
Abstract
There is now a large body of evidence linking inflammation to Alzheimer's disease (AD). This association manifests itself neuropathologically in the presence of activated microglia and astrocytes around neuritic plaques and increased levels of inflammatory mediators in the brains of AD patients. It is considered that amyloid-β peptide (Aβ), which is derived from the processing of the longer amyloid precursor protein (APP), could be the most important stimulator of this response, and therefore determining the role of the different secretases involved in its generation is essential for a better understanding of the regulation of inflammation in AD. The finding that certain non-steroidal anti-inflammatory drugs (NSAIDs) can affect the processing of APP by inhibiting β- and γ-secretases, together with recent revelations that these enzymes may be regulated by inflammation, suggest that they could be an interesting target for anti-inflammatory drugs. In this review we will discuss some of these issues and the role of the secretases in inflammation, independent of their effect on Aβ formation.
Similar content being viewed by others

Introduction
Alzheimer's disease (AD) is a devastating neurological disease affecting more than 26 million people around the world, and there are indications that this number will quadruple by 2050. Age is the most significant risk factor and it is estimated that 50% of people over 85 have either AD or Mild Cognitive Impairment (MCI). Brains of individuals with AD manifest massive neuronal and synaptic loss in certain areas which result in the memory impairment and disorientation associated with this disease. The neuroanatomical study of a typical AD brain reveals the presence of two characteristic lesions: extracellular amyloid (or senile plaques) and intracellular neurofibrillary tangles composed primarily of hyperphosphorylated tau protein [1]. Amyloid plaques contain small amyloid-β peptides (Aβ), which are toxic products from the catalytic cleavage of a larger amyloid precursor protein (APP) [1, 2].
In 1907 Alois Alzheimer described unique structures in the cerebral cortex of a 55 year old woman with progressive dementia that are now referred to as senile plaques [3]. Histological methods used to demonstrate plaques including thioflavine S, various silver stains, and immunocytochemical stains reveal various biochemical components of plaques. Aβ has been reported to have heterogeneous carboxyl termini, and Aβ1–40 and Aβ1–42 appear to be the major species in the parenchymal deposits. It is this C-terminal variation that has been most often associated with pathogenicity, with Aβ1–42 found to be the most toxic form [1, 2]. Aβ is found in normal cerebrospinal fluid and in conditioned media from various tissue culture cell lines [4–6], suggesting that it is produced and secreted constitutively.
Aβ deposition is considered a major pathogenic step in the development of AD. Evidence supporting the amyloid hypothesis has been extensively reviewed [7]. On the other hand, there is also the view that Aβ plaque formation is an epiphenomenon and its pathophysiological functions still remain unclear. In this regard, it has been shown that in people with AD the density of Aβ plaques correlates poorly with the severity of dementia [8]. There are, however, indications that dendritic and synaptic injury occur early in the course of the disease and that protofibrils and oligomers of Aβ40 and Aβ42 could cause neuronal dysfunction [9]. In addition, a recent study carried out using multiphoton laser confocal microscopy showed that in the early stages of the disease, microplaques can damage neighbouring axons and dendrites within days [10]. Furthermore, intraneuronal Aβ has also been implicated in the onset of cognitive dysfunction [11].
APP is a type I integral membrane protein [12] that resembles a signal-transduction receptor. It is synthesized in the ER, post-transcriptionally modified in the Golgi (including N- and O-linked glycosylation, sulfation and phosphorylation), and transported to the cell surface via the secretory pathway. APP is also endocytosed from the cell surface and processed in the endosomal-lysosomal pathway [13, 14], although autophagic vacuoles may also be a site for Aβ production [15]. Alternative processing pathways of APP have been described (Figure 1).

Proteolytic processing of APP. Proteolysis of APP by α-secretase or β-secretase leads to the secretion of αAPPs or βAPPs. Both secretases generate C-terminal fragments (CTF) of 10 kDa and 12 kDa respectively, which are inserted in the membrane (grey). These fragments can be cut by γ-secretase to release the peptides P3 and Aβ. Two further cleavage sites, termed ε and ζ, have been identified in the CTF.
Proteolysis of APP by α-secretase or β-secretase leads to the secretion of soluble α-APPs or β-APPs. This generates C-terminal fragments of 10 kDa and 12 kDa respectively, which are inserted in the membrane. These fragments can be cut by γ-secretase to release the peptides P3 and Aβ [16] and a cytoplasmic fragment identified as AICD (APP intracellular domain) [17]. Intriguingly, AICD starts at position 49/50 and would not correspond to the end of Aβ variants Aβ40 and Aβ42. Therefore this cleavage site, termed ε-cleavage, is topologically highly similar to the S3 cleavage of Notch [17, 18]. Recently, a new cleavage site was described for γ-secretase. The ξ-cleavage occurs between ε- and γ-cleavage sites and generates longer Aβ forms within cells and in the brain, including Aβ43, Aβ45, Aβ46, and Aβ48 [19, 20].
Inflammation and Alzheimer's disease
There is strong evidence that Aβ toxicity could be mediated through the induction of inflammatory events in the brain. Over the past decade it has been speculated that the inflammatory response associated with the presence of neuritic plaques could be involved in neuronal damage and contribute to the progression of the disease [21, 22]. A common feature in the brain of AD patients is the presence of astrocytes and microglia surrounding the senile amyloid plaques, already described by Alois Alzheimer in 1907.
The first evidence for an excessive inflammatory process in AD came from a study carried out in AD and Down syndrome brains that showed increased levels of S100 and IL-1 [23]. Since then, many of the cytokines and chemokines that have been studied in AD, including IL-1β, IL-6, TNF-α, IL-8, TGF-β and macrophage inflammatory protein-1α (MIP-1α) have been found to have altered expression compared with control individuals [22]. Animal models of Alzheimer's disease, such as the APP transgenic line Tg2567 carrying the Swedish mutation, also show enhanced levels for TNF-α, IL-1β, IL-1α, chemoattractant protein-1, COX-2 and complement component 1q [24, 25]. In addition, an increased risk of AD has been associated with several polymorphisms of proinflammatory genes, including IL-1 [26], IL-6 [27], TNF-α [28], and α1-antichymotrypsin [29].
Clinical investigation and studies in AD animal models have reinforced the suggestion that inflammatory changes in the AD brain are an early and prominent feature. In support of this hypothesis, imaging studies have detected microglial activation in patients at very early clinical stages of the disease [30]. Moreover, a recent study investigating predictors in plasma from MCI patients and those with pre-symptomatic AD that converted to AD identified factors associated with inflammation, such as TNFα, IL-3 IL-1α and IL-11 [31]. In animal models of AD, focal glial activation already takes place before amyloid plaque formation in APPV717 transgenic mice at 3 months of age [32]. These animals showed decreased LTP already at this age, which could be caused by impaired neuronal function derived from the increased secretion of pro-inflammatory cytokines [33]. Prominent microglial activation also preceded tangle formation in 3 month old P301S Tg mice [34]. Interestingly, neuroinflammation has been proposed to be the link between Aβ deposition and the formation of neurofibrillary tangles. Products of inflammation, such as pro-inflammatory cytokines might change the substrate specificity of kinases/phosphatases leading to tau phosphorylation at pathological sites [35].
