza L, Green E, Lo FS, Bui K, Mills J, Guido W: Structural and functional composition of the developing retinogeniculate pathway in the mouse. Vis Neurosci 2005, 22:661–676.
Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B, Prince DA: Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci USA 2010, 107:7975–7980.
Crane JW, Baiquni GP, Sullivan RK, Lee JD, Sah P, Taylor SM, Noakes PG, Woodruff TM: The C5a anaphylatoxin receptor CD88 is expressed in presynaptic terminals of hippocampal mossy fibres. J Neuroinflammation 2009, 6:34.
Pisalyaput K, Tenner AJ: Complement component C1q inhibits beta-amyloid- and serum amyloid P-induced neurotoxicity via caspase- and calpain-independent mechanisms. J Neurochem 2008, 104:696–707.
Benoit ME, Tenner AJ: Complement protein C1q-mediated neuroprotection is correlated with regulation of neuronal gene and microRNA expression. J Neurosci 2011, 31:3459–3469.
Yanamadala V, Friedlander RM: Complement in neuroprotection and neurodegeneration. Trends Mol Med 2010, 16:69–76.
Soane L, Rus H, Niculescu F, Shin ML: Inhibition of oligodendrocyte apoptosis by sublytic C5b-9 is associated with enhanced synthesis of bcl-2 and mediated by inhibition of caspase-3 activation. J Immunol 1999, 163:6132–6138.
Cudrici C, Niculescu F, Jensen T, Zafranskaia E, Fosbrink M, Rus V, Shin ML, Rus H: C5b-9 terminal complex protects oligodendrocytes from apoptotic cell death by inhibiting caspase-8 processing and up-regulating FLIP. J Immunol 2006, 176:3173–3180.
van Beek J, Nicole O, Ali C, Ischenko A, MacKenzie ET, Buisson A, Fontaine M: Complement anaphylatoxin C3a is selectively protective against NMDA-induced neuronal cell death. Neuroreport 2001, 12:289–293.
Heese K, Hock C, Otten U: Inflammatory signals induce neurotrophin expression in human microglial cells. J Neurochem 1998, 70:699–707.
Jauneau AC, Ischenko A, Chatagner A, Benard M, Chan P, Schouft MT, Patte C, Vaudry H, Fontaine M: Interleukin-1beta and anaphylatoxins exert a synergistic effect on NGF expression by astrocytes. J Neuroinflammation 2006, 3:8.
Osaka H, Mukherjee P, Aisen PS, Pasinetti GM: Complement-derived anaphylatoxin C5a protects against glutamate-mediated neurotoxicity. J Cell Biochem 1999, 73:303–311.
Mukherjee P, Pasinetti GM: Complement anaphylatoxin C5a neuroprotects through mitogen-activated protein kinase-dependent inhibition of caspase 3. J Neurochem 2001, 77:43–49.
Mukherjee P, Thomas S, Pasinetti GM: Complement anaphylatoxin C5a neuroprotects through regulation of glutamate receptor subunit 2 in vitro and in vivo. J Neuroinflammation 2008, 5:5.
Persson M, Pekna M, Hansson E, Ronnback L: The complement-derived anaphylatoxin C5a increases microglial GLT-1 expression and glutamate uptake in a TNF-alpha-independent manner. Eur J Neurosci 2009, 29:267–274.
Rahpeymai Y, Hietala MA, Wilhelmsson U, Fotheringham A, Davies I, Nilsson AK, Zwirner J, Wetsel RA, Gerard C, Pekny M, Pekna M: Complement: a novel factor in basal and ischemia-induced neurogenesis. EMBO J 2006, 25:1364–1374.
Benard M, Gonzalez BJ, Schouft MT, Falluel-Morel A, Vaudry D, Chan P, Vaudry H, Fontaine M: Characterization of C3a and C5a receptors in rat cerebellar granule neurons during maturation. Neuroprotective effect of C5a against apoptotic cell death. J Biol Chem 2004, 279:43487–43496.
Jurianz K, Ziegler S, Donin N, Reiter Y, Fishelson Z, Kirschfink M: K562 erythroleukemic cells are equipped with multiple mechanisms of resistance to lysis by complement. Int J Cancer 2001, 93:848–854.
Benard M, Raoult E, Vaudry D, Leprince J, Falluel-Morel A, Gonzalez BJ, Galas L, Vaudry H, Fontaine M: Role of complement anaphylatoxin receptors (C3aR, C5aR) in the development of the rat cerebellum. Mol Immunol 2008, 45:3767–3774.
