
Abstract
Background
These data compare the efficacy and safety of highly purified human-derived follicle-stimulating hormone (Bravelle(R)) and recombinant follitropin-β (Follistim(R)) in women undergoing in vitro fertilization.
Methods
This report describes the pooled data from two, nearly identical, randomized, controlled, parallel-group, multicenter studies conducted in a total of 19 academic and private IVF-ET centers in the United States. Infertile premenopausal women underwent pituitary down-regulation using leuprolide acetate followed by a maximum of 12 days of subcutaneous Bravelle(R) (n = 120) or Follistim(R) (n = 118), followed by administration of human chorionic gonadotropin, oocyte retrieval and embryo transfer. The primary efficacy measure was the mean number of oocytes retrieved; secondary efficacy measures included the total dose and duration of gonadotropin treatment; peak serum estradion levels; embryo transfer and implantation rates; chemical, clinical and continuing pregnancies; and live birth rates. All adverse events were recorded and injection site pain was recorded daily using a patient, self-assessment diary.
Results
Similar efficacy responses were observed for all outcome parameters in the two treatment groups. Although patients receiving Bravelle(R) consistently reported a greater number of chemical, clinical and continuing pregnancies, as well as an increased rate of live birth, the data did not attain statistical significance (P > 0.05). The overall incidence of adverse events was similar in both groups, but compared to Follistim(R), injections of Bravelle(R) were reported by patients to be significantly less painful (P < 0.001).
Conclusions
Bravelle(R) and Follistim(R) had comparable efficacy in controlled ovarian hyperstimulation in women undergoing IVF-ET. There were no differences in the nature or number of adverse events between the treatment groups although Bravelle(R) injections were reported to be significantly less painful.
Similar content being viewed by others


Introduction
Following their discovery in the 1920s, the pituitary gonadotropins, follicle-stimulating hormone (FSH) and luteinizing hormone (LH), were first used successfully for ovulation induction in 1958 [1]. Since then, exogenous hormone preparations of these gonadotropins have been widely used for the treatment of oligoanovulatory infertility, as well as for stimulation of multiple follicular development in assisted reproductive techniques.
The first commercially available gonadotropin preparation, human menopausal gonadotropin (hMG), was purified from the urine of postmenopausal women and contained equal amounts of FSH and LH activity [2]. With the development of sophisticated purification techniques [3], more purified FSH preparations containing small amounts of LH became available in 1986. Thereafter, a highly purified FSH preparation was developed and approved for use in 1996.
Advances in molecular biology techniques made it possible to produce recombinant FSH, which is secreted from genetically engineered Chinese hamster ovary cells [4]. Despite identical amino acid sequences, recombinant and natural FSH preparations have different glycosylation patterns, which are known to influence the isoform profile of each product [5, 6]. The clinical implications of differing isoform profiles for FSH products are unclear and there are no convincing data to suggest marked superiority of either human-derived or animal-derived FSH formulations for ovarian stimulation in terms of efficacy or safety [7–14]. Nonetheless, since subtle differences due exist between human derived and recombinant forms of FSH, efforts continue to identify clinically relevant differences between the two FSH preparations.
This report presents the pooled data from two independent trials, nearly identical in design, which compared the efficacy and safety of highly purified human-derived FSH (Bravelle®) with recombinant follitropin-β (Follistim®), administered subcutaneously in infertile women with regular ovulatory menstrual cycles undergoing in vitro fertilization procedures. The present data form part of the clinical development program describing the comparative efficacy and safety of Bravelle®, the newest approved, highly purified (96–98%), human-derived FSH which contains ≤ 2% LH activity.
Materials and Methods
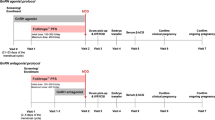
Two randomized, parallel group, clinical trials (one recently published [8]) compared single cycle treatment with subcutaneously administered, purified, human-derived FSH (Bravelle®; n = 120) vs. recombinant follitropin-β (Follistim®; n = 118) in infertile female patients undergoing IVF. Data were collected from 19 IVF-ET centers, each of which obtained institutional review board approval. Eleven centers participated in the first study while 13 centers participated in the second study. Both studies were conducted in accordance with Good Clinical Practice guidelines and written informed consent was obtained from all participants prior to screening and enrollment.