Further evidence for a link between inflammation and AD comes from observations in head-injured patients. A number of large epidemiological studies (e.g. the MIRAGE study) [36] have identified an increased risk of dementia in patients who have suffered a serious head injury during their life. At the more acute pathological level, in a cohort of patients who died within weeks of their head injury, approximately a third showed signs of Aβ deposition in their brains [37]. While there was a wide age range in this cohort and some of these Aβ deposits were almost certain to predate the trauma, the deposits seen in the younger cases are likely to have been generated de novo, as a result of the trauma. We have hypothesised that it is the inflammatory response generated as a result of the trauma which triggers the AD-type degenerative changes [38]. Traumatic brain injury in rats has been shown to increase BACE1 activity in the hippocampus with a concomitant increase in APP cleavage products [39]. Work in a porcine model of head injury has shown accumulation of APP and BACE in injured axons and it is suggested that the abnormal accumulation of these proteins may favour Aβ production [40]. At the molecular level acute hypoxia has been shown to increase the production of APP and subsequently Aβ. This action is thought to be mediated by the binding of HIF-1 to the promoter region of BACE1 mRNA resulting in increased levels of BACE1 protein in the tissue [41]. More recently it has been shown that brain injury in mice increases the expression of presenilin 1 and nicastrin, both components of the γ-secretase complex, in activated microglia and astrocytes [42].
Conversely, it has to be noted that inflammation is not only contributing to the disease progression, but could have beneficial effects. This may depend on the inflammatory elements activated, the time in the disease development and also whether the response is acute or chronic. Activated microglia can reduce Aβ accumulation by increasing its phagocytosis or extracellular degradation [43–45]. Microglia also release trophic factors such as the glia-derived neurotrophic factor (GDNF), which is neuroprotective [46]. In addition, certain cytokines have an anti-inflammatory effect, such as IL-1 receptor antagonist (IL-1Ra), IL-4, IL-10 and TGF-β [22, 47]. It was recently reported that the interaction between newly formed amyloid plaque and microglia shows that, unless further activated, microglia do not successfully clear plaques but may well restrict their growth, leading to a steady state of plaque size after initial formation [10].
Further support for the involvement of inflammation with the pathogenesis of AD came from the finding in epidemiological studies that treatment with non-steroidal anti-inflammatory drugs (NSAIDs) was associated with a reduced risk of developing AD. A meta-analysis of nine studies revealed that the benefit was greater with long-term use than with intermediate use [48]. Recently, a modified Mini-Mental State Examination reported that NSAIDs use prevented cognitive decline in older adults if started in midlife (prior to age 65) rather than late in life (after age 65). This effect was more pronounced in those who had one or more APOE e4 alleles [49]. However, the possible preventive effect of NSAIDs has not been yet confirmed in clinical trials. The failure of the trials may be attributed to the facts that the benefit of NSAIDs may only be observed in early phases of the disease, that is, they are preventive and not curative, and also to the choice of NSAID, mostly COX-2-specific inhibitors because (i) only a subset of NSAIDs are able to lower Aβ production and (ii) this capacity appears to be COX-2 independent [50]. In animals, the beneficial effects of NSAIDs have been confirmed, showing behavioural improvement and reductions in glial activation, in Aβ levels and plaque size [45, 51–55]. In vitro studies have revealed several potential targets for NSAIDs, including NFκB, Rho-GTPases, PPARγ and secretases [50]. However, the inhibition of the canonical targets of NSAIDs, cyclooxygenase-1 and -2 (COX-1 and COX-2), do not seem to be responsible for the protective effect of NSAIDs in AD. On the contrary, COX-2 inhibitors may raise Aβ1–42 secretion [56].
The finding that NSAIDs could affect APP processing and that certain NSAIDs can inhibit β and γ-secretases with the recent revelation that secretases may be influenced by inflammation indicates that they could be an interesting target for anti-inflammatory drugs [50].
Secretases and inflammation
α-Secretase
APP is cleaved by α-secretase in the centre of the Aβ domain. Three related metalloproteases of the ADAM (a disintegrin and metalloprotease) family, ADAM-9, ADAM-10 and ADAM-17, also termed TACE (tumor necrosis factor converting enzyme), appear to exert α-secretase activity [16, 57]. A confirmation that ADAM10 is involved in α-secretase activity came from studies in transgenic mice. A moderate neuronal over-expression of ADAM10 in mice transgenic for human APP([V717I]) showed increased secretion of the neurotrophic soluble α-secretase-released N-terminal APP domain (α-APPs), reduced formation of Aβ peptides, and prevention of their deposition in plaques [58]. Functionally, in these mice impaired long-term potentiation and cognitive deficits were alleviated. Expression of mutant catalytically inactive ADAM10 led to an enhancement of the number and size of amyloid plaques in the brains of double-transgenic mice. These results provided the first in vivo evidence for a proteinase of the ADAM family as an α-secretase of APP, revealed activation of ADAM10 as a promising therapeutic target, and supported the hypothesis that a decrease in α-secretase activity contributes to the development of AD [58]. Another candidate with α-secretase activity is BACE2, which likewise cleaves within the Aβ domain and abrogates Aβ formation [57] (Figure 1).
ADAM proteinases have emerged as the major protein family that mediates ectodomain shedding, the proteolytic release of extracellular domains from their membrane-bound precursors. Proteolytic cleavage or ectodomain shedding is an additional mechanism whereby cells can regulate the proteins expressed on their cell surface. In many cases, soluble ectodomains are biologically active as mediators of functions ascribed to their transmembrane counterparts [59]. ADAM-like sheddases activate, for instance, growth factors and cytokines, thus regulating signalling pathways that are important in development and pathological processes such as cancer [60]. ADAM17 and 10 appear to play a particularly prominent role in ectodomain shedding of inflammatory proteins at all stages of leukocyte recruitment. Soluble TNF-α is released from its membrane-bound precursor by shedding through ADAM17 [61]. In vivo studies have shown that many, but not all, of the inflammatory effects of TNF-α require cleavage and shedding from the cell surface. Besides TNF-α, ADAM17 cleaves ectodomains of other receptors and ligands, such as TNFR2 and L-selectin [62]. In addition, it was recently shown that IL-1R2 can be proteolytically processed in a manner similar to APP [63]. IL-1R2 undergoes ectodomain shedding in an α-secretase manner, resulting in the secretion of the IL-1R2 ectodomain and the generation of a C-terminal fragment.
That α-secretase is involved in inflammation is supported by the observation that ADAM-10 is expressed constitutively by astrocytes in the normal and inflamed human CNS [64]. ADAM10 and ADAM-17 are also enriched in microglia [65]. The functions of ADAMs in microglia are complex and they do not only have pro-inflammatory and neurotoxic properties but also reparative ones [66]. In fact, IL-1α has been reported to increase ADAM-17 and ADAM-10 levels and α-secretase activity in human astrocytic cultures [67]. More recently, it has been shown that the proinflammatory cytokines TNF-α, IFN-γ and IL-1β, as well as TGF-β and LPS, are able to increase ADAM10 activity, leading to a loss in E-cadherin expression, which is another substrate for this secretase [68].
On the other hand, it has been also reported that various NSAIDs (including nimesulide, ibuprofen and indomethacin) stimulate the non-amyloidogenic secretion of sAPPα from neuroblastoma cells [69]. Shedding of sAPPα induced by nimesulide and thalidomide was modulated by inhibitors of PKV and Erk MAPK, indicating that NSAIDs activate the Erk MAPK signalling cascade. However, these results have not been reproduced by other groups [70, 71].