Woodruff TM, Nandakumar KS, Tedesco F: Inhibiting the C5-C5a receptor axis. Mol Immunol 2011, 48:1631–1642.
Wagner E, Frank MM: Therapeutic potential of complement modulation. Nat Rev Drug Discov 2010, 9:43–56.
Morariu MA, Dalmasso AP: Experimental allergic encephalomyelitis in cobra venom factor-treated and C4-deficient guinea pigs. Ann Neurol 1978, 4:427–430.
Morgan BP, Campbell AK, Compston DA: Terminal component of complement (C9) in cerebrospinal fluid of patients with multiple sclerosis. Lancet 1984, 2:251–254.
Nataf S, Carroll SL, Wetsel RA, Szalai AJ, Barnum SR: Attenuation of experimental autoimmune demyelination in complement-deficient mice. J Immunol 2000, 165:5867–5873.
Niculescu T, Weerth S, Niculescu F, Cudrici C, Rus V, Raine CS, Shin ML, Rus H: Effects of complement C5 on apoptosis in experimental autoimmune encephalomyelitis. J Immunol 2004, 172:5702–5706.
Liu J, Miwa T, Hilliard B, Chen Y, Lambris JD, Wells AD, Song WC: The complement inhibitory protein DAF (CD55) suppresses T cell immunity in vivo. J Exp Med 2005, 201:567–577.
Rus H, Cudrici C, Niculescu F, Shin ML: Complement activation in autoimmune demyelination: dual role in neuroinflammation and neuroprotection. J Neuroimmunol 2006, 180:9–16.
Li Q, Nacion K, Bu H, Lin F: The complement inhibitor FUT-175 suppresses T cell autoreactivity in experimental autoimmune encephalomyelitis. Am J Pathol 2009, 175:661–667.
Ramaglia V, Hughes TR, Donev RM, Ruseva MM, Wu X, Huitinga I, Baas F, Neal JW, Morgan BP: C3-dependent mechanism of microglial priming relevant to multiple sclerosis. Proc Natl Acad Sci USA 2012, 109:965–970.
Ingram G, Hakobyan S, Robertson NP, Morgan BP: Complement in multiple sclerosis: its role in disease and potential as a biomarker. Clin Exp Immunol 2009, 155:128–139.
Zhou J, Fonseca MI, Pisalyaput K, Tenner AJ: Complement C3 and C4 expression in C1q sufficient and deficient mouse models of Alzheimer's disease. J Neurochem 2008, 106:2080–2092.
Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA: Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci 2008, 28:6333–6341.
Fonseca MI, Ager RR, Chu SH, Yazan O, Sanderson SD, LaFerla FM, Taylor SM, Woodruff TM, Tenner AJ: Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer's disease. J Immunol 2009, 183:1375–1383.
Veerhuis R: Histological and direct evidence for the role of complement in the neuroinflammation of AD. Curr Alzheimer Res 2011, 8:34–58.
Woodruff T, Crane J, Proctor L, Buller K, Shek A, de Vos K, Pollitt S, Williams H, Shiels I, Monk P, Taylor S: Therapeutic activity of C5a receptor antagonists in a rat model of neurodegeneration. FASEB J 2006, 20:1407–1417.
Moller T: Neuroinflammation in Huntington's disease. J Neural Transm 2010, 117:1001–1008.
Wang XJ, Yan ZQ, Lu GQ, Stuart S, Chen SD: Parkinson disease IgG and C5a-induced synergistic dopaminergic neurotoxicity: role of microglia. Neurochem Int 2007, 50:39–50.
Depboylu C, Schafer MK, Arias-Carrion O, Oertel WH, Weihe E, Hoglinger GU: Possible involvement of complement factor C1q in the clearance of extracellular neuromelanin from the substantia nigra in Parkinson disease. J Neuropathol Exp Neurol 2011, 70:125–132.
Rus H, Niculescu F: The complement system in central nervous system diseases. Immunol Res 2001, 24:79–86.
Woodruff TM, Costantini KJ, Crane JW, Atkin JD, Monk PN, Taylor SM, Noakes PG: The complement factor C5a contributes to pathology in a rat model of amyotrophic lateral sclerosis. J Immunol 2008, 181:8727–8734.
Woodruff TM, Costantini KJ, Taylor SM, Noakes PG: Role of complement in motor neuron disease: animal models and therapeutic potential of complement inhibitors. Adv Exp Med Biol 2008, 632:143–158.
