
Abstract
With the role of angiogenesis in tumor growth and progression firmly established, considerable effort has been directed to antiangiogenic therapy as a new modality to treat human cancers. Antiangiogenic agents have recently received much widespread attention but strategies for their optimal use are still being developed. Gene therapy represents an attractive alternative to recombinant protein administration for several reasons. This review evaluates the potential advantages of gene transfer for antiangiogenic cancer therapy and describes preclinical gene transfer work with endogenous angiogenesis inhibitors demonstrating the feasibility of effectively suppressing and even eradicating tumors in animal models. Additionally, we describe the advantages and disadvantages of currently available gene transfer vectors and update novel developments in this field. In conclusion, gene therapy holds great promise in advancing antiangiogenesis as an effective cancer therapy and will undoubtedly be evaluated in human clinical trials in the near future.
Similar content being viewed by others


Introduction
In 1971, Dr. Judah Folkman first proposed the hypothesis that tumor growth is angiogenesis dependent [1]. Angiogenesis, the growth of new capillary blood vessels from preexisting vasculature, has long been appreciated for its role in normal growth and development and now is widely recognized for its role in tumor progression and metastasis [2]. Angiogenesis is a multi-step process that includes endothelial cell (EC) proliferation, migration, basement membrane degradation, and new lumen organization. Within a given microenvironment, the angiogenic response is determined by a net balance between pro- and anti-angiogenic regulators released from activated ECs, monocytes, smooth muscle cells and platelets.
The principal growth factors driving angiogenesis are vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), and hepatocyte growth factor. Other positive regulators are angiotropin, angiogenin, epidermal growth factor, granulocyte colony-stimulating factor, interleukin-1 (IL-1), IL-6, IL-8, platelet-derived growth factor (PDGF), tumor necrosis factor-α (TNF-α), and matrix proteins such as collagen and the integrins. Several proteolytic enzymes critical to angiogenesis include cathepsin, urokinase-type plasminogen activator, gelatinases A/B, and stromelysin [3, 4].
Tumor Growth and Metastasis are Angiogenesis Dependent
Transformed cells do not become tumorigenic unless they acquire angiogenic potential [5]. The angiogenesis response occurs early in tumor development and is rate limiting for tumor progression [6]. A mutual stimulation occurs between tumor and ECs by paracrine mechanisms. Every increase in the tumor cell population must be preceded by an increase in new capillaries that converge upon the tumor [4]. An angiogenic phenotype able to support tumorigenicity can arise in a step-wise fashion in response to both oncogene activation and tumor suppressor gene loss and involve both a decrease in the secretion of inhibitors and the sequential upregulation of inducers of angiogenesis [7]. This phenomenon is nearly universal; most of the human solid tumors and hematopoietic malignancies are angiogenesis dependent [8]. Additionally, under the influence of endogenous angiogenesis inhibitors, metastases remain dormant and tumor cell proliferation is balanced by an equivalent rate of cell death [9].
Angiogenesis as a Target for Cancer Therapy
With the role of angiogenesis in tumor growth and progression firmly established, considerable efforts have been directed to antiangiogenic therapy as a new modality to treat human cancers. A broad classification of angiogenesis inhibitors is illustrated in Figure 1. Currently available antiangiogenic agents have recently received much widespread attention – in fact, antiangiogenesis therapies for cancer were honored as one of the ten runners – up for Breakthrough of the Year in Science [10]. At the time of this writing, there were over 60 angiogenesis inhibitors in clinical trials and a number of these agents are listed in Table 1[2].

Antiangiogenic Inhibitors. The flowchart depicts the two major groups of antiangiogenic inhibitors, direct and indirect. It highlights the major differences between the groups and shows some representative examples in each category.
These agents target ECs rather than the more conventional target – the tumor cell itself. In addition, these agents seem to preferentially target tumor endothelium versus normal. Reasons may include that ECs proliferate more rapidly in tumors than in normal tissues, activated tumor ECs show higher expression of certain surface markers than normal ECs, or that tumor vasculature is chaotic with interrupted basement membrane [11, 12]. Similar differences are seen across most human tumors. Also advantageously, slowly growing, poorly vascularized as well as rapidly growing highly vascularized tumors respond to antiangiogenic therapy. Additional advantages to antiangiogenic therapy include low toxicity, minimal drug resistance, and repeated cycles of antiangiogenic therapy may be followed by a prolonged tumor dormancy without further therapy [13].
Antiangiogenic agents work through different mechanisms, like inhibition of EC proliferation, migration, and EC apoptosis. When an angiogenesis inhibitor induces EC apoptosis in a microvessel, tumor cells supported by that vessel subsequently undergo apoptosis. It has been established that EC apoptosis precedes tumor cell apoptosis. Various mechanisms might be responsible for EC-induced tumor cell apoptosis. First, death of ECs limit the oxygen and nutrient supply to the surrounding tumor cells. Secondly, growth factors produced by ECs are no longer available for tumor cell growth [14]. Furthermore, even without changes in microvessel density (MVD), apoptosis of tumor cells occurs in proximity to endothelium following treatment with angiogenesis inhibitors [15]. Endothelial cell apoptosis also influences outcome of radiation and chemotherapy. Garcia-Barros and co-workers have shown that microvascular damage regulates tumor cell response to radiation therapy [16]. Also, an antiangiogenic schedule of cyclophosphamide can control tumor growth more effectively by sustained apoptosis of ECs within the vascular bed of a tumor [17, 18].
The more widely studied antiangiogenic agents include naturally occurring angiogenesis inhibitors (angiostatin, endostatin, thrombospondins, platelet factor-4, etc.); inhibitors of EC growth (TNP-470, thalidomide, interleukin-12 [IL-12] etc), inhibitors of proangiogenic molecules (antibodies, antisense and soluble receptors for FGF, VEGF); agents that interfere with basement membranes and extracellular matrix (tissue inhibitors of matrix metalloproteinases [TIMPs]); antibodies to adhesion molecules (αvβ3) and small inhibitors of receptor tyrosine kinases. TNF-α, transforming growth factor-beta, and IL-4 are bifunctional modulators. These molecules are either stimulators or inhibitors depending on the amount, the site, the microenvironment, and the presence of other cytokines. Despite the enthusiasm and wide variety of agents, results have been disappointing and strategies for their optimal use are still being developed.
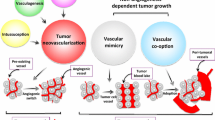
One obstacle in optimizing antiangiogenic strategies has been the unavailability of good surrogate markers to follow the success of antiangiogenic trials or to measure clinical response. The most widely used angiogenic marker is microvessel density (MVD). It has proven effective as a prognostic indicator in several types of malignant tumors including hematopoietic malignancies [19, 20]. However, the more recent thought is that MVD is not, by itself, an indicator of therapeutic efficacy nor should it be used to guide the stratification of patients for therapeutic trials [21]. Recently, it has been shown that circulating mature or bone marrow driven endothelial precursor cells play an important role in neovascularization. The release of angiogenic factors by tumor cells causes mobilization of bone marrow derived ECs and hematopoietic cells which promote tumor growth. There is a close relation between an increase in circulating ECs (CECs) and tumor progression, and evaluation of CECs could be used as a clinically relevant, non invasive angiogenesis marker [22].
New concepts such as vasculogenic mimicry and mosaic tumor vessels have also been examined as a marker for angiogenesis [23, 24]. Vasculogenic mimicry was first observed in melanoma and describes the ability of aggressive melanoma cells to express endothelium-associated genes and form extracellular matrix-rich vasculogenic-like networks in three-dimentional culture [25, 26]. They have been observed in other malignancies as well. A strong association between poor patient outcome and aggressive tumors that contain vasculogenic networks have been shown [27]. Individuals with melanomas that have undergone vasculogenic mimicry have a poor prognosis [25, 28]. Endostatin, a widely studied antiangiogenic agent, can inhibit endothelial cell-driven angiogenesis but not vasculogenic mimicry [25].
Antiangiogenic Gene Therapy
Several lessons learned from early clinical trials in antiangiogenic therapy would seem to support a role for antiangiogenic gene transfer strategies in the future. These lessons include:
(1) Genetic stability – Endothelial cells are much more genetically stable than tumor cells and are therefore less likely to accumulate mutations that confer early drug resistance [29]. Gene therapy strategies that result in constitutive expression of an antiangiogenic protein would be expected to be more effective in this setting than a gene therapy approach targeting a tumor cell that might quickly develop an escape mechanism.
(2) Low, continuous dosing – Constitutive expression of an antiangiogenic protein even at lower concentrations than bolus doses may be more effective than the intermittent peaks associated with repeated delivery of a recombinant protein. Some evidence for this thinking has been demonstrated in a mouse model [30].
(3) Angiogenic switch – Understanding the genetic and epigenetic events which transform a normal cell into a cancer cell has been one of the major advances in the field of tumor biology. However, in addition to these changes that occur during transformation, the induction of tumor vasculature, called the angiogenic switch, has increasingly become recognized as a critical step in tumor propagation and progression [31]. From this perspective, the body may harbor many in situ tumors yet the tumors do not progress to lethal tumors unless there is an imbalance between a tumor's pro-angiogenic output and the body's total angiogenic defense [32]. Gene therapy offers a strategy whereby an individual could boost their endogenous angiogenic defenses and tip the balance favorably.
(4) Cost of production – The production of functional proteins can be expensive and the availability of some of the recombinant proteins may become scarce. For example, production of two of the most well known and widely studied angiogenesis inhibitors – angiostatin and endostatin – has recently halted [33]. Gene therapy offers the opportunity for the patient to become his/her own source of production, an endogenous factory for antiangiogenic protein production.
(5) Multiple pathways – The ability to inhibit multiple angiogenic pathways could be made easier utilizing gene transfer strategies which could achieve prolonged, sustained levels of multiple therapeutic agents rather than repeated, systemic boluses of numerous antiangiogenic agents [34].
Candidate Genes for Antiangiogenic Gene Therapy
At this time, antiangiogenic gene therapy strategies remain in the preclinical stage and have not yet been tested in patients. We review several candidate genes below:
Thrombospondin-1 (THBS1)
Thrombospondin-1 was the first naturally occurring angiogenesis inhibitor to be identified and is a potent inhibitor of angiogenesis [35]. Its activity as an antiangiogenic antitumoral agent has been extensively demonstrated in cell culture and mouse model systems and has been reviewed extensively elsewhere [36]. The antiangiogenic effects of THBS1 appear to be localized to a peptide sequence containing the type I repeats domain, which binds CD36 on ECs [37]. However, the utility of THBS1 as a reproducible recombinant protein for clinical application has been limited by its large size (450 kd), poor bioavailability, and susceptibility to proteolytic breakdown [37]. Currently, there are several Phase II studies which are recruiting patients with advanced renal cell cancer, advanced non-small cell lung cancer, refractory lymphoma and advanced sarcoma to evaluate the safety and efficacy of a THBS1 mimetic called ABT-510.
Jin et al liposomally transfected THBS1 cDNA into DU145, an human prostate cancer cell line [38]. There was no significant inhibition of growth on the cell line in vitro. However, the TSP-1 overexpressing construct inhibited growth of DU-145 xenografts in Balb/c mice. More recently, investigators constructed a recombinant adenovirus expressing a portion of the THBS1 gene which has been shown to encode its antiangiogenic properties [39]. K562 human myelogenous leukemia cells were stably transduced and demonstrated similar results. There was little effect on in vitro growth but THBS1-transduced cell growth in xenografts was dramatically inhibited.
Endostatin
First discovered in 1997, endostatin, a 20 kD internal fragment of the alpha1 chain of type XVIII collagen, has become the most well characterized of the body's angiogenesis inhibitors [40]. Endostatin appears to inhibit ECs via several pathways, including binding alphaVbeta1integrin [41], inhibiting the receptor for VEGF [42], and inhibiting cyclin D1 [43]. However, the complete picture has yet to emerge. Endostatin was the first endogenous inhibitor studied in clinical trials [44]. To date, endostatin has been evaluated in phase I clinical trials only. Recombinant human endostatin appears to be essentially free of significant dose-limiting toxicity but also demonstrated no clinical responses in patients with various solid tumors [45, 46].
In our laboratory, we have evaluated endostatin in several preclinical gene therapy models. Using a recombinant adenovirus encoding murine endostatin, we were able to achieve high circulating levels of endostatin in mice after systemic delivery [47]. In this setting, there was marked growth inhibition of MC38 murine colon carcinoma cells implanted subcutaneously. We have also retrovirally transduced a murine liver cell line NMuLi with murine endostatin [48]. In vitro growth of these cells was not significantly impacted but subcutaneously implanted tumors were dramatically slowed and a survival advantage was demonstrated in mice given intraperitoneal injections of endostatin-producing clones versus controls.