β-Secretase
β-Secretase (BACE1 for β-site APP cleaving enzyme) was cloned and identified as a type I transmembrane aspartyl protease [72]. BACE1 cleaves APP at the N-terminal position of Aβ. BACE1 deficiency precludes Aβ formation in transgenic mice, and does not cause or promote any neurological or phenotypic abnormalities. However, it has been recently demonstrated that BACE1 knockout mice exhibit a number of schizophrenia-like behavioural traits, most likely because BACE1 is involved in the cleavage of neuregulin-1, which has been linked to the pathogenesis of schizophrenia and related psychiatric disorders [73]. Because BACE1 inactivation rescues memory deficits in transgenic mice [74], this strongly supports the importance of BACE1 as a therapeutic target in AD.
BACE1 is primarily expressed in neurons [72, 75, 76], but it can be also expressed in astrocytes under conditions of chronic stress [77] and in old transgenic mice [76, 32]. In addition, in young transgenic mice, neuronal BACE1 was induced in the proximity of activated microglia and astrocytes [32]. These observations lead to the conclusion that BACE1 expression is regulated by inflammatory events. BACE1 has also been found up-regulated in neuronal cultures upon exposure to pro-inflammatory cytokines [70, 78], under oxidative stress in NT2 neurons [79], and in the hippocampus of rats after experimental traumatic brain injury [39]. In addition, BACE1 protein levels, activity and the β-secretase product (β-CTF) are increased in brain of sporadic AD patients [80, 81] as well as in platelets and CSF from AD and MCI patients [82–84]. These alterations in BACE1 expression and activity indicate a transcriptional and/or translational regulation of BACE1 expression in the brain [85]. Interestingly, consensus binding sites for various transcription factors that are known to be regulated by inflammation (such as NFκB, PPARγ and STAT1) are present in the BACE1 promoter.
Transcriptional regulation
NFκB
NFκB sites are present in the promoters of APP [86], presenilin and BACE1 [87]. In neurons exposed to soluble Aβ peptides and in TNFα-activated glial cells the mutation of the BACE1 promoter NFκB site led to significant decreases in promoter activity, indicating an activating role for NFκB in BACE1 expression in Aβ [87]. In addition, some NSAIDs such as flurbiprofen and indomethacin, which target NFκB, have been shown to be effective at decreasing amyloid load in vitro and also in APP transgenic mice [54, 55, 88]. A recent report showed that deletion of TNFα1 death receptor (TNFα1R) in APP23 transgenic mice inhibited Aβ generation reducing BACE1 levels and activity via the NFκB pathway [89].
The effect of NFκB on BACE1 promoter could be direct or through changes in PPARγ, because PPARγ agonists can antagonize the activity of transcription factors such as NFκB [50].
PPARγ
PPARγ is a transcription factor that is involved in the regulation of the metabolism of glucose and lipids, in cellular differentiation as well as in the control of transcription of a wide range of inflammatory genes. A consensus binding site for PPARγ was found in the BACE1 promoter. PPARγ activation by agonists such as thiazolidinediones (TZD) and certain NSAIDs such as ibuprofen, indomethacin and naproxen results in a decrease of BACE1 transcription, expression and activity [70]. Furthermore, lack of PPARγ led to an increase of BACE1 promoter activity [90], which suggested that PPARγ could be a repressor of BACE1.
PPARγ levels are decreased in AD brain, indicating that inflammatory events may decrease PPARγ transcription. Furthermore, in vitro experiments have shown that inflammatory cytokines and oxidative stress decrease PPARγ levels. Therefore, these findings suggest the existence of a down-regulation of PPARγ under inflammatory conditions, which would result in an increase in BACE1 transcription and Aβ generation. This effect could be prevented by modulation of PPARγ activity by NSAIDs, which have been shown to increase the levels of PPARγ in adipocytes and neurons [90, 53].
STAT-1
STAT-1 can bind directly to a putative STAT1 binding sequence in the BACE1 promoter. A recent study showed that when STAT1 becomes phosphorylated by IFNγ-mediated activation of JAK2 and ERK1/2, STAT1 binds to BACE1 promoter region to increase BACE1 protein expression in astrocytes [91]. On the other hand, another report has demonstrated that IFNγ-receptor knockout mice crossed with Tg2576 mice expressing the human Swedish mutant APP had reduced gliosis and amyloid plaque compared with Tg2756 animals, apparently by decreasing TNFα secretion and the number of reactive astrocytes expressing BACE1 in cortex and hippocampus [78].
Post-transcriptional modifications
Evidence has been presented for regulation of BACE1 expression at the translational level. The untranslated 5'-BACE1 transcript leader contains upstream open reading frames (uORF) that can reduce the translation of the main open reading frame [92, 93]. It remains unclear as to whether BACE1 translation is constitutively repressed or whether the repression may be alleviated upon physiological and pathophysiological stimuli [85]. Potentially, the identification of proteins specifically binding to long, GC-rich 5'UTR may yield new insights into the possible regulation of BACE1 translation. Evidence for these mechanisms was shown by Zacchetti et al., who described that the 5'-UTR-dependent translational repression of BACE1 may be alleviated in activated astrocytes [94]. Therefore, it seems that inflammation may play also a role in the translational regulation of BACE1.
γ-Secretase
γ-Secretase is a protein complex of four essential membrane proteins called aph-1, pen-2, nicastrin and presenilin (PS). While aph-1, pen-2 and nicastrin function in the assembly and subcellular transport of γ-secretase and in the recognition of protein substrates [95–97]. PS proteins are members of an aspartyl protease family and represent the catalytically active components of the γ-secretase complex [98]. PS1 and PS2 proteins are homologous polytopic membrane proteins critically involved in intra-membraneous cleavage of APP and a number of other type I membrane proteins [98–100]. Accordingly, PS proteins have been implicated in different biological processes, including the regulation of cell differentiation and death, calcium homeostasis, cell adhesion, and subcellular trafficking of several membrane proteins. Mutations in PS are associated with familial forms of early onset Alzheimer's disease (AD) and increase the ratio of Aβ42/Aβ40 by either elevating production of the elongated, highly fibrillogenic Aβ42 or by decreasing the generation of Aβ40 [57, 101, 102].
γ-Secretase and inflammation
It was recently published that activated microglia and astrocytes have enhanced expression of PS and nicastrin following brain damage [42]. Although glial cells express PS1, it is not known if PS1 mutations alter glial cell functions. Carriers of PS FAD associated mutations not only show earlier deposition of Aβ in β-amyloid plaques, but also inflammatory processes in the brain [90]. On the other hand, it is unclear whether PS FAD associated mutations cause neuroinflammation by promoting formation and deposition of Aβ42 or by triggering other processes. In PS1 associated FAD brain, distinct 'inflammatory plaques' have been described, that lack reactivity for apolipoprotein E and Aβ in the core, but revealed association with reactive microglia and astrocytes, suggesting that mutations in PS1 could also induce Aβ independent neuroinflammation in Early Onset Alzheimer's disease (EOAD) [103]. In addition, studies using knock-in mice for PS1 FAD mutations have revealed an enhanced inflammatory cytokine response to immune challenge with bacterial lipopolysaccharide (LPS). LPS-induced levels of mRNAs encoding TNFα, IL-1α, IL-1β, IL-1 receptor antagonist, and IL-6 were significantly greater in the hippocampus and cerebral cortex of PS1 mutant mice as compared to wild-type mice. These findings demonstrate an adverse effect of PS1 mutations on microglial cells that results in their hyperactivation under pro-inflammatory conditions, which may, together with direct effects of mutant PS1 in neurons, contribute to the neurodegenerative process in AD [104].