Heurich B, El Idrissi NB, Donev RM, Petri S, Claus P, Neal J, Morgan BP, Ramaglia V: Complement upregulation and activation on motor neurons and neuromuscular junction in the SOD1 G93A mouse model of familial amyotrophic lateral sclerosis. J Neuroimmunol 2011, 235:104–109.
Gasque P, Dean YD, McGreal EP, VanBeek J, Morgan BP: Complement components of the innate immune system in health and disease in the CNS. Immunopharmacology 2000, 49:171–186.
Singhrao SK, Neal JW, Rushmere NK, Morgan BP, Gasque P: Spontaneous classical pathway activation and deficiency of membrane regulators render human neurons susceptible to complement lysis. Am J Pathol 2000, 157:905–918.
Agoropoulou C, Piddlesden SJ, Lachmann PJ, Wing MG: Neuronal protection of oligodendrocytes from antibody-independent complement lysis. Neuroreport 1998, 9:927–932.
Alexander JJ, Anderson AJ, Barnum SR, Stevens B, Tenner AJ: The complement cascade: Yin-Yang in neuroinflammation–neuro-protection and -degeneration. J Neurochem 2008, 107:1169–1187.
Galvan MD, Luchetti S, Burgos AM, Nguyen HX, Hooshmand MJ, Hamers FPT, Anderson AJ: Deficiency in complement C1q improves histological and functional locomotor outcome after spinal cord injury. J Neurosci 2008, 28:13876–13888.
Arumugam TV, Woodruff TM, Lathia JD, Selvaraj PK, Mattson MP, Taylor SM: Neuroprotection in stroke by complement inhibition and immunoglobulin therapy. Neuroscience 2009, 158:1074–1089.
Qiao F, Atkinson C, Song H, Pannu R, Singh I, Tomlinson S: Complement plays an important role in spinal cord injury and represents a therapeutic target for improving recovery following trauma. Am J Pathol 2006, 169:1039–1047.
Guo Q, Li S, Liang Y, Zhang Y, Zhang J, Wen C, Lin S, Wang H, Su B: Effects of C3 deficiency on inflammation and regeneration following spinal cord injury in mice. Neurosci Lett 2010, 485:32–36.
Kato H, Kogure K: Biochemical and molecular characteristics of the brain with developing cerebral infarction. Cell Mol Neurobiol 1999, 19:93–108.
Mocco J, Wilson DA, Komotar RJ, Sughrue ME, Coates K, Sacco RL, Elkind MS, Connolly ES: Alterations in plasma complement levels after human ischemic stroke. Neurosurgery 2006, 59:28–33. discussion 28–33
Pedersen ED, Loberg EM, Vege E, Daha MR, Maehlen J, Mollnes TE: In situ deposition of complement in human acute brain ischaemia. Scand J Immunol 2009, 69:555–562.
Cervera A, Planas AM, Justicia C, Urra X, Jensenius JC, Torres F, Lozano F, Chamorro A: Genetically-defined deficiency of mannose-binding lectin is associated with protection after experimental stroke in mice and outcome in human stroke. PLoS One 2010, 5:e8433.
Morrison H, Frye J, Davis-Gorman G, Funk J, McDonagh P, Stahl G, Ritter L: The contribution of mannose binding lectin to reperfusion injury after ischemic stroke. Curr Neurovasc Res 2011, 8:52–63.
Gesuete R, Storini C, Fantin A, Stravalaci M, Zanier ER, Orsini F, Vietsch H, Mannesse ML, Ziere B, Gobbi M, De Simoni MG: Recombinant C1 inhibitor in brain ischemic injury. Ann Neurol 2009, 66:332–342.
Schafer MK, Schwaeble WJ, Post C, Salvati P, Calabresi M, Sim RB, Petry F, Loos M, Weihe E: Complement C1q is dramatically up-regulated in brain microglia in response to transient global cerebral ischemia. J Immunol 2000, 164:5446–5452.
Mack WJ, Sughrue ME, Ducruet AF, Mocco J, Sosunov SA, Hassid BG, Silverberg JZ, Ten VS, Pinsky DJ, Connolly ES: Temporal pattern of C1q deposition after transient focal cerebral ischemia. J Neurosci Res 2006, 83:883–889.
Ten VS, Sosunov SA, Mazer SP, Stark RI, Caspersen C, Sughrue ME, Botto M, Connolly ES, Pinsky DJ: C1q-deficiency is neuroprotective against hypoxic-ischemic brain injury in neonatal mice. Stroke 2005, 36:2244–2250.