Tumstatin
Tumstatin, a cleavage fragment of the alpha3 chain of type IV collagen, represents an exciting potential target for gene therapy. Human tumstatin prevents angiogenesis via inhibition of EC proliferation and promotion of apoptosis; its activity appears to be mediated by alpha v beta 3 integrin [49]. Tumstatin also suppresses tumor growth in several different mouse models [50–52]. At this time, gene transfer experiments with tumstatin have not been published.
Arresten
Arresten, first identified in 2000, similarly represents a potent angiogenic protein derived from the vascular basement membrane [53]. This 26-kD NC1 domain of the alpha1 chain of type IV collagen, functions as an anti-angiogenic molecule by inhibiting EC proliferation, migration and tube formation. Its antiangiogenic activity appears to be mediated by alpha1beta1 integrins on ECs. Arresten inhibited the growth of two human xenograft tumors in nude mice and the development of tumor metastases in a murine model [53]. These results suggest that arresten represents an excellent target for future gene therapy experiments.
Canstatin
Canstatin, also identified in 2000, is an endogenous 24 kD fragment of the alpha2 chain of type IV collagen. It has been demonstrated to inhibit EC migration and tube formation and induce EC apoptosis selectively [54]. Canstatin suppressed tumor growth in human xenograft mouse models and demonstrated antivascular changes in these tumors [54]. In our laboratory, we have cloned canstatin into various gene transfer vectors and have achieved promising results in some in vivo tumor models (unpublished data).
Vastatin, Restin
Other basement membrane cleavage products include vastatin, a fragment of the NC1 domain of collagen VIII. Vastatin was shown to inhibit the proliferation of bovine aortic ECs and induce cell apoptosis [55]. Restin, a 22 kD fragment of human collagen XV, inhibits the migration of ECs and suppresses the growth of tumors in a xenograft renal carcinoma model [56].
Angiostatin
Angiostatin, a 38 kD internal fragment of plasminogen, is one of the most potent endogenous inhibitors of angiogenesis and has been shown to suppress tumor growth and metastases in murine tumor models [57–59]. It appears to exhibit its antiangiogenic activity via interaction with at least 3 potential receptors on the EC: ATP synthase, angiomotin, and alphavbeta3 [60]. Angiostatin has also been evaluated in Phase I clinical trials [61]. Again, no significant dose-limiting toxicity was seen in patients undergoing twice-daily subcutaneous injections and levels were reached in which significant effects were seen in preclinical models.
Investigators have studied angiostatin in a number of gene transfer modalities. Recently, using a recombinant adeno-associated virus (AAV) encoding kringles 1–3 of human angiostatin, scientists were able to achieve continuous and sustained expression of angiostatin in the sera of mice for more than 6 months following a single injection of recombinant vector [62]. AAV-mediated stable expression of angiostatin slowed tumor growth and prolonged survival in the highly aggressive B16F10 melanoma and Lewis lung carcinoma models of experimental metastasis.
Another interesting strategy that is increasingly being studied is the combination of chemotherapeutics with gene transfer. In the case of angiostatin, using the human prostate cancer line PC3, tumor bearing mice were treated with recombinant adenovirus encoding the kringle 1–3 region of angiostatin plus docetaxel versus adenovirus alone and controls [63]. Tumor regression was seen only in mice treated with the combination adenovirus chemotherapy strategy. This type of synergy may represent a major antiangiogenic strategy in the future.
16 kD Prolactin Fragment
Another cleavage fragment of potential interest is the 16 kD prolactin fragment. Clapp et al first demonstrated its antiangiogenic properties by demonstrating that it inhibits capillary EC proliferation, migration, and organization into microvessels [64]. To date, the receptor mediating human prolactin fragment activity remains unknown [65].
The 16 kD prolactin fragment was expressed and secreted from HCT116 human colon cancer cells stably transfected with an expression vector encoding the 16 kD prolactin fragement gene. Protein expression by the transfected HCT116 cells inhibited tumor growth and neovascularization when implanted subcutaneously in Rag1 mice [66]. In addition, using an adenovirus transfer vector, Kim et al have shown that expression of the16 kD prolactin fragment in prostate cancer cells markedly reduced their ability to form tumors in a xenograft murine model [66, 67].
Platelet Factor-4
The chemokine platelet factor 4 (PF4), first identified as an antiangiogenic agent in 1990 [68], has been shown to suppress tumor growth in vivo in murine melanoma and human colon carcinoma xenograft mouse models [69]. Both cell lines were not inhibited with PF4 administration in vitro, suggesting a selective antiangiogenic antitumor mechanism. In addition, Kolber et al demonstrated a dose-dependent suppression of tumor metastases in an experimental lung metastasis model [70]. Recombinant PF4 was evaluated in a Phase I trial of patients with metastatic colorectal cancer. PF4 was well tolerated in 11 patients though no clinical responses were noted [71].
Tanaka et al first reported effective antitumor activity of PF4 using a gene transfer strategy [72]. Using retroviral and adenoviral vectors to express a secretable form of platelet factor 4, they demonstrated growth inhibition of and hypovascularity in intracerebral gliomas in the PF4-transduced group. Animal survival was also prolonged. Additionally, Li et al retrovirally transduced KB cells, a human head and neck squamous carcinoma cell line. PF4-transduced cell growth was not inhibited in vitro but in vivo, but subcutaneously implanted tumor growth was inhibited. Anti-angiogenic changes were seen in xenograft vascular histochemistry. These findings supported a selective antiangiogenic antitumor mechanism [73].
Interferon-inducible protein-10 (IP-10)
IP-10, a member of the C-X-C chemokine family, has a multitude of biological functions, including potent immunomodulatory and antiangiogenic effects [74]. In our laboratory, we retrovirally transduced A375 human melanoma cells with the human IP-10 gene and inoculated nude mice subcutaneously. In vivo growth of IP-10-transduced melanoma cells was markedly reduced compared to controls and histologic analysis of these tumors demonstrated reduced MVD [75].
Angiopoietins
The angiopoietins are a family of structurally related proteins that specifically bind a common endothelial cell-specific receptor tyrosine kinase, Tie2 [76]. The angiopoietins play an essential and complex role in angiogenesis. Angiopoietin-1 appears to activate Tie2, evidenced by increased tyrosine phosphorylation of Tie2, whereas angiopoietin-2 seems to have both agonistic and antagonistic properties depending on the local environment of the Tie2 receptor [76]. The expression patterns of Ang-1 and Ang-2 in the tumor and tumor vessel microenvironment are complex and the precise role that each plays in tumor angiogenesis is still being avidly studied and debated [76].
Recently, Stoeltzing et al liposomally transfected human colon cancer cells with an Ang-1 construct. Cells were then injected directly into the liver of nude mice. Tumor burden and vessel counts were significantly lower in the Ang-1-transduced cells as compared to controls. Another approach targeting this complex pathway has been to block Tie2 activation by the presence of a recombinant, soluble Tie2 receptor. Lin et al constructed an adenoviral vector encoding the recombinant, soluble Tie2 receptor [77]. Administration of this vector to tumor bearing mice – murine mammary carcinoma and murine melanoma – significantly inhibited the growth rate of both tumors as compared to controls. Administration of this vector also slowed the development and progression of metastatic disease in experimental metastases models.
Interleukin-12 (IL-12)
IL-12 is a heterodimeric pro-inflammatory cytokine which has multiple functions, including the induction of interferon-gamma, activation of T helper and NK cells, and is a mediator between innate resistance and adaptive immunity [78]. Pertinent to this review, IL-12 has been identified as a potent antiangiogenic and antitumor agent [79, 80]. The exact mechanism is complex, including the induction of secondary cytokines such as interferon-gamma or chemokines such as interferon-inducible protein 10 – which may have direct cytotoxic effects and/or antiangiogenic mechanisms for tumor inhibition [78].
Interleukin-12 has been evaluated as a cancer therapeutic in Phase I trials. In Germany, recombinant human IL-12 was administered subcutaneously three times weekly to patients with advanced renal cell carcinoma [81]. Dose limiting toxicities included deterioration of performance status, fever, vomiting, mental depression, leucopenia, oral mucositis and elevation of hepatic enzymes. One patient had a partial response and seven had stable disease. In 28 patients, one patient was seen to have a partial response.
Additionally, recombinant human IL-12 (rhIL-12) was administered as a twice-weekly intravenous infusion for 6 weeks in patients with metastatic renal cell cancer and malignant melanoma [82]. Dose-limiting toxicities included elevated hepatic transaminases and cytopenias. There was one partial response at the maximum tolerated dose in a patient with renal cell cancer. At the MTD (n = 14), there was one partial response occurring after 6 cycles of rhIL-12 in a patient with renal cell cancer and 2 patients with renal cell cancer exhibited disease stabilization.
In a phase II study in the United States, rhIL-12 was administered intravenously to 28 patients with advanced ovarian cancer [83]. Again, there was one partial response. Toxicities included grade 4 myelotoxicity and capillary leak syndrome. Because of the toxicity of systemically delivered IL-12, there is much enthusiasm for gene therapy approaches which may allow achievement of high concentrations in the local tumor environment but low systemic levels. Several viral and nonviral vectors have been developed to transfer IL-12 into tumor cells and/or antigen presenting cells and have shown antitumor effects [84]. For example, Caruso et al directly injected recombinant adenovirus expressing murine IL-12 intratumorally in the MCA-26 tumor model [85]. Mice injected with IL-12 expressing adenovirus achieved significant survival advantage compared to controls.
Interleukin-18 (IL-18)
IL-18 has also been identified as an angiogenic inhibitor and tumor suppressor [86]. Cao et al demonstrated its antiangiogenic effects in a number of in vitro and in vivo angiogenesis assays [86]. Systemic and intralesional administrations of IL-18 produced significant suppression of murine T241 fibrosarcoma growth in C57Bl6/J and SCID mice. No inhibition of cell growth was seen in culture, suggestive of a specific antiangiogenic antitumor mechanism.
Nagai et al transfected B16F10 melanoma cells with a modified form of the IL-18 gene to enable tumor cells to secrete IL-18 [87]. Significant growth inhibition was observed in these modified melanoma cells as compared to controls. Histologic analysis demonstrated reduced vascularization. The antitumor effect appeared to be mediated through induction of interferon-gamma.
IL-18 may also act synergistically with other inhibitors of angiogenesis. Liu et al constructed two adenoviral vectors encoding IP-10 and IL-18 [88]. Direct intratumoral injection of either vector alone into J558 murine myeloma subcutaneous nodules delayed tumor growth to some extent whereas coinjection of both vectors in the same tumor nodule much more significantly delayed tumor growth and even cured established tumors in 8 of 10 mice.
Interferons
The interferons (IFN) have been widely used for a number of applications, including antiviral agents for hepatitis and as cytotoxic agents for certain leukemias and some bladder cancers [29]. They have also been shown to inhibit angiogenesis and to induce tumor regression through apparent antiangiogenic mechanisms [89, 90]. One mechanism by which the IFN may exhibit this action is by downregulation of pro-angiogenic factors secreted by the tumor cell [91]. Inhibition of bFGF is the primary pro-angiogenic factor that has been implicated [92].
Albini et al retrovirally transduced endothelial-like Eahy926 cells with IFN-alpha1 and interferon-beta murine cDNAs [93]. IFN-transduced cells demonstrated decreased migration, invasion, and capillary-like structure formation in matrigel. Additionally, co-inoculation of Kaposi's sarcoma cell line and IFN-transduced cells resulted in marked reduction of tumor growth in nude mice as compared to controls alone.
Endothelial-monocyte activating polypeptide-II (EMAP-II)
EMAP-II is a tumor-derived cytokine with potent effects on ECs, including induction of tissue factor, upregulation of TNF receptor 1, and upregulation of E-selectin and P-selectin [94]. EMAP-II inhibits EC proliferation but has little effect on tumor cell or fibroblast proliferation [94]. Inhibition appears to be mediated through pro-apoptotic pathways. Release of EMAP-II in melanoma cells appears to render the tumor-associated vasculature sensitive to TNF [95]. We used these findings to develop a gene transfer strategy utilizing EMAPII to sensitize tumors to TNF-α [96]. We constructed a recombinant vaccinia virus encoding the human EMAP-II gene and transfected a human melanoma cell line previously insensitive to TNF-α treatment. EMAP-II expressing human melanoma cells implanted in nude mice demonstrated significant tumor regression after treatment with systemic TNF-α. Control groups remained insensitive to TNF-α therapy.