Additional evidence for an Aβ independent role of PS proteins in inflammation comes from studies with conditional knockout mice lacking both PS genes in the postnatal forebrain. These mice display strong age-dependent neurodegeneration and impairment of cognitive function [105]. Gene profiling revealed up-regulation of several pro-inflammatory genes, including glial fibrillary acidic protein, complement component C1q, and cathepsin S. Also, activated microglia were detected in the brain of these mice. Since Aβ production is strongly reduced upon deletion of PS in forebrain neurons, these data indicate that impairment of PS function could also trigger inflammation independent of Aβ [105]. Analogously, the cleavage of other substrates for PS, such as Notch, could also have some effect on brain inflammation. It was shown that mice transgenic for antisense Notch and normal mice treated with inhibitors of γ-secretase showed reduced damage to brain cells and improved functional outcome in a model of focal ischemic stroke. These mice had a reduced number of activated microglial cells in the brain after ischemic perfusion [106].
NSAIDs and γ-secretase
As described above several NSAIDs decrease the risk for development of AD. Although the molecular mechanism(s) underlying this protective activity remain to be identified, certain NSAID could directly modulate γ-secretase activity [107]. Thus, besides suppression of inflammatory processes via inhibition of COX dependent biosynthesis of pro-inflammatory prostaglandins, the protective role of NSAIDs could also involve altered generation of Aβ. Indeed, several NSAIDs, including ibuprofen, sulindac sulphide, and indomethacin could decrease the levels of secreted Aβ42 [108, 54, 109]. Importantly, generation of Aβ40 is largely unaltered by these compounds, indicating that certain NSAIDs modulate rather than inhibit γ-secretase activity. The decrease in Aβ42 production was associated with increased generation of Aβ38 suggesting a shift in the cleavage specificity of the protease [107]. However, more recently it has been shown that the generation of Aβ38 and Aβ42 is independent and differently affected by FAD mutations in PS as well as by modulators of γ-secretase [110]. After the initial identification of ibuprofen, sulindac sulphide, and indomethacin as selective Aβ42 lowering drugs, several other NSAIDs, including flurbiprofen and fenoprofen have been shown to exert similar effects [54]. Because Aβ42 lowering activity was also observed with several derivatives of NSAIDs that do not inhibit COX, and was also present in COX1/2 deficient cell lines, the modulation of γ-secretase activity is unlikely to be mediated via COX enzymes [107].
Surprisingly, the PPARα antagonist fenofibrate and some COX-2 specific inhibitors could even increase Aβ42 production [56, 111, 112]. Whether additional effects of NSAIDs on NFκB and PPARα or PPARγ (see above) contribute to the Aβ42 lowering activity remains to be investigated in more detail. However, certain NSAIDs decrease Aβ42 production in vitro with purified γ-secretase complex, indicating NFκB and PPARγ independent effects [113, 114]. Interestingly, FAD associated mutations in PS1 could cause insensitivity of γ-secretase to Aβ42 lowering NSAIDs [115]. Because direct interaction of NSAIDs with the presenilins or other components of the γ-secretase complex has not been shown so far, the molecular mechanisms for the action of NSAIDs are unclear. Since NSAIDs reveal Aβ42 lowering activity at relatively high doses (IC50 values of 25 – 500 μM) at which they could also affect biophysical properties of biological membranes, the modulation of γ-secretase specificity might also involve interference with membrane fluidity and/or accessibility of substrate to the enzyme [116]. Nonetheless, since NSAIDs, even at higher concentrations did not inhibit cleavage of Notch and ErbB4 or alter AICD formation, this class of inhibitors might be valuable compounds to selectively decrease Aβ42 production without affecting important intracellular signaling pathways, thereby reducing potential side effects in clinical applications.
In preclinical studies, long-term treatment of APP transgenic Tg2576 mice with ibuprofen and indomethacin were effective in reducing the plaque pathology [51, 117]. Also, acute treatment with additional NSAIDs, sulindac sulfide, and flurbiprofen, selectively reduced Aβ42 levels without changing the concentration of Aβ40 [54, 108]. However, other studies did not reproduce these results [111, 118]. In clinical trials some positive effects on cognitive performance of AD patients were observed with indomethacin and the (R)-enantiomer of flurbiprofen, while other NSAIDs without Aβ42 reducing activity did not reveal beneficial effects [107] (see Table 1 for summary). Although this indicates that it would be relevant to evaluate Aβ42 lowering NSAIDs in further clinical trails, recent epidemiological studies revealed that the protective effect of NSAIDs seems to be independent of the Aβ42 suppressing activity of the NSAID [119, 120].
Conclusion
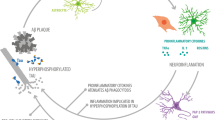
In summary, in this review we have tried to give a perspective on the wide variety of interactions between inflammatory mediators and APP secretases. On the one hand, pro-inflammatory cytokines are able to increase the levels and/or activity of some secretases, such as α-secretase and BACE1. On the other hand, NSAIDs are able to modulate the activity of the secretases by regulating their levels in the case of BACE1, or by directly shifting the cleavage site of γ-secretase (see Table 1). In addition it now seems that α-secretase plays a particularly prominent role in ectodomain shedding of inflammatory proteins, thus regulating the activity of cytokines such as TNFα. Bringing all this together, it is clear that the association between inflammation and AD, as suggested by the wealth of epidemiological, clinical and laboratory data, is based on a series of complex molecular interactions that we are only just beginning to understand in detail (Figure 2). The initial signs are encouraging but further work is needed in this area to determine whether modification of these interactions can provide a viable therapeutic target for the treatment of AD.

Schematic representation of the interactions between inflammatory processes and APP processing. Aβ generation by BACE1 and γ-secretase induces an inflammatory response, which involves the activation of microglia and astrocytes and the release of pro-inflammatory cytokines. This inflammatory response could be enhanced by brain trauma or by PS1 mutations, probably also by increased Aβ production. Inflammatory cytokines have been involved in the aggregation of Aβ [121]. Moreover, cytokines can affect the expression of secretases and APP, influencing their transcription, translation and/or activation [122]. Non-steroidal anti-inflammatory drug treatment could reverse the effect of inflammation on BACE1 transcription, modulate the cleavage site of γ-secretase and decrease the secretion of cytokines and the number of microglia and astrocytes. On the other hand, α-secretase has been involved in the shedding of certain cytokines, potentiating their activity. See text for further details of individual interactions. Numbers on the diagram correspond to the appropriate references in the review.
Abbreviations
- ADAM:
-
a disintegrin and metalloprotease
- ApoE:
-
Apolipoprotein E
- COX:
-
cyclooxygenase
- EOAD:
-
Early Onset Alzheimer's Disease
- FAD:
-
Familiar Alzheimer's Disease
- IL:
-
Interleukin
- iNOS:
-
inducible nitric oxide synthase
- MCI:
-
mild cognitive impairment
- NFκB:
-
nuclear factor kappa B
- NSAIDs:
-
Non-steroidal anti-inflammatory drugs
- PPAR:
-
Peroxisome Proliferator-activated receptor
- PS:
-
presenilin
- STAT:
-
Signal transducers and activators of transcription
- TNFα:
-
Transforming Necrosis Factor-α
- TZD:
-
thiazolidinedione
References
Selkoe DJ: Folding proteins in fatal ways. Nature. 2003, 42: 900-904. 10.1038/nature02264.
Selkoe DJ: Alzheimer's disease is a synaptic failure. Science. 2002, 298: 789-791. 10.1126/science.1074069.