Patients were non-smoking, infertile, premenopausal females, aged 18–39 years, with body mass index (BMI) ≤ 34 kg/m2 and regular ovulatory menstrual cycles (24–35 days). Causes of infertility were attributed to tubal factor, endometriosis (American Fertility Society revised stage I or II) or unexplained. Participants had ultrasound-confirmed normal ovaries with healthy uterus and adnexae. Further eligibility criteria included: unremarkable medical history and physical examination with normal baseline hematology and clinical chemistry; normal range serum estradiol (E2), follicle stimulating hormone (FSH), luteinizing hormone (LH), testosterone, dihydroepiandrosterone sulfate, prolactin and thyroid stimulating hormone. All patients needed to be seronegative for hepatitis B and C, HIV and syphilis and have a negative pregnancy test before starting treatment. Male partner or donor semen was required to meet World Health Organization criteria [15] for normal, within 6 months of study entry.
Patients who fulfilled screening criteria were instructed to inject leuprolide acetate (TAP Pharmaceuticals, Deerfield, IL), 0.5 mg subcutaneously each morning, for pituitary down-regulation. Leuprolide acetate was injected into the thigh or arm, beginning 7 days before the anticipated onset of menses and administration continued for ≤ 20 days or until their serum E2 concentration was ≤ 45 pg/mL and endometrial thickness was ≤ 7 mm on transvaginal ultrasound. If no menstrual bleeding occurred during this time, the patient was withdrawn from the trial, otherwise, leuprolide acetate was continued at the same dose until the day before hCG (Novarel™, Ferring Pharmaceuticals, Inc, Suffern, New York) administration.
Patients who met pituitary down-regulation requirements were randomized, using randomization codes generated with SAS® Proc Plan, to receive Bravelle® (n = 120; Ferring Pharmaceuticals, Inc, Suffern, New York) or Follistim® (n = 118; Organon Inc., West Orange, New Jersey) once daily, in a dose of 225 IU, subcutaneously, for 5 days. Study center staff administered the first dose of gonadotropin and provided guidance on abdominal injection technique, including alternation of injection sites, for subsequent dosing at home. Patients were instructed to maintain a daily, gonadotropin diary of the injection site and the presence and intensity of any pain.
After the initial 5 days of FSH treatment, ovarian response was evaluated by transvaginal ultrasound and serum E2 concentrations. If necessary, the dose of gonadotropin was increased in increments of 75–150 IU/day on alternate days, to a maximum of 450 IU/day. The duration of the controlled ovarian hyperstimulation cycle was not to exceed 12 days. At their discretion, investigators could reduce the dose or discontinue treatment at any time if concerned about patient safety.
Eligibility criteria for Novarel™ administration were the presence of at least three follicles with a diameter of ≥ 16 mm, calculated as the mean of three perpendicular planes by transvaginal ultrasound, along with a serum E2 level that was judged by the investigator to be appropriate for the number of follicles. A single dose of 10,000 IU USP Novarel™ was administered intramuscularly, 1 day after the final dose of gonadotropin. Patients who had not achieved eligibility criteria for Novarel™ treatment following a maximum of 12 days of gonadotropin stimulation were allowed to 'coast' for a further 2 days. If the follicular response remained suboptimal, participants were considered non-responders and were withdrawn from the trial.