Tissue Inhibitors of Metalloproteinases (TIMPs)
Matrix metalloproteinases (MMPs) play a critical role in extracellular matrix remodeling, an essential component of physiologic and pathologic angiogenesis and pathologic tumor growth and metastasis [97]. TIMPs function to block the activity of MMPs and their role in tumor growth and tumor angiogenesis is being actively studied.
The dominant paradigm has been that TIMPs serve an important role in suppressing tumor invasion and metastasis [97]. Numerous in vivo and in vitro studies have supported this assumption and have been extensively reviewed [97]. For example, in a gene transfer strategy, Wang et al, transfected full-length TIMP-4 cDNA into a human breast cancer cell line. TIMP-4 transfected cells demonstrated decreased invasiveness in an in vitro assay and, in a nude mouse model, decreased growth and metastatic burden [98].
As the understanding of TIMPs evolves, however, it seems that TIMPs play a much more complex role in tumor angiogenesis and tumor growth and metastastic spread. In fact, research has suggested that TIMPs, in certain scenarios, may be proangiogenic and favor tumor growth [97]. This complexity may underpin some of the disappointing results seen in clinical trials. Thus, a more thorough understanding of TIMPs may be necessary before realizing the potential of these agents as effective antitumor therapies.
Tumor Necrosis Factor Alpha (TNF-α)
One of the most heralded cytokines of the last century has been TNF-α. TNF is a homotrimeric complex of 52 kD which is produced by many cell types and has been shown to have profound antivascular and antitumor effects [99]. Unfortunately, phase I/II trials have been disappointing, largely due to profound dose-limiting toxicity, most notably sepsis-like hypotension. The maximal tolerated dose in clinical trials is about 1/50 of the effective dose in murine tumor models [99]. Thus, TNF-α may represent an ideal agent for localized gene transfer strategies, which could allow for high locoregional doses but low systemic levels.
One such effort has involved a replication deficient adenoviral vector called TNFerade. This vector expresses human TNF-α and contains a radiation-inducible Egr-1 promoter. TNFerade was recently evaluated in a phase I, dose escalation trial in patients with treatment refractory solid tumors [100]. TNFerade was injected intratumorally once weekly for 6 weeks with concomitant radiation. TNFerade-related toxicities included fever, injection site pain, and chills but dose-limiting toxicities were not seen. Overall, 21 of 30 patients demonstrated an objective response (5 CRs, 9 PRs) and, interestingly, in patients with synchronous lesions, a more favorable tumor response was seen in lesions treated with TNFerade + radiation as compared to lesions treated with radiation alone (4 of 5 patients). Currently, Phase II randomized trials with TNFerade are open for patients with rectal cancer and unresectable pancreatic cancer. A single arm Phase II trial is open for patients with locally advanced esophageal cancer.
p53
Recently, attention has been focused on the role of the p53 gene in angiogenesis. The p53 gene is an important tumor suppressor gene and inactivated in over 50% of all human cancers. Mutant p53 correlates with reduced expression of thrombospondin-1, increased angiogenesis, and malignant progression in melanoma [101]. Transfection of wild type p53 back into glioblastoma cells leads to angiosuppression [102]. It has been shown that as nonmalignant fibroblasts progress to tumorigenicity, cells become fully angiogenic in two steps. First, there is loss of both alleles of wild-type p53, which causes a 20-fold drop in secreted TSP and a fourfold increase in secreted VEGF [7]. Second, angiogenic activity increases again on transformation by activated ras oncogene due to a further twofold increase in secreted levels of angiogenic proteins. Thus, there is a step-wise change in the angiogenic phenotype in response to oncogene activation and tumor suppressor gene loss involving a decrease in the secretion of inhibitors and the sequential up-regulation of inducers of angiogenesis [7]. By inhibiting angiogenesis, p53 indirectly induces apoptosis and can revert tumors to a dormant phenotype [103]. PTEN and phosphatidylinositol 3'-kinase inhibitors up-regulate p53 and block tumor-induced angiogenesis in human brain ECs [104]. Thus, restoration of p53 tumor suppressor gene may be an important antiangiogenic treatment modality.
Gene Therapy Vectors
Clearly, the body of preclinical work in antiangiogenic gene therapy demonstrates the potential to effectively eradicate or control tumor burden in mouse models. The major shortcoming for human application remains the vectors used for gene transfer. A variety of nonviral vectors – such as naked DNA, antisense RNA, small interfering (Si) RNA, cationic liposomes – and viral vectors – including adenoviruses, adeno-associated viruses, retroviruses, lentiviruses, and bacteriophage vectors – have been used. The following section summarizes the variety of nonviral and viral vectors that have been utilized in gene therapy and describes recent developments that may hold promise for more effective therapies in the future.
Nonviral vectors
Naked Plasmid DNA
The simplest non-viral gene delivery system uses naked expression vector DNA. Direct injection of free DNA into certain tissues, particularly muscle, has been shown to produce surprisingly high levels of gene expression, and the simplicity of this approach has led to its adoption in a number of clinical protocols. However, naked DNA and peptides have a very short half life due to in vivo enzymatic degradation. Plasmid DNA suffers from low transfection efficiency. However, there are reports showing efficacy of naked plasmid DNA administration. Intra-tumoral administration of naked plasmid DNA encoding mouse endostatin inhibits renal carcinoma growth [105]. Intramuscular administration of the endostatin gene could significantly retard the growth of metastatic brain tumors [106]. A single intramuscular administration of the endostatin gene could secret endostatin for up to 2 weeks and could inhibit systemic angiogenesis [107]. Injected endostatin gene also inhibited both the growth of primary tumors and the development of metastatic lesions. Thus, these results demonstrate the potential utility of intramuscular delivery of an antiangiogenic gene for treatment of disseminated cancers. However, in comparison to naked DNA, complexes of plasmid DNA with liposomes are relatively more stable with higher potency for transfection [108].
Cationic Liposomes
Liposomes are microscopic spherical vesicles of phospholipids and cholesterol. Recently, liposomes have been evaluated as delivery systems for drugs and have been loaded with a great variety of molecules such as small drug molecules, proteins, nucleotides and even plasmids. The advantages of using liposomes as drug carriers are that they can be injected intravenously and when they are modified with lipids which render their surface more hydrophilic, their circulation time in the bloodstream can be increased significantly. They can be targeted to the tumor cells by conjugating them to specific molecules like antibodies, proteins, and small peptides [109]. The cationic liposomes can significantly improve systemic delivery and gene expression of DNA [110]. Systemic, liposome-mediated administration of angiostatin could suppress the growth of melanoma tumors in mice [111]. Similar findings were observed by Chen and co-workers with angiostatin and endostatin [112]. It has been shown that angiogenic ECs take 15-33-fold more cationic liposome:DNA complexes than corresponding normal ECs. Thus, preferential uptake of cationic liposomes could be used to target diagnostic or therapeutic agents selectively to angiogenic blood vessels in tumors [113]. Liposomes modified with angiogenic homing peptide for ECs can strongly suppress tumor growth compared to unmodified liposomes [114]. Janssen et al showed that the coupling of cyclic RGD-peptides to the surface of PEG-liposomes can target to tumor endothelium [115]. Tumor vessel-targeted liposomes can also be used to efficiently deliver therapeutic doses of chemotherapy [116].
Antisense RNA
Antisense oligodeoxynucleotides (ODNs) are synthetic molecules that block mRNA translation. They can be used as a tool to inhibit mRNA translation of a diseased gene. There are reports demonstrating use of VEGF and VEGFR antisense RNA in preclinical models. Angiogenesis and tumorigenicity (as measured by MVD and tumor volume, respectively) of human esophageal squamous cell carcinoma can be effectively inhibited by VEGF165 antisense RNA [117]. Im and co-workers used an adenovirus to transfer antisense VEGF sequence into glioma cells in vitro and in vivo [118]. The treatment resulted in reduction of the level of the endogenous VEGF mRNA and protein and inhibited growth of glioma tumors. VEGF mediated neovascularization can also be inhibited by combination of antisense oligonucleotides to VEGFR1 and VEGFR2 [119]. Expression of antisense RNA to Ang1 could reduce tumor volume, decrease MVD and increase apoptosis in nude mice [120]. Expression of antisense to integrin subunit beta 3 inhibits microvascular EC capillary tube formation in fibrin, with the extent of down-regulation correlating with the extent of tube formation inhibition [121].
Small Interfering RNA (SiRNA)
The ability of small dsRNA to suppress the expression of a gene corresponding to its own sequence is called RNA interference (RNAi). The discovery of RNAi has added a promising tool to the field of molecular biology. Introducing the SiRNA corresponding to a particular gene will knock out the cell's own expression of that gene. The application of SiRNA to silence gene expression has profound implications for the intervention of human diseases including cancer. There are published reports using SiRNA to silence expression of angiogenic genes. SiRNA targeted to either subunit of the alpha6beta4 (a laminin adhesion receptor) integrin reduced its cell surface expression and resulted in decreased invasion of MDA-MB-231 breast carcinoma cells [122]. Knockdown of Her-2/neu expression by siRNA is associated with increased expression of the anti-angiogenic factor THBS-1 and decreased expression of VEGF in human breast and ovarian cancer. Thus, siRNA-mediated gene silencing may be a useful therapeutic strategy for Her-2/neu-over-expressing breast or ovarian cancer [123].
The disadvantage to simply introducing dsRNA fragments into a cell is that gene expression is only temporarily reduced. However, Brummelkamp et. al. developed a new vector system, named pSUPER, which directs the synthesis of siRNA in mammalian cells [124]. The authors have shown that siRNA expression mediated by this vector causes persistent and specific down-regulation of gene expression, resulting in functional inactivation of the targeted gene over longer periods of time. VEGF carries out multifaceted functions in tumor development. DNA-vector based RNAi, in which RNAi sequences targeting VEGF isoforms, has potential applications in isoform-specific knock-down of VEGF [125].
However, the disadvantages of non viral vectors – stability, non-specific uptake by various tissues, poor adsorption, short half life in the circulation, aggregate formation, and low in-vivo potency for cell transfection – continue to limit its use.
Viral Vectors
Delivery of genes expressing inhibitors of angiogenesis using viral vectors represents a more effective strategy, as it can produce stable and higher quantities of gene product compared to the systemic infusion of antiangiogenic DNA/protein. The main characteristics of any viral vector are easy purification into high titers, to mediate targeted gene delivery and prolonged gene expression with minimal side effects. There are 5 main classes of clinically applicable viral vectors; adenoviruses, adeno-associated viruses (AAVs), retroviruses, lentiviruses, and herpes simplex-1 viruses (HSV-1s) [126]. The main difference between these vectors is retroviruses and lentiviruses can integrate into the genome, whereas the other three classes predominantly persist as extrachromosomal episomes. Although the advantage of chromosomal integration is long term transgene expression, these vectors can infect dividing cells only. The non-integrated viral vectors can infect non-dividing cells, but are not a favorable choice to bring about stable genetic change.
Adenoviruses
Adenovirus is a double stranded DNA virus. It binds initially to the target cell through the viral fiber protein; however, the subsequent cell entry involves interaction between the viral capsid penton proteins and integrins on the target cell. Adenoviruses can be produced in high titers and can efficiently deliver the therapeutic gene. Replication-defective adenoviral vectors are indeed particularly well suited for cancer gene therapy as they lead to a transient, but robust, expression of the transgene, and efficient in vivo gene transfer has been reported especially in the liver after systemic injection. However, they do not integrate into the host genome and the gene expression is transient. Also, adenovirus elicits an immune response resulting in an elimination of vector expressing cells. Despite these apparent drawbacks, the adenovirus remains a popular vector for gene therapy due to its high gene transfer efficiency and high level of expression in a wide variety of cell types. Adenovirus can be targeted to pulmonary endothelium by complexing it with a bispecific antibody to viral particle and angiotensin-converting enzyme [127]. Bilamellar cationic liposomes can also be used to encapsulate adenovirus. The encapsulated adenovirus can transduce CAR negative cells and is resistant to the neutralizing anti-adenoviral antibodies, allowing the readministration of the adenovirus [128].