Alzheimer A: Ueber eine eigenartige Erkrankung der Hirnrinde. Centralblatt fur Nervenheilkunde un Psychiatrie. 1907, 30: 177-179.
Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, Cai XD, McKay DM, Tintner R, Frangione B, Younkin SG: Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science. 1992, 258: 126-129. 10.1126/science.1439760.
Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB, Selkoe DJ: Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature. 1992, 359: 322-325. 10.1038/359322a0.
Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C, McCormack R, Wolfert R, Selkoe D, Lieberburg I, Schenk D: Isolation and quantification of soluble Alzheimer's beta-peptide from biological fluids. Nature. 1992, 359: 325-327. 10.1038/359325a0.
Walsh DM, Selkoe DJ: Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron. 2004, 44: 181-193. 10.1016/j.neuron.2004.09.010.
Braak H, Braak E: Evolution of neuronal changes in the course of Alzheimer's disease. J Neural Transm. 1998, 127-140. Suppl 53
Walsh D, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ: Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002, 416: 535-539. 10.1038/416535a.
Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT: Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. 2008, 451: 720-724. 10.1038/nature06616.
Billings LM, Oddo S, Green KN, McGaugh JL, Laferla FM: Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005, 45: 675-688. 10.1016/j.neuron.2005.01.040.
Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Mueller-Hill B: The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987, 325: 733-736. 10.1038/325733a0.
Koo EH, Squazzo SL: Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem. 1994, 269: 17386-17389.
Bossy-Wetzel E, Schwarzenbacher R, Lipton SA: Molecular pathways to neurodegeneration. Nat Med. 2004, 10: S2-9. 10.1038/nm1067.
Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Näslund J, Mathews PM, Cataldo AM, Nixon RA: Macroautophagy – a novel Beta-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol. 2005, 171: 87-98. 10.1083/jcb.200505082.
Walter J, Kaether C, Steiner H, Haass C: The cell biology of Alzheimer's disease: uncovering the secrets of secretases. Curr Opin Neurobiol. 2001, 11: 585-590. 10.1016/S0959-4388(00)00253-1.
Sastre M, Steiner H, Fuchs K, Capell A, Multhaup G, Condron MM, Teplow DB, Haass C: Presenilin-dependent γ-secretase processing of β-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Reports. 2001, 2: 835-841. 10.1093/embo-reports/kve180.
Weidemann A, Eggert S, Reinhard FB, Vogel M, Paliga K, Baier G, Masters CL, Beyreuther K, Evin G: A novel epsilon-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with Notch processing. Biochemistry. 2002, 41: 2825-2835. 10.1021/bi015794o.
Zhao G, Mao G, Tan J, Dong Y, Cui MZ, Kim SH, Xu X: Identification of a New Presenilin-Dependent ζ-Cleavage Site within the Transmembrane Domain of Amyloid Precursor Protein. J Biol Chem. 2004, 279: 50647-50650. 10.1074/jbc.C400473200.
Qi-Takahara Y, Morishima-Kawashima M, Tanimura Y, Dolios G, Hirotani N, Horikoshi Y, Kametani F, Maeda M, Saido TC, Wang R, Ihara Y: Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by gamma-secretase. J Neurosci. 2005, 25: 436-445. 10.1523/JNEUROSCI.1575-04.2005.
Aisen PS: Inflammation and Alzheimer's disease: mechanisms and therapeutic strategies. Gerontology. 1997, 43: 143-149.
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T: Inflammation and Alzheimer's disease. Neurobiol Aging. 2000, 21: 383-421. 10.1016/S0197-4580(00)00124-X.
Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL, Araoz C: Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA. 1989, 86: 7611-7615. 10.1073/pnas.86.19.7611.
Benzing WC, Wujek JR, Ward EK, Shaffer D, Ashe KH, Younkin SG, Brunden KR: Evidence for glial-mediated inflammation in aged APPSw transgenic mice. Neurobiol Aging. 2000, 16: 523-530.
Matsuoka Y, Picciano M, Malester B, LaFrancois J, Zehr C, Daeschner JM, Olschowka JA, Fonseca MI, O'Banion MK, Tenner AJ, Lemer CA, Duff K: Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer's disease. Am J Path. 2001, 158: 1345-1354.
Nicoll JA, Mrak RE, Graham DI, Stewart J, Wilcock G, MacGowan S, Esiri MM, Murray LS, Dewar D, Love S, Moss T, Griffin WS: Association of interleukin-1 gene polymorphisms with Alzheimer's disease. Ann Neurol. 2000, 47: 365-368. 10.1002/1531-8249(200003)47:3<365::AID-ANA13>3.0.CO;2-G.
Papassotiropoulos A, Bagli M, Jessen F, Bayer TA, Maier W, Rao ML, Heun R: A genetic variation of the inflammatory cytokine interleukin-6 delays the initial onset and reduces the risk for sporadic Alzheimer's disease. Ann Neurol. 1999, 45: 666-668. 10.1002/1531-8249(199905)45:5<666::AID-ANA18>3.0.CO;2-3.
McCusker SM, Curran MD, Dynan KB, McCullagh CD, Urquhart DD, Middleton D, Patterson CC, McIlroy SP, Passmore AP: Association between polymorphism in regulatory region of gene encoding tumour necrosis factor alpha and risk of Alzheimer's disease and vascular dementia: a case-control study. Lancet. 2001, 357: 436-439. 10.1016/S0140-6736(00)04008-3.
Kamboh MI, Sanghera DK, Ferrell RE, DeKosky ST: APOE*4-associated Alzheimer's disease risk is modified by alpha 1-antichymotrypsin polymorphism. Nat Genet. 1995, 10: 486-488. 10.1038/ng0895-486.
Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB: In-vivo measurement of activated microglia in dementia. Lancet. 2001, 358: 461-467. 10.1016/S0140-6736(01)05625-2.
Motta M, Imbesi R, Di Rosa M, Stivala F, Malaguarnera L: Altered plasma cytokine levels in Alzheimer's disease: correlation with the disease progression. Immunol Lett. 2007, 114: 46-51. 10.1016/j.imlet.2007.09.002.
Heneka MT, Sastre M, Dumitrescu-Ozimek L, Dewachter I, Walter J, Klockgether T, Van Leuven : Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP(V717I) transgenic mice. J Neuroinflammation. 2005, 2: 22-10.1186/1742-2094-2-22.
Tancredi V, D'Arcangelo G, Grassi F, Tarroni P, Palmieri G, Santoni A, Eusebi F: Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neurosci Lett. 1992, 146: 176-178. 10.1016/0304-3940(92)90071-E.
Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM: Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007, 53: 337-351. 10.1016/j.neuron.2007.01.010.
Arnaud L, Robakis NK, Figueiredo-Pereira ME: It may take inflammation, phosphorylation and ubiquitination to 'tangle' in Alzheimer's disease. Neurodegener Dis. 2006, 3: 313-319. 10.1159/000095638.
Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, Chui H, Green RC, Sadovnick AD, Duara R, DeCarli C, Johnson K, Go RC, Growdon JH, Haines JL, Kukull WA, Farrer LA: Head injury and the risk of AD in the MIRAGE study. Neurology. 2000, 54: 1316-23.
Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI: Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J Neurol Neurosurg Psychiatry. 1994, 57: 419-425.
Gentleman SM, Leclercq PD, Moyes L, Graham DI, Smith C, Griffin WS, Nicoll JA: Long-term intracerebral inflammatory response after traumatic brain injury. Forensic Sci Int. 2004, 146: 97-104. 10.1016/j.forsciint.2004.06.027.