Mocco J, Mack WJ, Ducruet AF, Sosunov SA, Sughrue ME, Hassid BG, Nair MN, Laufer I, Komotar RJ, Claire M, Holland H, Pinsky DJ, Connolly ES: Complement component c3 mediates inflammatory injury following focal cerebral ischemia. Circ Res 2006, 99:209–217.
Reichwald J, Danner S, Wiederhold KH, Staufenbiel M: Expression of complement system components during aging and amyloid deposition in APP transgenic mice. J Neuroinflammation 2009, 6:35.
De Simoni MG, Rossi E, Storini C, Pizzimenti S, Echart C, Bergamaschini L: The powerful neuroprotective action of C1-inhibitor on brain ischemia-reperfusion injury does not require C1q. Am J Pathol 2004, 164:1857–1863.
Akita N, Nakase H, Kaido T, Kanemoto Y, Sakaki T: Protective effect of C1 esterase inhibitor on reperfusion injury in the rat middle cerebral artery occlusion model. Neurosurgery 2003, 52:395–400. discussion 400–1
Kishimoto T, Akira S, Narazaki M, Taga T: Interleukin-6 family of cytokines and gp130. Blood 1995, 86:1243–1254.
Xing Z, Gauldie J, Cox G, Baumann H, Jordana M, Lei XF, Achong MK: IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest 1998, 101:311–320.
Storini C, Rossi E, Marrella V, Distaso M, Veerhuis R, Vergani C, Bergamaschini L, De Simoni MG: C1-inhibitor protects against brain ischemia-reperfusion injury via inhibition of cell recruitment and inflammation. Neurobiol Dis 2005, 19:10–17.
Huang J, Kim LJ, Mealey R, Marsh HC, Zhang Y, Tenner AJ, Connolly ES, Pinsky DJ: Neuronal protection in stroke by an sLex-glycosylated complement inhibitory protein. Science 1999, 285:595–599.
Huang Y, Qiao F, Atkinson C, Holers VM, Tomlinson S: A novel targeted inhibitor of the alternative pathway of complement and its therapeutic application in ischemia/reperfusion injury. J Immunol 2008, 181:8068–8076.
Mocco J, Mack WJ, Ducruet AF, King RG, Sughrue ME, Coon AL, Sosunov SA, Sciacca RR, Zhang Y, Marsh HC, Pinsky DJ, Connolly ES: Preclinical evaluation of the neuroprotective effect of soluble complement receptor type 1 in a nonhuman primate model of reperfused stroke. J Neurosurg 2006, 105:595–601.
Atkinson C, He S, Morris K, Qiao F, Casey S, Goddard M, Tomlinson S: Targeted complement inhibitors protect against posttransplant cardiac ischemia and reperfusion injury and reveal an important role for the alternative pathway of complement activation. J Immunol 2010, 185:7007–7013.
Arumugam TV, Tang SC, Lathia JD, Cheng A, Mughal MR, Chigurupati S, Magnus T, Chan SL, Jo DG, Ouyang X, Fairlie DP, Granger DN, Vortmeyer A, Basta M, Mattson MP: Intravenous immunoglobulin (IVIG) protects the brain against experimental stroke by preventing complement-mediated neuronal cell death. Proc Natl Acad Sci USA 2007, 104:14104–14109.
Costa C, Zhao L, Shen Y, Su X, Hao L, Colgan SP, Stahl GL, Zhou T, Wang Y: Role of complement component C5 in cerebral ischemia/reperfusion injury. Brain Res 2006, 1100:142–151.
Pavlovski D, Thundyil J, Monk PN, Wetsel RA, Taylor SM, Woodruff TM: Generation of complement component C5a by ischemic neurons promotes neuronal apoptosis. FASEB J 2012,:. in press
Ten VS, Yao J, Ratner V, Sosunov S, Fraser DA, Botto M, Sivasankar B, Morgan BP, Silverstein S, Stark R, Polin R, Vannucci SJ, Pinsky D, Starkov AA: Complement component c1q mediates mitochondria-driven oxidative stress in neonatal hypoxic-ischemic brain injury. J Neurosci 2010, 30:2077–2087.
Harhausen D, Khojasteh U, Stahel PF, Morgan BP, Nietfeld W, Dirnagl U, Trendelenburg G: Membrane attack complex inhibitor CD59a protects against focal cerebral ischemia in mice. J Neuroinflammation 2010, 7:15.