Retroviral Vectors
Retroviruses are a class of enveloped viruses containing a single stranded RNA molecule as the genome. Retrovirus vectors have been used in the majority of human gene transfers. They are able to efficiently integrate permanently into the human genome where they provide the basis for permanent expression of up to 8–9 kb of foreign DNA. Simple retroviruses, such as murine leukemia virus (MLV), and the vectors derived from them, require cell division for infection and thus possess a degree of inherent specificity for the rapidly dividing cells of neoplastic tissue [129]. Though transgene expression is usually adequate in vitro, prolonged expression is difficult to attain in vivo. Also, retroviruses are inactivated by complement proteins and inflammatory IFN, specifically IFN-alpha and IFN-gamma [130, 131]. A major shortcoming of retrovirus-derived vectors is their tendency to revert to replication-competent retrovirus (RCR), which could lead to fatal neoplasms. With the use of the latest packaging cell lines and vectors, the risk of RCR-generation has been drastically reduced. Currently, the greatest safety concern of using retroviral vectors is related to the risk of malignant transformation following oncogene activation due to random retroviral genomic integration and will be discussed in more detail below.
Adeno-associated virus (AAV)
The adeno-associated virus (AAV) is a vector that combines some of the advantages of both the adenoviral and retroviral vectors. It can efficiently transfer genes to a number of different cell types. The broad host range, low level of immune response, and longevity of gene expression observed with these vectors has enabled the initiation of a number of clinical trials using this gene delivery system [132]. A potential barrier, however, is the low transduction efficiencies of recombinant AAV vectors. AAV tropism can be genetically engineered by use of phage display-derived peptides to generate vectors that are selective for the vasculature [133]. The journal Nature recently reported concerns that there is a possibility that recombinant AAV vectors may cause or contribute to cancer in gene therapy subjects [134]. However, because of infrequent integration efficiency of AAV, the risk of cancer in current AAV trials is negligible [135].
Lentiviral Vectors
Lentiviral vectors represent a new vector system that can achieve permanent integration of the gene into non-dividing cells. Gene transfer can be achieved in very quiescent cells, nondividing or terminally differentiated cells such as neurons. Lentiviral vectors are especially useful in transducing cells which lack receptors for adenoviruses. A broad tissue tropism for lentivirus can be achieved using variety of viral envelopes [136]. So far, lentiviral vectors expressing matrix metalloproteinase-2 (MMP-2), angiostatin and endostatin have been developed [136, 137]. However, lentivirus has a low transduction efficiency for ECs and may result in significant vector-associated cytotoxicity [136].
Herpes Simplex Virus
The herpes simplex virus (HSV) thymidine kinase gene (tk) therapy with ganciclovir forms the basis of a widely used strategy for suicide gene therapy [138]. HSV can also be used to deliver a therapeutic gene of interest, especially to the nervous system. The advantages in using HSV include its wide host range, its ability to accommodate large genes, and its ability to establish long-lived asymptomatic infections in neuronal cells. However, the virus's ability to replicate lytically in the brain, under some circumstances causing encephalitis, has led to fears about its potential safety for ultimate use in humans.
Targeted Gene Therapy
The systemic delivery of viral vectors can lead to non specific uptake by various different tissues and hence result into systemic toxicity because of transgene expression. Local delivery can circumvent this problem; however, many times the tumor is not accessible for local injection. Hence, transgene expression in the targeted tissue is very important. Targeting tumor vasculature by gene therapy represents an ideal target, as tumor blood vessels are easily accessible to systemically applied vectors. Certain considerations such as vascular dependency of a tumor, the differences between tumor associated and normal vasculature, and accessibility of tumor vasculature to circulating vectors must be addressed.
Targeted viral vectors can be constructed in several ways. The first approach is pseudotyping, in which one species of virus is made to incorporate the envelope protein of another virus. Adeno-associated virus genome with its inverted terminal repeats can be packaged in the capsids of different serotypes which enables transduction with broad specificity [139]. The second approach is to genetically modify the viral capsid protein to incorporate a small peptide coding for a specific receptor and hence, allow targeting by ligand-receptor internalization. Tissue specific targeting can also be achieved by conjugating specific antibodies to receptors onto the viral capsids [140].
Various transductional and transcriptional approaches have also been devised to improve targeting of adenoviral vectors. Using this approach, Reynolds et al increased the selectivity of transgene expression by 300,000 for lung versus liver, the usual site of vector sequestration [141]. Another strategy is to use vectors guided by EC specific promoters, such as the promoters for endoglin, endothelin-1 gene, and von Willebrand factor. The murine preproendothelin-1 promoter is highly specific for ECs and could be use to achieve very high levels of gene expression in vascularized tumor metastases compared to normal or less vascularized primary tumor [142].
Whereas most antiangiogenic agents prevent new blood vessel formation, vascular targeting agents (VTAs) destroy pre-existing blood vessels of solid tumors. VTAs can be more active in large tumors and produce a characteristic pattern of widespread central necrosis. Combination of VTAs and angiogenic inhibitors can lead to synergistic effects. Two major types of VTAs are ligand-directed VTAs to deliver gene product to tumor endothelium and small molecule VTAs that exploit pathophysiological differences between tumor and normal endothelium [143]. The ligand-directed VTAs use ligands specifically expressed on the tumor endothelium, eg. VEGF receptors, αvβ3 integrins, tissue factor, and cell adhesion molecules like VCAM-1. A small molecule VTA described thus far is the tubulin binding agent combretastatin, which destabilizes the tubulin cytoskeleton. It has antiproliferative and cytotoxic effects on both proliferating tumor and ECs. It results in extensive and prolonged shut-down of blood flow in established tumor blood vessels, with much less effect in normal tissues [144]. Recently, bacteriophage vectors have been developed for targeted gene delivery of antiangiogenic VTAs.
Phage Vectors
Phage display represents a novel method of individually displaying up to tens of billions of peptides, proteins, including human antibodies and enzymes, on the surface of a small bacterial virus called a phage. Phage display allows producing and searching through large libraries of peptides and proteins to rapidly identify those compounds that bind with high affinity and high specificity to targets of interest. Pasqualini and Ruoslahti first distinguished active proliferating microvascular ECs and quiescent nonproliferating ECs using an in vivo phage display technique [145]. The phage display peptide libraries have been used to identify peptides that home to tumors through the circulation and that specifically bind to the tumor ECs or lymphatic cells [146, 147]. Phage displaying an Arg-Gly-Asp (RGD)-containing peptide binds to alpha v integrins with high affinity and homes to tumors when injected intravenously into tumor-bearing mice. Phage displaying the cyclic peptide His-Try-Gly-Phe (HWGF) specifically targets angiogenic blood vessels in vivo and specifically inhibits MMP-2 and MMP-9 metalloproteinases [148]. MMP-2 can directly bind to αvβ3 integrin on tumor cells [149]. These tumor endothelial specific signatures were later described by Dr. Folkman as an angiogenic zip codes [150].
The picture of integrins and their ligands is very complex. The αvβ3 integrin is expressed on proliferating ECs such as those present in growing tumors of various origins. VEGF-induced EC migration requires interaction between VEGF receptor2 (VEGFR2) and αvβ3 to drive the activation of downstream mitogenic pathways [151]. Several studies have demonstrated that αvβ3 is involved in melanoma cell invasion and promotes metastasis (reviewed in [152]). Although the precise mechanisms of αvβ3-promoted tumor progression are not clear, various studies suggest roles for αvβ3 in selective tumor cell migration, generation of growth and survival signals, intracellular signaling and generation of MMPs [152]. It has been observed that antibody to αvβ3 integrin can block angiogenesis without affecting the normal vasculature [115, 153]. The αvβ3 integrin can also be blocked with small peptides containing Arg-Gly-Asp (RGD) amino acid sequence. Exposure of human EC to TNF and interferon-γ decreases αvβ3-dependent EC adhesion and survival [154].
Other integrins such as αvβ5, and αvβ1 are also considered very significant for the regulation of angiogenesis. The β3 chain play a significant role in promoting angiogenesis, although the lack of it can also promote angiogenesis, given that the angiogenesis inhibitor tumstatin binds through this receptor [41]. Also, the expression of integrin αvβ6, a fibronectin receptor, promotes migration and invasion in squamous carcinoma cells [155].
Bacteriophages expressing ligands for specific receptors like integrins, growth factors, and antibody can be made to infect mammalian cells [156–158]. The phage vectors are an attractive alternative to existing animal viral vectors because they lack intrinsic tropism for mammalian cells and can be produced in bacteria in large titers. Currently, phage vector transduction efficiency (1–4%) is considerably lower than most viral vectors. Burg et al tested 14 human cancer cell lines from different tissues, showing viral transduction efficiency varies from 0.001 to 10%. However the transduction efficiency can be improved substancially by treating cells with camptothecin [159]. Thus, using specific targets in the tumor endothelium, it is possible to target gene therapy specifically to the tumor endothelium.
Nanoparticles
In recent years, significant effort has been devoted to developing nanotechnology for drug delivery since it offers a suitable means of delivering small molecular weight drugs as well as macromolecules such as proteins, peptides or genes [160]. These systems in general can be used to provide targeted (cellular/tissue) delivery of drugs, to improve oral bioavailability, to sustain drug/gene effect in target tissue, to solubilize drugs for intravascular delivery, and to improve the stability of therapeutic agents against enzymatic degradation (nucleases and proteases), especially of protein, peptide, and nucleic acids drugs [161]. Nanoparticles (NPs) are submicron-sized polymeric colloidal particles with a therapeutic agent of interest encapsulated within their polymeric matrix or adsorbed or conjugated onto the surface [162]. NPs have in general relatively higher intracellular uptake because of their smaller size. The therapeutic efficacy of the NPs could be due to their ability to protect the therapeutic agent from degradation due to lysosomal enzymes and sustained intracellular retention. Nanoparticles conjugated to the contrast agent like gadolinium can be used in magnetic resonance based imaging techniques [163, 164]. They can be targeted to tumor endothelium by covalently coupling to different ligands or antibodies [165]. David Cheresh's group at the Scripps Research Institute showed that NPs coupled to an integrin αvβ3-targeting ligand can deliver genes selectively to angiogenic blood vessels in tumor-bearing mice with no detectable levels in the other tissues [165]. Further, the authors showed that NPs conjugated to a mutant Raf gene blocks endothelial signaling and angiogenesis in response to multiple growth factors.
Current State of Gene Therapy Clinical Trials
Despite the tantalizing promise of gene therapy, there has been much recent alarm in the field and no review of the current state of and potential future role of gene therapy would be complete without noting the major current controversy in the field. The first true successes of gene therapy in clinical trials were reported in 2000 and 2002, in which scientists successfully treated children suffering from SCIDs by retrovirally transducing hematopoietic stem cells with the gamma-c gene [32]. Unfortunately, 2 of the 10 children subsequently developed leukemia-like conditions, eventually attributed to retroviral vector integration in proximity to the LMO2 proto-oncogene promoter, leading to aberrant transcription and expression of LMO2 [166]. These cases garnered enormous attention from scientists, regulators and the general public [167]. Reaction varied but some countries even imposed a general moratorium on trials involving retroviral gene transfer. Though in most countries trials have now been allowed to resume, the reaction has thrown the field of gene therapy into recession and has discouraged many scientists from starting new clinical trials [167].
Future Directions
As addressed above, the development of better vectors remains the cornerstone for advancing the field of antiangiogenic gene therapy of cancer. Important concerns about oncogene activation by retroviral integrating vector systems remain. Also, currently available nonviral vectors are not very efficient.
Future areas of investigation also include:
Combination Therapy
Studies have shown advantages to combining antiangiogenic agents with each other, as well as with chemotherapy and radiation [168]. Combining agents that target a number of different and synergistic pathways may yield improved antitumor effects and long term suppression of metastatic progression. For example, the combination of SU5416, VEGFR2 tyrosine kinase inhibitor and low-dose endostatin reduced tumor growth more efficiently than monotherapy alone [169]. Whereas SU5416 specifically inhibits vascular endothelial growth factor signaling, low-dose endostatin is able to inhibit a broader spectrum of diverse angiogenic pathways directly in the endothelium. Gene therapy strategies certainly need to be further evaluated in these combination settings.
Preventive Therapy
More specific to the field of antiangiogenic gene therapy, there is much enthusiasm for the role that antiangiogenic agents may play in preventive therapy. Because antiangiogenic therapy is generally less toxic and less susceptible to acquired resistance, angiogenesis inhibitors have been considered an ideal prophylactic agent in patients at high risk for cancer or for recurrence of cancer [29]. At our institution, we are currently recruiting patients with previously resected recurrent or metastatic colorectal cancer and randomizing these patients to receive thalidomide or placebo. Gene therapy offers an ideal strategy for long term, continuous production of antiangiogenic agents that may prevent development of primary or recurrent disease.