Blasko I, Beer R, Bigl M, Apelt J, Franz G, Rudzki D, Ransmayr G, Kampfl A, Schliebs R: Experimental traumatic brain injury in rats stimulates the expression, production and activity of Alzheimer's disease β-secretase (BACE-1). J Neural Transm. 2004, 111: 523-536. 10.1007/s00702-003-0095-6.
Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH: Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am J Pathol. 2004, 165: 357-371.
Zhang X, Zhou K, Wang R, Cui J, Lipton SA, Liao FF, Xu H, Zhang YW: Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J Biol Chem. 2007, 282: 10873-10880. 10.1074/jbc.M608856200.
Nadler Y, Alexandrovich A, Grigoriadis N, Hartmann T, Rao KSJ, Shohami E, Stein R: Increased expression of the γ-secretase components presenilin-1 and nicastrin in activated astrocytes and microglia following traumatic brain injury. Glia. 2008, 56: 552-567. 10.1002/glia.20638.
Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM: Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998, 152: 307-317.
Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB, Rosner MR, Safavi A, Hersh LB, Selkoe DJ: Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J Biol Chem. 1998, 273: 32730-32738. 10.1074/jbc.273.49.32730.
Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, Citron M, Landreth G: Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer's disease. J Neurosci. 2003, 23: 7504-7509.
Liu B, Hong JS: Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther. 2003, 304: 1-7. 10.1124/jpet.102.035048.
Heneka MT, Sastre M, Huell M: Inflammatory processes in Alzheimer's disease. Glia and inflammation in neurodegenerative disease 2006. Edited by: Yenari MA, Giffard RG. 2006, Nova Science Publishers Inc, 117.
Etminan M, Gill S, Samii A: Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer's disease: systematic review and meta-analysis of observational studies. BMJ. 2003, 327: 128-10.1136/bmj.327.7407.128.
Hayden KM, Zandi PP, Khachaturian AS, Szekely CA, Fotuhi M, Norton MC, Tschanz JT, Pieper CF, Corcoran C, Lyketsos CG, Breitner JC, Welsh-Bohmer KA, Cache County Investigators: Does NSAID use modify cognitive trajectories in the elderly? The Cache County study. Neurology. 2007, 69: 275-282. 10.1212/01.wnl.0000265223.25679.2a.
Lleo A, Galea E, Sastre M: Molecular targets of non-steroidal anti-inflammatory drugs in neurodegenerative diseases. Cell Mol Life Sci. 2007, 64: 1403-1418. 10.1007/s00018-007-6516-1.
Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM: Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci. 2000, 20: 5709-5714.
Lim GP, Yang F, Chu T, Gahtan E, Ubeda O, Beech W, Overmier JB, Hsiao-Ashec K, Frautschy SA, Cole GM: Ibuprofen effects on Alzheimer pathology and open field activity in APPsw transgenic mice. Neurobiol Aging. 2001, 22: 983-991. 10.1016/S0197-4580(01)00299-8.
Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, O'banion K, Klockgether T, Van Leuven F, Landreth GE: Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1–42 levels in APPV717I transgenic mice. Brain. 2005, 128: 1442-1453. 10.1093/brain/awh452.
Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, Ozols VV, Jessing KW, Zavitz KH, Koo EH, Golde TE: NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Aβ42 in vivo. J Clin Invest. 2003, 112: 440-449.
Sung S, Yang H, Uryu K, Lee EB, Zhao L, Shineman D, Trojanowski JQ, Lee V, Pratico D: Modulation of nuclear factor-kappa B activity by indomethacin influences Abeta levels but not A beta precursor protein metabolism in a model of Alzheimer's disease. Am J Pathol. 2004, 165: 2197-2206.
Kukar T, Murphy MP, Eriksen JL, Sagi SA, Weggen S, Smith TE, Ladd T, Khan MA, Kache R, Beard J, Dodson M, Merit S, Ozols VV, Anastasiadis PZ, Das P, Fauq A, Koo EH, Golde TE: Diverse compounds mimic Alzheimer disease-causing mutations by augmenting Aβ42 production. Nat Med. 2005, 11: 545-550. 10.1038/nm1235.
Tanzi RE, Bertram L: Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005, 120: 545-455. 10.1016/j.cell.2005.02.008.
Postina R, Schroeder A, Dewachter I, Bohl J, Schmitt U, Kojro E, Prinzen C, Endres K, Hiemke C, Blessing M, Flamez P, Dequenne A, Godaux E, van Leuven F, Fahrenholz F: A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J Clin Invest. 2004, 113: 1456-1464.
Garton KJ, Gough PJ, Raines EW: Emerging roles for ectodomain shedding in the regulation of inflammatory responses. J Leukoc Biol. 2006, 79: 1105-1116. 10.1189/jlb.0106038.
Huovila AP, Turner AJ, Pelto-Huikko M, Kärkkäinen I, Ortiz RM: Shedding light on ADAM metalloproteinases. Trends Biochem Sci. 2005, 30: 413-422. 10.1016/j.tibs.2005.05.006.
Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP: A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997, 385: 729-733. 10.1038/385729a0.
Black RA, White JM: ADAMs: focus on the protease domain. Curr Opin Cell Biol. 1998, 10: 654-659. 10.1016/S0955-0674(98)80042-2.
Kuhn PH, Marjaux E, Imhof A, De Strooper B, Haass C, Lichtenthaler SF: Regulated intramembrane proteolysis of the interleukin-1 receptor II by alpha-, beta-, and gamma-secretase. J Biol Chem. 2007, 282: 11982-11995. 10.1074/jbc.M700356200.
Kieseier BC, Pischel H, Neuen-Jacob E, Tourtellotte WW, Hartung HP: ADAM-10 and ADAM-17 in the inflamed human CNS. Glia. 2003, 42: 398-405. 10.1002/glia.10226.
Nuttall RK, Silva C, Hader W, Bar-Or A, Patel KD, Edwards DR, Yong VW: Metalloproteinases are enriched in microglia compared with leukocytes and they regulate cytokine levels in activated microglia. Glia. 2007, 55: 516-526. 10.1002/glia.20478.
Yong VW: Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci. 2005, 6: 931-944. 10.1038/nrn1807.
Bandyopadhyay S, Hartley DM, Cahill CM, Lahiri DK, Chattopadhyay N, Rogers JT: Interleukin-1alpha stimulates non-amyloidogenic pathway by alpha-secretase (ADAM-10 and ADAM-17) cleavage of APP in human astrocytic cells involving p38 MAP kinase. J Neurosci Res. 2006, 84: 106-118. 10.1002/jnr.20864.
Maretzky T, Scholz F, Köten B, Proksch E, Saftig P, Reiss K: ADAM10-Mediated E-Cadherin Release Is Regulated by Proinflammatory Cytokines and Modulates Keratinocyte Cohesion in Eczematous Dermatitis. J Invest Dermatol. 2008, 128: 1737-1746. 10.1038/sj.jid.5701242.
Avramovich Y, Amit T, Youdim MB: Non-steroidal anti-inflammatory drugs stimulate secretion of non-amyloidogenic precursor protein. J Biol Chem. 2002, 277: 31466-31473. 10.1074/jbc.M201308200.
Sastre M, Dewachter I, Landreth GE, Willson TM, Klockgether T, van Leuven F, Heneka MT: Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-γ agonists modulate immunostimulated processing of amyloid precursor protein through regulation of β-secretase. J Neurosci. 2003, 23: 9796-9804.