Nishino H, Czurko A, Fukuda A, Hashitani T, Hida H, Karadi Z, Lenard L: Pathophysiological process after transient ischemia of the middle cerebral artery in the rat. Brain Res Bull 1994, 35:51–56.
Barnum SR, Ames RS, Maycox PR, Hadingham SJ, Meakin J, Harrison D, Parsons AA: Expression of the complement C3a and C5a receptors after permanent focal ischemia: An alternative interpretation. Glia 2002, 38:169–173.
Ducruet AF, Hassid BG, Mack WJ, Sosunov SA, Otten ML, Fusco DJ, Hickman ZL, Kim GH, Komotar RJ, Mocco J, Connolly ES: C3a receptor modulation of granulocyte infiltration after murine focal cerebral ischemia is reperfusion dependent. J Cereb Blood Flow Metab 2008, 28:1048–1058.
Proctor LM, Arumugam TV, Shiels I, Reid RC, Fairlie DP, Taylor SM: Comparative anti-inflammatory activities of antagonists to C3a and C5a receptors in a rat model of intestinal ischaemia/reperfusion injury. Br J Pharmacol 2004, 142:756–764.
Mathieu MC, Sawyer N, Greig GM, Hamel M, Kargman S, Ducharme Y, Lau CK, Friesen RW, O'Neill GP, Gervais FG, Therien AG: The C3a receptor antagonist SB 290157 has agonist activity. Immunol Lett 2005, 100:139–145.
Therien AG: Agonist activity of the small molecule C3aR ligand SB 290157. J Immunol 2005, 174:7479–7480.
Kim GH, Mocco J, Hahn DK, Kellner CP, Komotar RJ, Ducruet AF, Mack WJ, Connolly ES: Protective effect of C5a receptor inhibition after murine reperfused stroke. Neurosurgery 2008, 63:122–125. discussion 125–6
Bellander B-M, Singhrao SK, Ohlsson M, Mattsson P, Svensson M: Complement activation in the human brain after traumatic head injury. J Neurotrauma 2001, 18:1295–1311.
Stahel PF, Morganti-Kossmann MC, Kossmann T: The role of the complement system in traumatic brain injury. Brain Res Brain Res Rev 1998, 27:243–256.
Kossmann T, Stahel PF, Morganti-Kossmann MC, Jones JL, Barnum SR: Elevated levels of the complement components C3 and factor B in ventricular cerebrospinal fluid of patients with traumatic brain injury. J Neuroimmunol 1997, 73:63–69.
Stahel PF, Morganti-Kossmann MC, Perez D, Redaelli C, Gloor B, Trentz O, Kossmann T: Intrathecal levels of complement-derived soluble membrane attack complex (sC5b-9) correlate with blood–brain barrier dysfunction in patients with traumatic brain injury. J Neurotrauma 2001, 18:773–781.
Bellander BM, von Holst H, Fredman P, Svensson M: Activation of the complement cascade and increase of clusterin in the brain following a cortical contusion in the adult rat. J Neurosurg 1996, 85:468–475.
Leinhase I, Holers VM, Thurman JM, Harhausen D, Schmidt OI, Pietzcker M, Taha ME, Rittirsch D, Huber-Lang M, Smith WR, Ward PA, Stahel PF: Reduced neuronal cell death after experimental brain injury in mice lacking a functional alternative pathway of complement activation. BMC Neurosci 2006, 7:55.
Leinhase I, Rozanski M, Harhausen D, Thurman JM, Schmidt OI, Hossini AM, Taha ME, Rittirsch D, Ward PA, Holers VM, Ertel W, Stahel PF: Inhibition of the alternative complement activation pathway in traumatic brain injury by a monoclonal anti-factor B antibody: a randomized placebo-controlled study in mice. J Neuroinflammation 2007, 4:13.
Longhi L, Perego C, Ortolano F, Zanier ER, Bianchi P, Stocchetti N, McIntosh TK, De Simoni MG: C1-inhibitor attenuates neurobehavioral deficits and reduces contusion volume after controlled cortical impact brain injury in mice. Crit Care Med 2009, 37:659–665.
Yang S, Nakamura T, Hua Y, Keep RF, Younger JG, He Y, Hoff JT, Xi G: The role of complement C3 in intracerebral hemorrhage-induced brain injury. J Cereb Blood Flow Metab 2006, 26:1490–1495.