Chronic Disease Treatment
Additionally, many leaders in the field of angiogenesis now believe that some of the most important future cancer therapies may not completely eradicate all tumor cells in an individual but, instead, may turn cancer into a chronic manageable disease [32]. Gene therapy strategies leading to increased production of endogenous angiogenesis inhibitors would seem perfectly suited to support such an approach by tipping the balance toward a more antiangiogenic state.
Conclusions
Enthusiasm for antiangiogenic approaches to cancer therapy has never been higher, given the recent approval by the FDA for the first angiogenesis inhibitor and given the recent encouraging clinical trial data [170]. The versatility shown by the field of antiangiogenesis has enabled many human cancers to be realistically considered for therapeutic intervention and has considerably broadened the scope of gene therapy. Because of the difficulties and high costs of manufacturing numerous endogenous inhibitors of angiogenesis and because of the need for chronic administration of these agents, gene therapy remains an exciting strategy to circumvent these difficulties. Although no clinical gene therapy trials to date have utilized a focused antiangiogenic strategy, an abundance of preclinical research demonstrates that gene therapy-based antiangiogenesis approaches are an effective means of controlling and even eradicating tumor growth in animal models. As better vectors are developed, combination strategies continue to evolve, and increased understanding of the complex role that endogenous angiogenesis inhibitors play in tumor growth and progression takes place, antiangiogenic gene therapy will certainly be evaluated in future clinical trials.

Antiangiogenic Gene Therapy: Recent Developments. The figure depicts different modes of gene therapy directed to tumor endothelial cell (EC) and its microenvironment. The expression of EC specific cell surface molecules like, vascular endothelial growth factor receptor (VEGFR), E selectin or angiogenic growth factors (VEGF, FGF, PDGF) produced by tumor cells can be inhibited by specific antibodies or antisense RNA or using gene specific SiRNA. The targeted gene therapy can be achieved by using either viral vectors or nanoparticles carrying ligand (RGD) to endothelial cell surface specific receptors (αvβ5, αvβ3) to target antiangiogenic genes to tumor vasculature. The inhibitors can also selectively express in EC using endothelial cell specific promoters like endothelin 1 (EDT1). Ad: adenovirus; AAV: adeno-associated virus; Retro: retrovirus; VEGF: vascular endothelial growth factor; FGF: fibroblast growth factor; PDGF: platelet derived growth factor; TNF: tumor necrosis factor; RGD: Arginine-glycine-aspartic acid; SiRNA: small interfering RNA
References
Folkman J: Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971, 285: 1182-1186.
Satchi-Fainaro R, Puder M, Davies JW, Tran HT, Sampson DA, Greene AK, Corfas G, Folkman J: Targeting angiogenesis with a conjugate of HPMA copolymer and TNP-470. Nat Med. 2004, 10: 255-261. 10.1038/nm1002.
Ribatti D, Vacca A, Presta M: The discovery of angiogenic factors: a historical review. Gen Pharmacol. 2000, 35: 227-231. 10.1016/S0306-3623(01)00112-4.
Folkman J, Klagsbrun M: Angiogenic factors. Science. 1987, 235: 442-447.
Dameron KM, Volpert OV, Tainsky MA, Bouck N: Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science. 1994, 265: 1582-1584.
Skobe M, Rockwell P, Goldstein N, Vosseler S, Fusenig NE: Halting angiogenesis suppresses carcinoma cell invasion. Nat Med. 1997, 3: 1222-1227.
Volpert OV, Dameron KM, Bouck N: Sequential development of an angiogenic phenotype by human fibroblasts progressing to tumorigenicity. Oncogene. 1997, 14: 1495-1502. 10.1038/sj.onc.1200977.
Bertolini F, Mancuso P, Gobbi A, Pruneri G: The thin red line: angiogenesis in normal and malignant hematopoiesis. Exp Hematol. 2000, 28: 993-1000. 10.1016/S0301-472X(00)00508-7.
Holmgren L, O'Reilly MS, Folkman J: Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med. 1995, 1: 149-153.
Breakthrough of the year. The runners-up. Science. 2003, 302: 2039-2045. 10.1126/science.302.5653.2039.
Sedlacek HH: Pharmacological aspects of targeting cancer gene therapy to endothelial cells. Crit Rev Oncol Hematol. 2001, 37: 169-215. 10.1016/S1040-8428(00)00113-X.
Vartanian RK, Weidner N: Correlation of intratumoral endothelial cell proliferation with microvessel density (tumor angiogenesis) and tumor cell proliferation in breast carcinoma. Am J Pathol. 1994, 144: 1188-1194.
Boehm T, Folkman J, Browder T, O'Reilly MS: Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997, 390: 404-407. 10.1038/37126.
Dixelius J, Larsson H, Sasaki T, Holmqvist K, Lu L, Engstrom A, Timpl R, Welsh M, Claesson-Welsh L: Endostatin-induced tyrosine kinase signaling through the Shb adaptor protein regulates endothelial cell apoptosis. Blood. 2000, 95: 3403-3411.
Bergers G, Javaherian K, Lo KM, Folkman J, Hanahan D: Effects of angiogenesis inhibitors on multistage carcinogenesis in mice. Science. 1999, 284: 808-812. 10.1126/science.284.5415.808.
Garcia-Barros M, Paris F, Cordon-Cardo C, Lyden D, Rafii S, Haimovitz-Friedman A, Fuks Z, Kolesnick R: Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science. 2003, 300: 1155-1159. 10.1126/science.1082504.
Browder T, Butterfield CE, Kraling BM, Shi B, Marshall B, O'Reilly MS, Folkman J: Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000, 60: 1878-1886.
Folkman J: Angiogenesis and apoptosis. Semin Cancer Biol. 2003, 13: 159-167. 10.1016/S1044-579X(02)00133-5.
Gasparini G: Clinical significance of determination of surrogate markers of angiogenesis in breast cancer. Crit Rev Oncol Hematol. 2001, 37: 97-114. 10.1016/S1040-8428(00)00105-0.
Munshi NC, Wilson C: Increased bone marrow microvessel density in newly diagnosed multiple myeloma carries a poor prognosis. Semin Oncol. 2001, 28: 565-569. 10.1053/sonc.2001.28954.
Hlatky L, Hahnfeldt P, Folkman J: Clinical application of antiangiogenic therapy: microvessel density, what it does and doesn't tell us. J Natl Cancer Inst. 2002, 94: 883-893. 10.1093/jnci/94.12.883.
Mancuso P, Calleri A, Cassi C, Gobbi A, Capillo M, Pruneri G, Martinelli G, Bertolini F: Circulating endothelial cells as a novel marker of angiogenesis. Adv Exp Med Biol. 2003, 522: 83-97.
Chang YS, di Tomaso E, McDonald DM, Jones R, Jain RK, Munn LL: Mosaic blood vessels in tumors: frequency of cancer cells in contact with flowing blood. Proc Natl Acad Sci U S A. 2000, 97: 14608-14613. 10.1073/pnas.97.26.14608.
Hendrix MJ, Seftor EA, Meltzer PS, Gardner LM, Hess AR, Kirschmann DA, Schatteman GC, Seftor RE: Expression and functional significance of VE-cadherin in aggressive human melanoma cells: role in vasculogenic mimicry. Proc Natl Acad Sci U S A. 2001, 98: 8018-8023. 10.1073/pnas.131209798.
Hendrix MJ, Seftor EA, Hess AR, Seftor RE: Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat Rev Cancer. 2003, 3: 411-421. 10.1038/nrc1092.
Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe'er J, Trent JM, Meltzer PS, Hendrix MJ: Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999, 155: 739-752.
Thies A, Mangold U, Moll I, Schumacher U: PAS-positive loops and networks as a prognostic indicator in cutaneous malignant melanoma. J Pathol. 2001, 195: 537-542. 10.1002/path.988.
Makitie T, Summanen P, Tarkkanen A, Kivela T: Microvascular loops and networks as prognostic indicators in choroidal and ciliary body melanomas. J Natl Cancer Inst. 1999, 91: 359-367. 10.1093/jnci/91.4.359.
Kerbel R, Folkman J: Clinical translation of angiogenesis inhibitors. Nat Rev Cancer. 2002, 2: 727-739. 10.1038/nrc905.
Kisker O, Becker CM, Prox D, Fannon M, D'Amato R, Flynn E, Fogler WE, Sim BK, Allred EN, Pirie-Shepherd SR, Folkman J: Continuous administration of endostatin by intraperitoneally implanted osmotic pump improves the efficacy and potency of therapy in a mouse xenograft tumor model. Cancer Res. 2001, 61: 7669-7674.
Bergers G, Benjamin LE: Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003, 3: 401-410. 10.1038/nrc1093.
Folkman J, Kalluri R: Cancer without disease. Nature. 2004, 427: 787-10.1038/427787a.
Marx J: Angiogenesis. A boost for tumor starvation. Science. 2003, 301: 452-454. 10.1126/science.301.5632.452.
Liau G, Su EJ, Dixon KD: Clinical efforts to modulate angiogenesis in the adult: gene therapy versus conventional approaches. Drug Discov Today. 2001, 6: 689-697. 10.1016/S1359-6446(01)01809-8.
Volpert OV, Alani RM: Wiring the angiogenic switch: Ras, Myc, and Thrombospondin-1. Cancer Cell. 2003, 3: 199-200. 10.1016/S1535-6108(03)00056-4.
de Fraipont F, Nicholson AC, Feige JJ, Van Meir EG: Thrombospondins and tumor angiogenesis. Trends Mol Med. 2001, 7: 401-407. 10.1016/S1471-4914(01)02102-5.
Vailhe B, Feige JJ: Thrombospondins as anti-angiogenic therapeutic agents. Curr Pharm Des. 2003, 9: 583-588.
Jin RJ, Kwak C, Lee SG, Lee CH, Soo CG, Park MS, Lee E, Lee SE: The application of an anti-angiogenic gene (thrombospondin-1) in the treatment of human prostate cancer xenografts. Cancer Gene Ther. 2000, 7: 1537-1542. 10.1038/sj.cgt.7700266.
Liu P, Wang Y, Li YH, Yang C, Zhou YL, Li B, Lu SH, Yang RC, Cai YL, Tobelem G, Caen J, Han ZC: Adenovirus-mediated gene therapy with an antiangiogenic fragment of thrombospondin-1 inhibits human leukemia xenograft growth in nude mice. Leuk Res. 2003, 27: 701-708. 10.1016/S0145-2126(02)00346-6.
O'Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J: Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997, 88: 277-285. 10.1016/S0092-8674(00)81848-6.
Hamano Y, Zeisberg M, Sugimoto H, Lively JC, Maeshima Y, Yang C, Hynes RO, Werb Z, Sudhakar A, Kalluri R: Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell. 2003, 3: 589-601. 10.1016/S1535-6108(03)00133-8.
Kim YM, Hwang S, Pyun BJ, Kim TY, Lee ST, Gho YS, Kwon YG: Endostatin blocks vascular endothelial growth factor-mediated signaling via direct interaction with KDR/Flk-1. J Biol Chem. 2002, 277: 27872-27879. 10.1074/jbc.M202771200.
Hanai J, Dhanabal M, Karumanchi SA, Albanese C, Waterman M, Chan B, Ramchandran R, Pestell R, Sukhatme VP: Endostatin causes G1 arrest of endothelial cells through inhibition of cyclin D1. J Biol Chem. 2002, 277: 16464-16469. 10.1074/jbc.M112274200.
Folkman J: Angiogenesis inhibitors: a new class of drugs. Cancer Biol Ther. 2003, 2: S127-33.
Herbst RS, Hess KR, Tran HT, Tseng JE, Mullani NA, Charnsangavej C, Madden T, Davis DW, McConkey DJ, O'Reilly MS, Ellis LM, Pluda J, Hong WK, Abbruzzese JL: Phase I study of recombinant human endostatin in patients with advanced solid tumors. J Clin Oncol. 2002, 20: 3792-3803. 10.1200/JCO.2002.11.061.
Thomas JP, Arzoomanian RZ, Alberti D, Marnocha R, Lee F, Friedl A, Tutsch K, Dresen A, Geiger P, Pluda J, Fogler W, Schiller JH, Wilding G: Phase I pharmacokinetic and pharmacodynamic study of recombinant human endostatin in patients with advanced solid tumors. J Clin Oncol. 2003, 21: 223-231. 10.1200/JCO.2003.12.120.
Feldman AL, Restifo NP, Alexander HR, Bartlett DL, Hwu P, Seth P, Libutti SK: Antiangiogenic gene therapy of cancer utilizing a recombinant adenovirus to elevate systemic endostatin levels in mice. Cancer Res. 2000, 60: 1503-1506.