Leuchtenberger S, Maler J, Czirr E, Ness J, Lichtenthaler SF, Esselmann H, Pietrzik CU, Wiltfang J, Weggen S: Nonsteroidal Anti-Inflammatory Drugs and Ectodomain Shedding of the Amyloid Precursor Protein. Neurodegener Dis. 2008.
Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M: Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999, 286: 735-741. 10.1126/science.286.5440.735.
Savonenko AV, Melnikova T, Laird FM, Stewart KA, Price DL, Wong PC: Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc Natl Acad Sci USA. 2008, 105: 5585-5590. 10.1073/pnas.0710373105.
Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF: BACE1 Deficiency Rescues Memory Deficits and Cholinergic Dysfunction in a Mouse Model of Alzheimer's Disease. Neuron. 2004, 41: 27-33. 10.1016/S0896-6273(03)00810-9.
Bigl M, Apelt J, Luschekina EA, Lange-Dohna C, Rossner S, Schliebs R: Expression of beta-secretase mRNA in transgenic Tg2576 mouse brain with Alzheimer plaque pathology. Neurosci Lett. 2000, 292: 107-110. 10.1016/S0304-3940(00)01452-X.
Rossner S, Apelt J, Schliebs R, Perez-Polo JR, Bigl V: Neuronal and glial beta-secretase (BACE) protein expression in transgenic Tg2576 mice with amyloid plaque pathology. J Neurosci Res. 2001, 64: 437-446. 10.1002/jnr.1095.
Hartlage-Rübsamen M, Zeitschel U, Apelt J, Gärtner U, Franke H, Stahl T, Günther A, Schliebs R, Penkowa M, Bigl V, Rossner S: Astrocytic expression of the Alzheimer's disease beta-secretase (BACE1) is stimulus-dependent. Glia. 2003, 41: 169-179. 10.1002/glia.10178.
Yamamoto M, Kiyota T, Horiba M, Buescher JL, Walsh SM, Gendelman HE, Ikezu T: Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007, 170: 680-692. 10.2353/ajpath.2007.060378.
Tamagno E, Bardini P, Obbili A, Vitali A, Borghi R, Zaccheo D, Pronzato MA, Danni O, Smith MA, Perry G, Tabato M: Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis. 2002, 10: 279-288. 10.1006/nbdi.2002.0515.
Holsinger RM, McLean CA, Beyreuther K, Masters CL, Evin G: Increased expression of the amyloid precursor β-secretase in Alzheimer's disease. Ann Neurol. 2002, 51: 783-786. 10.1002/ana.10208.
Yang L-B, Lindholm K, Yan R, Citron M, Xia W, Yang X-L, Beach T, Sue L, Wong P, Price D, Li R, Shen Y: Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nature Med. 2003, 9: 3-4. 10.1038/nm0103-3.
Colciaghi F, Marcello E, Borroni B, Zimmermann M, Caltagirone C, Cattabeni F, Padovani A, Di Luca M: Platelet APP, ADAM 10 and BACE alterations in the early stages of Alzheimer disease. Neurology. 2004, 62: 498-501.
Holsinger RM, Lee JS, Boyd A, Masters CL, Collins SJ: CSF BACE1 activity is increased in CJD and Alzheimer disease versus [corrected] other dementias. Neurology. 2006, 67: 710-720. 10.1212/01.wnl.0000229925.52203.4c.
Zhong Z, Ewers M, Teipel S, Bürger K, Wallin A, Blennow K, He P, McAllister C, Hampel H, Shen Y: Levels of beta-secretase (BACE1) in cerebrospinal fluid as a predictor of risk in mild cognitive impairment. Arch Gen Psychiatry. 2007, 64: 718-726. 10.1001/archpsyc.64.6.718.
Rossner S, Sastre M, Bourne K, Lichtenthaler SF: Transcriptional and translational regulation of BACE1 expression – implications for Alzheimer's disease. Prog Neurobiol. 2006, 79: 95-111. 10.1016/j.pneurobio.2006.06.001.
Grilli M, Ribola M, Alberici A, Valerio A, Memo M, Spano P: Identification and characterization of a kappa B/Rel binding site in the regulatory region of the amyloid precursor protein gene. J Biol Chem. 1995, 270: 26774-26777. 10.1074/jbc.270.45.26774.
Bourne KZ, Ferrari DC, Lange-Dohna C, Rossner S, Wood TG, Perez-Polo JR: Differential regulation of BACE1 promoter activity by nuclear factor-kappaB in neurons and glia upon exposure to beta-amyloid peptides. J Neurosci Res. 2007, 85: 1194-1204. 10.1002/jnr.21252.
Kukar T, Prescott S, Eriksen JL, Holloway V, Murphy MP, Koo EH, Golde TE, Nicolle MM: Chronic administration of R-flurbiprofen attenuates learning impairments in transgenic amyloid precursor protein mice. BMC Neurosci. 2007, 8: 54-10.1186/1471-2202-8-54.
He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, Shen Y: Deletion of tumor necrosis factor death receptor inhibits amyloid {beta} generation and prevents learning and memory deficits in Alzheimer's mice. J Cell Biol. 2007, 178: 829-841. 10.1083/jcb.200705042.
Sastre M, Dewachter I, Rossner S, Bogdanovic N, Rosen E, Borghgraef P, Evert BO, Dumitrescu-Ozimek L, Thal DR, Landreth G, Walter J, Klockgether T, van Leuven F, Heneka MT: NSAIDs repress BACE1 gene promoter activity by activation of the peroxisome proliferator-activated receptor-γ (PPARγ). Proc Natl Acad Sci USA. 2006, 103: 443-448. 10.1073/pnas.0503839103.
Cho HJ, Kim SK, Jin SM, Hwang EM, Kim YS, Huh K, Mook-Jung I: IFN-gamma-induced BACE1 expression is mediated by activation of JAK2 and ERK1/2 signaling pathways and direct binding of STAT1 to BACE1 promoter in astrocytes. Glia. 2007, 55: 253-262. 10.1002/glia.20451.
Lammich S, Schöbel S, Zimmer AK, Lichtenthale SF, Haass C: Expression of the Alzheimer protease BACE1 is suppressed via its 5'-untranslated region. EMBO Rep. 2004, 5: 620-625. 10.1038/sj.embor.7400166.
Rogers G, Edelman GM, Mauro VP: Differential utilization of upstream AUGs in the beta-secretase mRNA suggests that a shunting mechanism regulates translation. Proc Natl Acad Sci USA. 2004, 101: 2794-2799. 10.1073/pnas.0308576101.
De Pietri Tonelli D, Mihailovich M, Di Cesare A, Codazzi F, Grohovaz F, Zacchetti D: Translational regulation of BACE-1 expression in neuronal and non-neuronal cells. Nucleic Acids Res. 2004, 32: 1808-1817. 10.1093/nar/gkh348.
Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T: The role of presenilin cofactors in the gamma-secretase complex. Nature. 2003, 422: 438-441. 10.1038/nature01506.
Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, Supala A, Levesque L, Yu H, Yang DS, Holmes E, Milman P, Liang Y, Zhang DM, Xu DH, Sato C, Rogaev E, Smith M, Janus C, Zhang Y, Aebersold R, Farrer LS, Sorbi S, Bruni A, Fraser P, St George-Hyslop P: Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature. 2000, 407: 48-54. 10.1038/35024009.
Shah S, Lee SF, Tabuchi K, Hao YH, Yu C, LaPlant Q, Ball H, Dann CE, Sudhof T, Yu G: Nicastrin functions as a gamma-secretase-substrate receptor. Cell. 2005, 122: 435-447. 10.1016/j.cell.2005.05.022.