Sewell DL, Nacewicz B, Liu F, Macvilay S, Erdei A, Lambris JD, Sandor M, Fabry Z: Complement C3 and C5 play critical roles in traumatic brain cryoinjury: blocking effects on neutrophil extravasation by C5a receptor antagonist. J Neuroimmunol 2004, 155:55–63.
Rancan M, Morganti-Kossmann MC, Barnum SR, Saft S, Schmidt OI, Ertel W, Stahel PF: Central nervous system-targeted complement inhibition mediates neuroprotection after closed head injury in transgenic mice. J Cereb Blood Flow Metab 2003, 23:1070–1074.
Leinhase I, Schmidt OI, Thurman JM, Hossini AM, Rozanski M, Taha ME, Scheffler A, John T, Smith WR, Holers VM, Stahel PF: Pharmacological complement inhibition at the C3 convertase level promotes neuronal survival, neuroprotective intracerebral gene expression, and neurological outcome after traumatic brain injury. Exp Neurol 2006, 199:454–464.
Stahel PF, Flierl MA, Morgan BP, Persigehl I, Stoll C, Conrad C, Touban BM, Smith WR, Beauchamp K, Schmidt OI, Ertel W, Leinhase I: Absence of the complement regulatory molecule CD59a leads to exacerbated neuropathology after traumatic brain injury in mice. J Neuroinflammation 2009, 6:2.
Garrett MC, Otten ML, Starke RM, Komotar RJ, Magotti P, Lambris JD, Rynkowski MA, Connolly ES: Synergistic neuroprotective effects of C3a and C5a receptor blockade following intracerebral hemorrhage. Brain Res 2009, 1298:171–177.
Hausmann ON: Post-traumatic inflammation following spinal cord injury. Spinal Cord 2003, 41:369–378.
Dumont RJO, David O, Subodh Verma , Hurlbert , John R, Boulos , Paul T, Ellegala , Dilantha B, Dumont , Aaron S: Acute spinal cord injury, part i: pathophysiologic mechanisms. Clin Neuropharmacol 2001, 24:254–264.
Reynolds DN, Smith SA, Zhang YP, Mengsheng Q, Lahiri DK, Morassutti DJ, Shields CB, Kotwal GJ: Vaccinia virus complement control protein reduces inflammation and improves spinal cord integrity following spinal cord injury. Ann N Y Acad Sci 2004, 1035:165–178.
Li LM, Li JB, Zhu Y, Fan GY: Soluble complement receptor type 1 inhibits complement system activation and improves motor function in acute spinal cord injury. Spinal Cord 2009, 48:105–111.
Banda NK, Hyatt S, Antonioli AH, White JT, Glogowska M, Takahashi K, Merkel TJ, Stahl GL, Mueller-Ortiz S, Wetsel R, Arend WP, Holers VM: Role of C3a receptors, C5a receptors, and complement protein C6 deficiency in collagen antibody-induced arthritis in mice. J Immunol 2012, 188:1469–1478.
Rossignol S, Frigon A: Recovery of locomotion after spinal cord injury: some facts and mechanisms. Annu Rev Neurosci 2011, 34:413–440.
Rice CE: The interchangeability of the complement components of different animal species; literature survey. Can J Comp Med Vet Sci 1950, 14:369–379.
Drake WP, Ungaro PC, Mardiney MR: The measurement and manipulation of hemolytic complement levels in tumor bearing C57BL/6 mice. Biomedicine 1973, 18:284–289.
Grant CK: Complement origin determines lytic activity of antibodies to nucleated target cells. Comparison of common complement sources. Transplantation 1976, 21:323–330.
Ebanks RO, Isenman DE: Mouse complement component C4 is devoid of classical pathway C5 convertase subunit activity. Mol Immunol 1996, 33:297–309.
Ong GL, Mattes MJ: Mouse strains with typical mammalian levels of complement activity. J Immunol Methods 1989, 125:147–158.
Acknowledgements
FHB is supported by an Australian Postgraduate Award (Australian Government) and MJR by a SpinalCure Australia Career Development Fellowship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors do not have any competing interests to declare.
Authors’ contributions
FHB and MJR drafted the manuscript. AJA, SMT and TMW contributed intellectually to interpretation, critical evaluation of content and manuscript revision. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Brennan, F.H., Anderson, A.J., Taylor, S.M. et al. Complement activation in the injured central nervous system: another dual-edged sword? . J Neuroinflammation 9, 137 (2012). https://doi.org/10.1186/1742-2094-9-137
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-2094-9-137