Feldman AL, Alexander HR, Hewitt SM, Lorang D, Thiruvathukal CE, Turner EM, Libutti SK: Effect of retroviral endostatin gene transfer on subcutaneous and intraperitoneal growth of murine tumors. J Natl Cancer Inst. 2001, 93: 1014-1020. 10.1093/jnci/93.13.1014.
Kalluri R: Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer. 2003, 3: 422-433. 10.1038/nrc1094.
Maeshima Y, Colorado PC, Torre A, Holthaus KA, Grunkemeyer JA, Ericksen MB, Hopfer H, Xiao Y, Stillman IE, Kalluri R: Distinct antitumor properties of a type IV collagen domain derived from basement membrane. J Biol Chem. 2000, 275: 21340-21348. 10.1074/jbc.M001956200.
Maeshima Y, Colorado PC, Kalluri R: Two RGD-independent alpha vbeta 3 integrin binding sites on tumstatin regulate distinct anti-tumor properties. J Biol Chem. 2000, 275: 23745-23750. 10.1074/jbc.C000186200.
Maeshima Y, Manfredi M, Reimer C, Holthaus KA, Hopfer H, Chandamuri BR, Kharbanda S, Kalluri R: Identification of the anti-angiogenic site within vascular basement membrane-derived tumstatin. J Biol Chem. 2001, 276: 15240-15248. 10.1074/jbc.M007764200.
Colorado PC, Torre A, Kamphaus G, Maeshima Y, Hopfer H, Takahashi K, Volk R, Zamborsky ED, Herman S, Sarkar PK, Ericksen MB, Dhanabal M, Simons M, Post M, Kufe DW, Weichselbaum RR, Sukhatme VP, Kalluri R: Anti-angiogenic cues from vascular basement membrane collagen. Cancer Res. 2000, 60: 2520-2526.
Kamphaus GD, Colorado PC, Panka DJ, Hopfer H, Ramchandran R, Torre A, Maeshima Y, Mier JW, Sukhatme VP, Kalluri R: Canstatin, a novel matrix-derived inhibitor of angiogenesis and tumor growth. J Biol Chem. 2000, 275: 1209-1215. 10.1074/jbc.275.2.1209.
Xu R, Yao ZY, Xin L, Zhang Q, Li TP, Gan RB: NC1 domain of human type VIII collagen (alpha 1) inhibits bovine aortic endothelial cell proliferation and causes cell apoptosis. Biochem Biophys Res Commun. 2001, 289: 264-268. 10.1006/bbrc.2001.5970.
Ramchandran R, Dhanabal M, Volk R, Waterman MJ, Segal M, Lu H, Knebelmann B, Sukhatme VP: Antiangiogenic activity of restin, NC10 domain of human collagen XV: comparison to endostatin. Biochem Biophys Res Commun. 1999, 255: 735-739. 10.1006/bbrc.1999.0248.
O'Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Cao Y, Moses M, Lane WS, Sage EH, Folkman J: Angiostatin: a circulating endothelial cell inhibitor that suppresses angiogenesis and tumor growth. Cold Spring Harb Symp Quant Biol. 1994, 59: 471-482.
O'Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, Lane WS, Cao Y, Sage EH, Folkman J: Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994, 79: 315-328. 10.1016/0092-8674(94)90200-3.
O'Reilly MS, Holmgren L, Chen C, Folkman J: Angiostatin induces and sustains dormancy of human primary tumors in mice. Nat Med. 1996, 2: 689-692.
Wahl ML, Moser TL, Pizzo SV: Angiostatin and Anti-angiogenic Therapy in Human Disease. Recent Prog Horm Res. 2004, 59: 73-104. 10.1210/rp.59.1.73.
Beerepoot LV, Witteveen EO, Groenewegen G, Fogler WE, Sim BK, Sidor C, Zonnenberg BA, Schramel F, Gebbink MF, Voest EE: Recombinant human angiostatin by twice-daily subcutaneous injection in advanced cancer: a pharmacokinetic and long-term safety study. Clin Cancer Res. 2003, 9: 4025-4033.
Lalani AS, Chang B, Lin J, Case SS, Luan B, Wu-Prior WW, VanRoey M, Jooss K: Anti-tumor efficacy of human angiostatin using liver-mediated adeno-associated virus gene therapy. Mol Ther. 2004, 9: 56-66. 10.1016/j.ymthe.2003.10.001.
Galaup A, Opolon P, Bouquet C, Li H, Opolon D, Bissery MC, Tursz T, Perricaudet M, Griscelli F: Combined effects of docetaxel and angiostatin gene therapy in prostate tumor model. Mol Ther. 2003, 7: 731-740. 10.1016/S1525-0016(03)00121-7.
Clapp C, Martial JA, Guzman RC, Rentier-Delure F, Weiner RI: The 16-kilodalton N-terminal fragment of human prolactin is a potent inhibitor of angiogenesis. Endocrinology. 1993, 133: 1292-1299. 10.1210/en.133.3.1292.
Tabruyn SP, Sorlet CM, Rentier-Delrue F, Bours V, Weiner RI, Martial JA, Struman I: The antiangiogenic factor 16K human prolactin induces caspase-dependent apoptosis by a mechanism that requires activation of nuclear factor-kappaB. Mol Endocrinol. 2003, 17: 1815-1823. 10.1210/me.2003-0132.
Kim J, Luo W, Chen DT, Earley K, Tunstead J, Yu-Lee LY, Lin SH: Antitumor activity of the 16-kDa prolactin fragment in prostate cancer. Cancer Res. 2003, 63: 386-393.
Bentzien F, Struman I, Martini JF, Martial J, Weiner R: Expression of the antiangiogenic factor 16K hPRL in human HCT116 colon cancer cells inhibits tumor growth in Rag1(-/-) mice. Cancer Res. 2001, 61: 7356-7362.
Maione TE, Gray GS, Petro J, Hunt AJ, Donner AL, Bauer SI, Carson HF, Sharpe RJ: Inhibition of angiogenesis by recombinant human platelet factor-4 and related peptides. Science. 1990, 247: 77-79.
Sharpe RJ, Byers HR, Scott CF, Bauer SI, Maione TE: Growth inhibition of murine melanoma and human colon carcinoma by recombinant human platelet factor 4. J Natl Cancer Inst. 1990, 82: 848-853.
Kolber DL, Knisely TL, Maione TE: Inhibition of development of murine melanoma lung metastases by systemic administration of recombinant platelet factor 4. J Natl Cancer Inst. 1995, 87: 304-309.
Belman N, Bonnem EM, Harvey HA, Lipton A: Phase I trial of recombinant platelet factor 4 (rPF4) in patients with advanced colorectal carcinoma. Invest New Drugs. 1996, 14: 387-389.
Tanaka T, Manome Y, Wen P, Kufe DW, Fine HA: Viral vector-mediated transduction of a modified platelet factor 4 cDNA inhibits angiogenesis and tumor growth. Nat Med. 1997, 3: 437-442.
Li Y, Jin Y, Chen H, Jie G, Tobelem G, Caen JP, Han ZC: Suppression of tumor growth by viral vector-mediated gene transfer of N-terminal truncated platelet factor 4. Cancer Biother Radiopharm. 2003, 18: 829-840. 10.1089/108497803770418373.
Neville LF, Mathiak G, Bagasra O: The immunobiology of interferon-gamma inducible protein 10 kD (IP-10): a novel, pleiotropic member of the C-X-C chemokine superfamily. Cytokine Growth Factor Rev. 1997, 8: 207-219. 10.1016/S1359-6101(97)00015-4.
Feldman AL, Friedl J, Lans TE, Libutti SK, Lorang D, Miller MS, Turner EM, Hewitt SM, Alexander HR: Retroviral gene transfer of interferon-inducible protein 10 inhibits growth of human melanoma xenografts. Int J Cancer. 2002, 99: 149-153. 10.1002/ijc.10292.
Jones PF: Not just angiogenesis--wider roles for the angiopoietins. J Pathol. 2003, 201: 515-527. 10.1002/path.1452.
Lin P, Buxton JA, Acheson A, Radziejewski C, Maisonpierre PC, Yancopoulos GD, Channon KM, Hale LP, Dewhirst MW, George SE, Peters KG: Antiangiogenic gene therapy targeting the endothelium-specific receptor tyrosine kinase Tie2. Proc Natl Acad Sci U S A. 1998, 95: 8829-8834. 10.1073/pnas.95.15.8829.
Trinchieri G: Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003, 3: 133-146. 10.1038/nri1001.
Voest EE, Kenyon BM, O'Reilly MS, Truitt G, D'Amato RJ, Folkman J: Inhibition of angiogenesis in vivo by interleukin 12. J Natl Cancer Inst. 1995, 87: 581-586.
Brunda MJ, Luistro L, Warrier RR, Wright RB, Hubbard BR, Murphy M, Wolf SF, Gately MK: Antitumor and antimetastatic activity of interleukin 12 against murine tumors. J Exp Med. 1993, 178: 1223-1230. 10.1084/jem.178.4.1223.
Portielje JE, Kruit WH, Schuler M, Beck J, Lamers CH, Stoter G, Huber C, de Boer-Dennert M, Rakhit A, Bolhuis RL, Aulitzky WE: Phase I study of subcutaneously administered recombinant human interleukin 12 in patients with advanced renal cell cancer. Clin Cancer Res. 1999, 5: 3983-3989.
Gollob JA, Mier JW, Veenstra K, McDermott DF, Clancy D, Clancy M, Atkins MB: Phase I trial of twice-weekly intravenous interleukin 12 in patients with metastatic renal cell cancer or malignant melanoma: ability to maintain IFN-gamma induction is associated with clinical response. Clin Cancer Res. 2000, 6: 1678-1692.
Hurteau JA, Blessing JA, DeCesare SL, Creasman WT: Evaluation of recombinant human interleukin-12 in patients with recurrent or refractory ovarian cancer: a gynecologic oncology group study. Gynecol Oncol. 2001, 82: 7-10. 10.1006/gyno.2001.6255.
Mazzolini G, Prieto J, Melero I: Gene therapy of cancer with interleukin-12. Curr Pharm Des. 2003, 9: 1981-1991.
Caruso M, Pham-Nguyen K, Kwong YL, Xu B, Kosai KI, Finegold M, Woo SL, Chen SH: Adenovirus-mediated interleukin-12 gene therapy for metastatic colon carcinoma. Proc Natl Acad Sci U S A. 1996, 93: 11302-11306. 10.1073/pnas.93.21.11302.
Cao R, Farnebo J, Kurimoto M, Cao Y: Interleukin-18 acts as an angiogenesis and tumor suppressor. Faseb J. 1999, 13: 2195-2202.
Nagai H, Hara I, Horikawa T, Oka M, Kamidono S, Ichihashi M: Gene transfer of secreted-type modified interleukin-18 gene to B16F10 melanoma cells suppresses in vivo tumor growth through inhibition of tumor vessel formation. J Invest Dermatol. 2002, 119: 541-548. 10.1046/j.1523-1747.2002.01866.x.
Liu Y, Huang H, Saxena A, Xiang J: Intratumoral coinjection of two adenoviral vectors expressing functional interleukin-18 and inducible protein-10, respectively, synergizes to facilitate regression of established tumors. Cancer Gene Ther. 2002, 9: 533-542. 10.1038/sj.cgt.7700466.
Sidky YA, Borden EC: Inhibition of angiogenesis by interferons: effects on tumor- and lymphocyte-induced vascular responses. Cancer Res. 1987, 47: 5155-5161.
Dvorak HF, Gresser I: Microvascular injury in pathogenesis of interferon-induced necrosis of subcutaneous tumors in mice. J Natl Cancer Inst. 1989, 81: 497-502.
Singh RK, Gutman M, Bucana CD, Sanchez R, Llansa N, Fidler IJ: Interferons alpha and beta down-regulate the expression of basic fibroblast growth factor in human carcinomas. Proc Natl Acad Sci U S A. 1995, 92: 4562-4566.
Lindner DJ: Interferons as antiangiogenic agents. Curr Oncol Rep. 2002, 4: 510-514.
Albini A, Marchisone C, Del Grosso F, Benelli R, Masiello L, Tacchetti C, Bono M, Ferrantini M, Rozera C, Truini M, Belardelli F, Santi L, Noonan DM: Inhibition of angiogenesis and vascular tumor growth by interferon-producing cells: A gene therapy approach. Am J Pathol. 2000, 156: 1381-1393.