Haass C, Steiner H: Alzheimer disease gamma-secretase: a complex story of GxGD-type presenilin proteases. Trends Cell Biol. 2002, 12: 556-562. 10.1016/S0962-8924(02)02394-2.
Annaert W, De Strooper B: A cell biological perspective on Alzheimer's disease. Annu Rev Cell Dev Biol. 2002, 18: 25-51. 10.1146/annurev.cellbio.18.020402.142302.
Selkoe DJ: Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001, 81: 741-766.
Younkin SG: The role of A beta 42 in Alzheimer's disease. J Physiol Paris. 1998, 92: 289-292. 10.1016/S0928-4257(98)80035-1.
Kennedy JL, Farrer LA, Andreasen NC, Mayeux R, George-Hyslop P: The genetics of adult-onset neuropsychiatric disease: complexities and conundra?. Science. 2003, 302: 822-826. 10.1126/science.1092132.
Shepherd CE, Gregory GC, Vickers JC, Halliday GM: Novel 'inflammatory plaque' pathology in presenilin-1 Alzheimer's disease. Neuropathol Appl Neurobiol. 2005, 31: 503-511. 10.1111/j.1365-2990.2005.00667.x.
Lee J, Chan SL, Mattson MP: Adverse effect of a presenilin-1 mutation in microglia results in enhanced nitric oxide and inflammatory cytokine responses to immune challenge in the brain. Neuromolecular Med. 2002, 2: 29-45. 10.1385/NMM:2:1:29.
Beglopoulos V, Sun X, Saura CA, Lemere CA, Kim RD, Shen J: Reduced beta-amyloid production and increased inflammatory responses in presenilin conditional knock-out mice. J Biol Chem. 2004, 279: 46907-46914. 10.1074/jbc.M409544200.
Arumugam TV, Chan SL, Jo DG, Yilmaz G, Tang SC, Cheng A, Gleichmann M, Okun E, Dixit VD, Chigurupati S, Mughal MR, Ouyang X, Miele L, Magnus T, Poosala S, Granger DN, Mattson MP: Gamma secretase-mediated Notch signaling worsens brain damage and functional outcome in ischemic stroke. Nat Med. 2006, 12: 621-623. 10.1038/nm1403.
Czirr E, Weggen S: Gamma-secretase modulation with Abeta42-lowering nonsteroidal anti-inflammatory drugs and derived compounds. Neurodegener Dis. 2006, 3: 298-304. 10.1159/000095270.
Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH: A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001, 414: 212-216. 10.1038/35102591.
Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Golde TE, Koo EH: Abeta42-lowering nonsteroidal anti-inflammatory drugs preserve intramembrane cleavage of the amyloid precursor protein (APP) and ErbB-4 receptor and signaling through the APP intracellular domain. J Biol Chem. 2003, 278: 30748-30754. 10.1074/jbc.M304824200.
Page RM, Baumann K, Tomioka M, Pérez-Revuelta BI, Fukumori A, Jacobsen H, Flohr A, Luebbers T, Ozmen L, Steiner H, Haass C: Generation of Abeta38 and Abeta42 is independently and differentially affected by familial Alzheimer disease-associated presenilin mutations and gamma-secretase modulation. J Biol Chem. 2008, 283: 677-683. 10.1074/jbc.M708754200.
Gasparini L, Ongini E, Wenk G: Non-steroidal anti-inflammatory drugs (NSAIDs) in Alzheimer's disease: old and new mechanisms of action. J Neurochem. 2004, 91: 521-536. 10.1111/j.1471-4159.2004.02743.x.
Gasparini L, Ongini E, Wilcock D, Morgan D: Activity of flurbiprofen and chemically related anti-inflammatory drugs in models of Alzheimer's disease. Brain Res Brain Res Rev. 2005, 48: 400-408. 10.1016/j.brainresrev.2004.12.029.
Beher D, Clarke EE, Wrigley JD, Martin AC, Nadin A, Churcher I, Shearman MS: Selected non-steroidal anti-inflammatory drugs and their derivatives target gamma-secretase at a novel site. J Biol Chem. 2004, 279: 43419-43426. 10.1074/jbc.M404937200.
Clarke EE, Churcher I, Ellis S, Wrigley JD, Lewis HD, Harrison T, Shearman MS, Beher D: Intra- or intercomplex binding to the gamma-secretase enzyme. A model to differentiate inhibitor classes. J Biol Chem. 2006, 281: 31279-31289. 10.1074/jbc.M605051200.
Czirr E, Leuchtenberger S, Dorner-Ciossek C, Schneider A, Jucker M, Koo EH, Pietrzik CU, Baumann K, Weggen S: Insensitivity to Abeta42-lowering nonsteroidal anti-inflammatory drugs and gamma-secretase inhibitors is common among aggressive presenilin-1 mutations. J Biol Chem. 2007, 282: 24504-24513. 10.1074/jbc.M700618200.
Gamerdinger M, Clement AB, Behl C: Cholesterol-like effects of selective cyclooxygenase inhibitors and fibrates on cellular membranes and amyloid-beta production. Mol Pharmacol. 2007, 72: 141-151. 10.1124/mol.107.034009.
Jantzen PT, Connor KE, DiCarlo G, Wenk GL, Wallace JL, Rojiani AM, Coppola D, Morgan D, Gordon MN: Microglial activation and beta-amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J Neurosci. 2002, 22: 2246-2254.
Lanz TA, Fici GJ, Merchant KM: Lack of specific amyloid-beta(1–42) suppression by nonsteroidal anti-inflammatory drugs in young, plaque-free Tg2576 mice and in guinea pig neuronal cultures. J Pharmacol Exp Ther. 2005, 312: 399-406. 10.1124/jpet.104.073965.
Vlad SC, Miller DR, Kowall NW, Felson DT: Protective effects of NSAIDs on the development of Alzheimer disease. Neurology. 2008, 70: 1672-1677. 10.1212/01.wnl.0000311269.57716.63.
Szekely CA, Green RC, Breitner JC, Ostbye T, Beiser AS, Corrada MM, Dodge HH, Ganguli M, Kawas CH, Kuller LH, Psaty BM, Resnick SM, Wolf PA, Zonderman AB, Welsh-Bohmer KA, Zandi PP: No advantage of A{beta}42-lowering NSAIDs for prevention of Alzheimer dementia in six pooled cohort studies. Neurology. 2008, 70: 2291-2298. 10.1212/01.wnl.0000313933.17796.f6.
Guo JT, Yu J, Grass D, de Beer FC, Kindy MS: Inflammation dependent cerebral deposition of serum amyloid a protein in a mouse model of amyloidosis. J Neurosci. 2002, 22: 5900-5909.
Rogers JT, Leiter LM, McPhee J, Cahill CM, Zhan SS, Potter H, Nilsson LN: Translation of the Alzheimer amyloid precursor protein mRNA is up-regulated by interleukin-1 through 50-unstranslated region sequences. J Biol Chem. 1999, 274: 6231-6421.
Acknowledgements
SMG is funded by NIH grant AG12411.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MS wrote most of the first draft, JW wrote the part of γ-secretase and SMG wrote the section on trauma. All authors were involved in the design of the figures and editing of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Sastre, M., Walter, J. & Gentleman, S.M. Interactions between APP secretases and inflammatory mediators. J Neuroinflammation 5, 25 (2008). https://doi.org/10.1186/1742-2094-5-25
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-2094-5-25