Berger AC, Alexander HR, Tang G, Wu PS, Hewitt SM, Turner E, Kruger E, Figg WD, Grove A, Kohn E, Stern D, Libutti SK: Endothelial monocyte activating polypeptide II induces endothelial cell apoptosis and may inhibit tumor angiogenesis. Microvasc Res. 2000, 60: 70-80. 10.1006/mvre.2000.2249.
Wu PC, Alexander HR, Huang J, Hwu P, Gnant M, Berger AC, Turner E, Wilson O, Libutti SK: In vivo sensitivity of human melanoma to tumor necrosis factor (TNF)-alpha is determined by tumor production of the novel cytokine endothelial-monocyte activating polypeptide II (EMAPII). Cancer Res. 1999, 59: 205-212.
Gnant MF, Berger AC, Huang J, Puhlmann M, Wu PC, Merino MJ, Bartlett DL, Alexander H. R., Jr., Libutti SK: Sensitization of tumor necrosis factor alpha-resistant human melanoma by tumor-specific in vivo transfer of the gene encoding endothelial monocyte-activating polypeptide II using recombinant vaccinia virus. Cancer Res. 1999, 59: 4668-4674.
Jiang Y, Goldberg ID, Shi YE: Complex roles of tissue inhibitors of metalloproteinases in cancer. Oncogene. 2002, 21: 2245-2252. 10.1038/sj.onc.1205291.
Wang M, Liu YE, Greene J, Sheng S, Fuchs A, Rosen EM, Shi YE: Inhibition of tumor growth and metastasis of human breast cancer cells transfected with tissue inhibitor of metalloproteinase 4. Oncogene. 1997, 14: 2767-2774. 10.1038/sj.onc.1201245.
ten Hagen TL, Eggermont AM: Solid tumor therapy: manipulation of the vasculature with TNF. Technol Cancer Res Treat. 2003, 2: 195-203.
Senzer N, Mani S, Rosemurgy A, Nemunaitis J, Cunningham C, Guha C, Bayol N, Gillen M, Chu K, Rasmussen C, Rasmussen H, Kufe D, Weichselbaum R, Hanna N: TNFerade biologic, an adenovector with a radiation-inducible promoter, carrying the human tumor necrosis factor alpha gene: a phase I study in patients with solid tumors. J Clin Oncol. 2004, 22: 592-601. 10.1200/JCO.2004.01.227.
Grant SW, Kyshtoobayeva AS, Kurosaki T, Jakowatz J, Fruehauf JP: Mutant p53 correlates with reduced expression of thrombospondin-1, increased angiogenesis, and metastatic progression in melanoma. Cancer Detect Prev. 1998, 22: 185-194. 10.1046/j.1525-1500.1998.0oa18.x.
Van Meir EG, Polverini PJ, Chazin VR, Su Huang HJ, de Tribolet N, Cavenee WK: Release of an inhibitor of angiogenesis upon induction of wild type p53 expression in glioblastoma cells. Nat Genet. 1994, 8: 171-176.
Holmgren L, Jackson G, Arbiser J: p53 induces angiogenesis-restricted dormancy in a mouse fibrosarcoma. Oncogene. 1998, 17: 819-824. 10.1038/sj.onc.1201993.
Su JD, Mayo LD, Donner DB, Durden DL: PTEN and phosphatidylinositol 3'-kinase inhibitors up-regulate p53 and block tumor-induced angiogenesis: evidence for an effect on the tumor and endothelial compartment. Cancer Res. 2003, 63: 3585-3592.
Szary J, Szala S: Intra-tumoral administration of naked plasmid DNA encoding mouse endostatin inhibits renal carcinoma growth. Int J Cancer. 2001, 91: 835-839. 10.1002/1097-0215(200002)9999:9999<::AID-IJC1123>3.3.CO;2-K.
Oga M, Takenaga K, Sato Y, Nakajima H, Koshikawa N, Osato K, Sakiyama S: Inhibition of metastatic brain tumor growth by intramuscular administration of the endostatin gene. Int J Oncol. 2003, 23: 73-79.
Blezinger P, Wang J, Gondo M, Quezada A, Mehrens D, French M, Singhal A, Sullivan S, Rolland A, Ralston R, Min W: Systemic inhibition of tumor growth and tumor metastases by intramuscular administration of the endostatin gene. Nat Biotechnol. 1999, 17: 343-348. 10.1038/7895.
Barron LG, Uyechi LS, Szoka F. C., Jr.: Cationic lipids are essential for gene delivery mediated by intravenous administration of lipoplexes. Gene Ther. 1999, 6: 1179-1183. 10.1038/sj.gt.3300929.
Marty C, Odermatt B, Schott H, Neri D, Ballmer-Hofer K, Klemenz R, Schwendener RA: Cytotoxic targeting of F9 teratocarcinoma tumours with anti-ED-B fibronectin scFv antibody modified liposomes. Br J Cancer. 2002, 87: 106-112. 10.1038/sj.bjc.6600423.
Templeton NS, Lasic DD, Frederik PM, Strey HH, Roberts DD, Pavlakis GN: Improved DNA: liposome complexes for increased systemic delivery and gene expression. Nat Biotechnol. 1997, 15: 647-652.
Rodolfo M, Cato EM, Soldati S, Ceruti R, Asioli M, Scanziani E, Vezzoni P, Parmiani G, Sacco MG: Growth of human melanoma xenografts is suppressed by systemic angiostatin gene therapy. Cancer Gene Ther. 2001, 8: 491-496. 10.1038/sj.cgt.7700331.
Chen QR, Kumar D, Stass SA, Mixson AJ: Liposomes complexed to plasmids encoding angiostatin and endostatin inhibit breast cancer in nude mice. Cancer Res. 1999, 59: 3308-3312.
Thurston G, McLean JW, Rizen M, Baluk P, Haskell A, Murphy TJ, Hanahan D, McDonald DM: Cationic liposomes target angiogenic endothelial cells in tumors and chronic inflammation in mice. J Clin Invest. 1998, 101: 1401-1413.
Kondo M, Asai T, Katanasaka Y, Sadzuka Y, Tsukada H, Ogino K, Taki T, Baba K, Oku N: Anti-neovascular therapy by liposomal drug targeted to membrane type-1 matrix metalloproteinase. Int J Cancer. 2004, 108: 301-306. 10.1002/ijc.11526.
Janssen AP, Schiffelers RM, ten Hagen TL, Koning GA, Schraa AJ, Kok RJ, Storm G, Molema G: Peptide-targeted PEG-liposomes in anti-angiogenic therapy. Int J Pharm. 2003, 254: 55-58. 10.1016/S0378-5173(02)00682-8.
Pastorino F, Brignole C, Marimpietri D, Cilli M, Gambini C, Ribatti D, Longhi R, Allen TM, Corti A, Ponzoni M: Vascular damage and anti-angiogenic effects of tumor vessel-targeted liposomal chemotherapy. Cancer Res. 2003, 63: 7400-7409.
Gu ZP, Wang YJ, Li JG, Zhou YA: VEGF165 antisense RNA suppresses oncogenic properties of human esophageal squamous cell carcinoma. World J Gastroenterol. 2002, 8: 44-48.
Im SA, Gomez-Manzano C, Fueyo J, Liu TJ, Ke LD, Kim JS, Lee HY, Steck PA, Kyritsis AP, Yung WK: Antiangiogenesis treatment for gliomas: transfer of antisense-vascular endothelial growth factor inhibits tumor growth in vivo. Cancer Res. 1999, 59: 895-900.
Marchand GS, Noiseux N, Tanguay JF, Sirois MG: Blockade of in vivo VEGF-mediated angiogenesis by antisense gene therapy: role of Flk-1 and Flt-1 receptors. Am J Physiol Heart Circ Physiol. 2002, 282: H194-204.
Shim WS, Teh M, Mack PO, Ge R: Inhibition of angiopoietin-1 expression in tumor cells by an antisense RNA approach inhibited xenograft tumor growth in immunodeficient mice. Int J Cancer. 2001, 94: 6-15. 10.1002/ijc.1428.
Dallabrida SM, De Sousa MA, Farrell DH: Expression of antisense to integrin subunit beta 3 inhibits microvascular endothelial cell capillary tube formation in fibrin. J Biol Chem. 2000, 275: 32281-32288. 10.1074/jbc.M001446200.
Lipscomb EA, Dugan AS, Rabinovitz I, Mercurio AM: Use of RNA interference to inhibit integrin (alpha6beta4)-mediated invasion and migration of breast carcinoma cells. Clin Exp Metastasis. 2003, 20: 569-576. 10.1023/A:1025819521707.
Yang G, Cai KQ, Thompson-Lanza JA, Bast R. C., Jr., Liu J: Inhibition of Breast and Ovarian Tumor Growth through Multiple Signaling Pathways by Using Retrovirus-mediated Small Interfering RNA against Her-2/neu Gene Expression. J Biol Chem. 2004, 279: 4339-4345. 10.1074/jbc.M311153200.
Brummelkamp TR, Bernards R, Agami R: A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002, 296: 550-553. 10.1126/science.1068999.
Zhang L, Yang N, Mohamed-Hadley A, Rubin SC, Coukos G: Vector-based RNAi, a novel tool for isoform-specific knock-down of VEGF and anti-angiogenesis gene therapy of cancer. Biochem Biophys Res Commun. 2003, 303: 1169-1178. 10.1016/S0006-291X(03)00495-9.
Thomas CE, Ehrhardt A, Kay MA: Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003, 4: 346-358. 10.1038/nrg1066.
Reynolds PN, Zinn KR, Gavrilyuk VD, Balyasnikova IV, Rogers BE, Buchsbaum DJ, Wang MH, Miletich DJ, Grizzle WE, Douglas JT, Danilov SM, Curiel DT: A targetable, injectable adenoviral vector for selective gene delivery to pulmonary endothelium in vivo. Mol Ther. 2000, 2: 562-578. 10.1006/mthe.2000.0205.
Yotnda P, Chen DH, Chiu W, Piedra PA, Davis A, Templeton NS, Brenner MK: Bilamellar cationic liposomes protect adenovectors from preexisting humoral immune responses. Mol Ther. 2002, 5: 233-241. 10.1006/mthe.2002.0545.
Logg CR, Kasahara N: Retrovirus-mediated gene transfer to tumors: utilizing the replicative power of viruses to achieve highly efficient tumor transduction in vivo. Methods Mol Biol. 2004, 246: 499-525.
Rollins SA, Birks CW, Setter E, Squinto SP, Rother RP: Retroviral vector producer cell killing in human serum is mediated by natural antibody and complement: strategies for evading the humoral immune response. Hum Gene Ther. 1996, 7: 619-626.
Ghazizadeh S, Carroll JM, Taichman LB: Repression of retrovirus-mediated transgene expression by interferons: implications for gene therapy. J Virol. 1997, 71: 9163-9169.
Smith-Arica JR, Bartlett JS: Gene therapy: recombinant adeno-associated virus vectors. Curr Cardiol Rep. 2001, 3: 43-49.
White SJ, Nicklin SA, Buning H, Brosnan MJ, Leike K, Papadakis ED, Hallek M, Baker AH: Targeted gene delivery to vascular tissue in vivo by tropism-modified adeno-associated virus vectors. Circulation. 2004, 109: 513-519. 10.1161/01.CIR.0000109697.68832.5D.
Giles J: Protests win reprieve for renowned medical lab. Nature. 2003, 423: 573-
Kay MA, Nakai H: Looking into the safety of AAV vectors. Nature. 2003, 424: 251-10.1038/424251b.
Shichinohe T, Bochner BH, Mizutani K, Nishida M, Hegerich-Gilliam S, Naldini L, Kasahara N: Development of lentiviral vectors for antiangiogenic gene delivery. Cancer Gene Ther. 2001, 8: 879-889. 10.1038/sj.cgt.7700388.
Pfeifer A, Kessler T, Silletti S, Cheresh DA, Verma IM: Suppression of angiogenesis by lentiviral delivery of PEX, a noncatalytic fragment of matrix metalloproteinase 2. Proc Natl Acad Sci U S A. 2000, 97: 12227-12232. 10.1073/pnas.220399597.
Latchman DS: Herpes simplex virus vectors for gene therapy. Mol Biotechnol. 1994, 2: 179-195.
Rabinowitz JE, Rolling F, Li C, Conrath H, Xiao W, Xiao X, Samulski RJ: Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J Virol. 2002, 76: 791-801. 10.1128/JVI.76.2.791-801.2002.
Khare PD, Liao S, Hirose Y, Kuroki M, Fujimura S, Yamauchi Y, Miyajima-Uchida H: Tumor growth suppression by a retroviral vector displaying scFv antibody to CEA and carrying the iNOS gene. Anticancer Res. 2002, 22: 2443-2446.
Reynolds PN, Nicklin SA, Kaliberova L, Boatman BG, Grizzle WE, Balyasnikova IV, Baker AH, Danilov SM, Curiel DT: Combined transductional and transcriptional targeting improves the specificity of transgene expression in vivo. Nat Biotechnol. 2001, 19: 838-842. 10.1038/nbt0901-838.
Varda-Bloom N, Shaish A, Gonen A, Levanon K, Greenbereger S, Ferber S, Levkovitz H, Castel D, Goldberg I, Afek A, Kopolovitc Y, Harats D: Tissue-specific gene therapy directed to tumor angiogenesis. Gene Ther. 2001, 8: 819-827. 10.1038/sj.gt.3301472.
Thorpe PE: Vascular targeting agents as cancer therapeutics. Clin Cancer Res. 2004, 10: 415-427.
Tozer GM, Kanthou C, Parkins CS, Hill SA: The biology of the combretastatins as tumour vascular targeting agents. Int J Exp Pathol. 2002, 83: 21-38. 10.1046/j.1365-2613.2002.00211.x.
Pasqualini R, Koivunen E, Ruoslahti E: Alpha v integrins as receptors for tumor targeting by circulating ligands. Nat Biotechnol. 1997, 15: 542-546.
Arap W, Pasqualini R, Ruoslahti E: Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 1998, 279: 377-380. 10.1126/science.279.5349.377.
Laakkonen P, Porkka K, Hoffman JA, Ruoslahti E: A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nat Med. 2002, 8: 751-755.
Koivunen E, Arap W, Valtanen H, Rainisalo A, Medina OP, Heikkila P, Kantor C, Gahmberg CG, Salo T, Konttinen YT, Sorsa T, Ruoslahti E, Pasqualini R: Tumor targeting with a selective gelatinase inhibitor. Nat Biotechnol. 1999, 17: 768-774. 10.1038/11703.
Brooks PC, Stromblad S, Sanders LC, von Schalscha TL, Aimes RT, Stetler-Stevenson WG, Quigley JP, Cheresh DA: Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell. 1996, 85: 683-693. 10.1016/S0092-8674(00)81235-0.
Folkman J: Angiogenic zip code. Nat Biotechnol. 1999, 17: 749-10.1038/11676.
Masson-Gadais B, Houle F, Laferriere J, Huot J: Integrin alphavbeta3, requirement for VEGFR2-mediated activation of SAPK2/p38 and for Hsp90-dependent phosphorylation of focal adhesion kinase in endothelial cells activated by VEGF. Cell Stress Chaperones. 2003, 8: 37-52. 10.1379/1466-1268(2003)8<37:IVRFVA>2.0.CO;2.
Marshall JF, Hart IR: The role of alpha v-integrins in tumour progression and metastasis. Semin Cancer Biol. 1996, 7: 129-138. 10.1006/scbi.1996.0018.
Eliceiri BP, Cheresh DA: The role of alphav integrins during angiogenesis: insights into potential mechanisms of action and clinical development. J Clin Invest. 1999, 103: 1227-1230.
Ruegg C, Yilmaz A, Bieler G, Bamat J, Chaubert P, Lejeune FJ: Evidence for the involvement of endothelial cell integrin alphaVbeta3 in the disruption of the tumor vasculature induced by TNF and IFN-gamma. Nat Med. 1998, 4: 408-414.
Thomas GJ, Lewis MP, Whawell SA, Russell A, Sheppard D, Hart IR, Speight PM, Marshall JF: Expression of the alphavbeta6 integrin promotes migration and invasion in squamous carcinoma cells. J Invest Dermatol. 2001, 117: 67-73. 10.1046/j.0022-202x.2001.01379.x.
Larocca D, Kassner PD, Witte A, Ladner RC, Pierce GF, Baird A: Gene transfer to mammalian cells using genetically targeted filamentous bacteriophage. Faseb J. 1999, 13: 727-734.
Poul MA, Marks JD: Targeted gene delivery to mammalian cells by filamentous bacteriophage. J Mol Biol. 1999, 288: 203-211. 10.1006/jmbi.1999.2678.
Di Giovine M, Salone B, Martina Y, Amati V, Zambruno G, Cundari E, Failla CM, Saggio I: Binding properties, cell delivery, and gene transfer of adenoviral penton base displaying bacteriophage. Virology. 2001, 282: 102-112. 10.1006/viro.2000.0809.
Burg MA, Jensen-Pergakes K, Gonzalez AM, Ravey P, Baird A, Larocca D: Enhanced phagemid particle gene transfer in camptothecin-treated carcinoma cells. Cancer Res. 2002, 62: 977-981.
Moghimi SM, Hunter AC, Murray JC: Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev. 2001, 53: 283-318.
Vinogradov SV, Bronich TK, Kabanov AV: Nanosized cationic hydrogels for drug delivery: preparation, properties and interactions with cells. Adv Drug Deliv Rev. 2002, 54: 135-147. 10.1016/S0169-409X(01)00245-9.
Panyam J, Labhasetwar V: Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003, 55: 329-347. 10.1016/S0169-409X(02)00228-4.
Anderson SA, Rader RK, Westlin WF, Null C, Jackson D, Lanza GM, Wickline SA, Kotyk JJ: Magnetic resonance contrast enhancement of neovasculature with alpha(v)beta(3)-targeted nanoparticles. Magn Reson Med. 2000, 44: 433-439. 10.1002/1522-2594(200009)44:3<433::AID-MRM14>3.0.CO;2-9.
Artemov D, Mori N, Okollie B, Bhujwalla ZM: MR molecular imaging of the Her-2/neu receptor in breast cancer cells using targeted iron oxide nanoparticles. Magn Reson Med. 2003, 49: 403-408. 10.1002/mrm.10406.
Hood JD, Bednarski M, Frausto R, Guccione S, Reisfeld RA, Xiang R, Cheresh DA: Tumor regression by targeted gene delivery to the neovasculature. Science. 2002, 296: 2404-2407. 10.1126/science.1070200.
Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, Sorensen R, Forster A, Fraser P, Cohen JI, de Saint Basile G, Alexander I, Wintergerst U, Frebourg T, Aurias A, Stoppa-Lyonnet D, Romana S, Radford-Weiss I, Gross F, Valensi F, Delabesse E, Macintyre E, Sigaux F, Soulier J, Leiva LE, Wissler M, Prinz C, Rabbitts TH, Le Deist F, Fischer A, Cavazzana-Calvo M: LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003, 302: 415-419. 10.1126/science.1088547.
Cavazzana-Calvo M, Thrasher A, Mavilio F: The future of gene therapy. Nature. 2004, 427: 779-781. 10.1038/427779a.
Scappaticci FA: Mechanisms and future directions for angiogenesis-based cancer therapies. J Clin Oncol. 2002, 20: 3906-3927. 10.1200/JCO.2002.01.033.
Abdollahi A, Lipson KE, Sckell A, Zieher H, Klenke F, Poerschke D, Roth A, Han X, Krix M, Bischof M, Hahnfeldt P, Grone HJ, Debus J, Hlatky L, Huber PE: Combined therapy with direct and indirect angiogenesis inhibition results in enhanced antiangiogenic and antitumor effects. Cancer Res. 2003, 63: 8890-8898.
Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, Topalian SL, Steinberg SM, Chen HX, Rosenberg SA: A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med. 2003, 349: 427-434. 10.1056/NEJMoa021491.
Abdollahi A, Hahnfeldt P, Maercker C, Grone HJ, Debus J, Ansorge W, Folkman J, Hlatky L, Huber PE: Endostatin's Antiangiogenic Signaling Network. Mol Cell. 2004, 13: 649-663. 10.1016/S1097-2765(04)00102-9.
Sudhakar A, Sugimoto H, Yang C, Lively J, Zeisberg M, Kalluri R: Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc Natl Acad Sci U S A. 2003, 100: 4766-4771. 10.1073/pnas.0730882100.
Panka DJ, Mier JW: Canstatin inhibits Akt activation and induces Fas-dependent apoptosis in endothelial cells. J Biol Chem. 2003, 278: 37632-37636. 10.1074/jbc.M307339200.
Xu R, Xin L, Fan Y, Meng HR, Li ZP, Gan RB: Mouse restin inhibits bovine aortic endothelial cell proliferation and causes cell apoptosis. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao (Shanghai). 2002, 34: 138-142.
Corbacho AM, Martinez De La Escalera G, Clapp C: Roles of prolactin and related members of the prolactin/growth hormone/placental lactogen family in angiogenesis. J Endocrinol. 2002, 173: 219-238.
Bikfalvi A, Gimenez-Gallego G: The control of angiogenesis and tumor invasion by platelet factor-4 and platelet factor-4-derived molecules. Semin Thromb Hemost. 2004, 30: 137-144. 10.1055/s-2004-822978.
Locksley RM, Killeen N, Lenardo MJ: The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001, 104: 487-501. 10.1016/S0092-8674(01)00237-9.
O'Reilly MS, Pirie-Shepherd S, Lane WS, Folkman J: Antiangiogenic activity of the cleaved conformation of the serpin antithrombin. Science. 1999, 285: 1926-1928. 10.1126/science.285.5435.1926.
Zhang M, Volpert O, Shi YH, Bouck N: Maspin is an angiogenesis inhibitor. Nat Med. 2000, 6: 196-199. 10.1038/72303.
Dawson DW, Volpert OV, Gillis P, Crawford SE, Xu H, Benedict W, Bouck NP: Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science. 1999, 285: 245-248. 10.1126/science.285.5425.245.
Zhai Y, Yu J, Iruela-Arispe L, Huang WQ, Wang Z, Hayes AJ, Lu J, Jiang G, Rojas L, Lippman ME, Ni J, Yu GL, Li LY: Inhibition of angiogenesis and breast cancer xenograft tumor growth by VEGI, a novel cytokine of the TNF superfamily. Int J Cancer. 1999, 82: 131-136. 10.1002/(SICI)1097-0215(19990702)82:1<131::AID-IJC22>3.3.CO;2-F.
Yu J, Tian S, Metheny-Barlow L, Chew LJ, Hayes AJ, Pan H, Yu GL, Li LY: Modulation of endothelial cell growth arrest and apoptosis by vascular endothelial growth inhibitor. Circ Res. 2001, 89: 1161-1167.
Mabjeesh NJ, Escuin D, LaVallee TM, Pribluda VS, Swartz GM, Johnson MS, Willard MT, Zhong H, Simons JW, Giannakakou P: 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell. 2003, 3: 363-375. 10.1016/S1535-6108(03)00077-1.
Folkman J, Ingber DE: Angiostatic steroids. Method of discovery and mechanism of action. Ann Surg. 1987, 206: 374-383.
Sage EH, Reed M, Funk SE, Truong T, Steadele M, Puolakkainen P, Maurice DH, Bassuk JA: Cleavage of the matricellular protein SPARC by matrix metalloproteinase 3 produces polypeptides that influence angiogenesis. J Biol Chem. 2003, 278: 37849-37857. 10.1074/jbc.M302946200.
Brekken RA, Sage EH: SPARC, a matricellular protein: at the crossroads of cell-matrix. Matrix Biol. 2000, 19: 569-580. 10.1016/S0945-053X(00)00105-0.
Colman RW, Jameson BA, Lin Y, Johnson D, Mousa SA: Domain 5 of high molecular weight kininogen (kininostatin) down-regulates endothelial cell proliferation and migration and inhibits angiogenesis. Blood. 2000, 95: 543-550.
Guo YL, Wang S, Colman RW: Kininostatin, an angiogenic inhibitor, inhibits proliferation and induces apoptosis of human endothelial cells. Arterioscler Thromb Vasc Biol. 2001, 21: 1427-1433.
Zhang JC, Donate F, Qi X, Ziats NP, Juarez JC, Mazar AP, Pang YP, McCrae KR: The antiangiogenic activity of cleaved high molecular weight kininogen is mediated through binding to endothelial cell tropomyosin. Proc Natl Acad Sci U S A. 2002, 99: 12224-12229. 10.1073/pnas.192668299.
Author information
Authors and Affiliations
Corresponding author
Additional information
Anita Tandle, Dan G Blazer III contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Tandle, A., Blazer, D.G. & Libutti, S.K. Antiangiogenic gene therapy of cancer: recent developments. J Transl Med 2, 22 (2004). https://doi.org/10.1186/1479-5876-2-22
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1479-5876-2-22