Standard site-specific procedures were used for oocyte retrieval and IVF-ET. Intracytoplasmic sperm injection (ICSI) and assisted hatching were prohibited. A maximum of four embryos were allowed to be transferred to each patient. Progesterone for luteal phase support was either self-administered (Crinone™ 8%, 90 mg q.d., Serono Laboratories Inc, Randolph, MA) or administered in the form of progesterone in oil (50 mg, i.m., q.d., Schein Pharmaceuticals, Florham Park, NJ), beginning 2 or 3 days after oocyte retrieval. Luteal phase support was maintained at least until there was fetal heart motion in an intrauterine pregnancy or a negative pregnancy test (β-hCG) was documented. The first serum pregnancy test was performed 14 days after embryo transfer and, if positive, was followed by a confirmatory β-hCG measurement 2 days later. Transvaginal ultrasound was performed approximately 2 weeks later to verify a clinical pregnancy (gestational sac and fetal heart motion) and repeated 1 week later to confirm a continuing pregnancy.
The primary efficacy parameter for this single-cycle analysis was the number of oocytes retrieved. Secondary efficacy variables included the percentage of cycles with oocyte retrieval; duration of gonadotropin therapy; total FSH dose; peak serum E2 levels; embryo transfer and implantation rates; chemical pregnancies (positive serum β-hCG); clinical pregnancies (intrauterine gestational sac with fetal heart motion); continuing pregnancies and live birth rates.
Statistical Analysis
The validity of pooling data from the two independent studies was confirmed by statistical examination, which concluded that the two studies were homogeneous by excluding treatment-by-study interaction for the number of oocytes retrieved, using an ANOVA model with factors for 'treatment', 'study' and 'treatment-by-study' interaction. The treatment-by-study interaction was not statistically significant (P = 0.87) and a fixed effects approach was therefore appropriate for data analysis. Data were pooled and adjusted using the 'study' variable as a fixed effect, to account for between-study variability.
Safety was evaluated by analysis of the nature and incidence of adverse events, irrespective of their relation to study medications. Injection site pain was self-rated using a daily diary with an analog scale of 1 (no pain) to 10 (severe or unbearable pain).
For the first study, sample size calculations were based on the clinical assumption that 10 oocytes per patient would be retrieved in the reference group. Power calculations were based on an α level of 0.05 (two-tailed test) with 80% power to detect a 30% difference with 50 evaluable patients per group. A sample of 60 patients per group was selected. For the second study, the assumption for the number of oocytes retrieved from the reference group was changed to 13.7, based on the results of the first study. The power calculation was based on an α level of 0.025 (one-tailed test) with 80% power to detect a 30% difference with 56 evaluable patients per group.
Continuous variables, including primary and secondary outcome parameters, were analyzed using a two-way ANOVA model with treatment and study as fixed effects. In addition, an analysis of covariance (ANCOVA) model was used to compare primary and secondary efficacy variables after correcting for baseline age and BMI. The difference between treatments (Bravelle® vs. Follistim®) was evaluated using a one-sided, 95% confidence interval. Bravelle® could be declared equivalent in terms of efficacy if the lower limit of the 95% confidence interval was greater than a difference of -2.2 oocytes.
Categorical variables, including adverse events, were compared using Cochran-Mantel-Haenszel (CMH) analyses, stratified by study, and a logistic regression model using treatment, study, baseline age and BMI as covariates.
Injection site pain scores on each day were compared using the two-way ANOVA model with treatment and study as fixed effects and also using a linear mixed model to compare data across all time points. The latter allowed analysis of continuous data to account for intrasubject variability. The Restricted Maximum Likelihood Method was used with a compound symmetric within-subject covariant structure, and the sandwich variance estimator.
Results
A total of 323 patients were enrolled in the two studies and received leuprolide acetate for pituitary down-regulation, 297 were randomized between the study arms. The most common reasons for non-randomization was failure to down-regulate to the protocol-specified criteria or patients had a positive pregnancy test prior to down-regulation on LA therapy. The initial study contained a third arm consisting of 59 patients, which are not included in this pooled analysis. The remaining 238 patients were randomized to receive Bravelle® (n = 120) or Follistim® (n = 118).
In the Bravelle and Follistim groups, 94.2% and 97.5% of women respectively, received Novarel™ and underwent oocyte retrieval. Since nearly all randomized patients received Novarel™, there were no clinically or statistically meaningful differences between the intent-to-treat analysis and the primary efficacy responders (those who received Novarel™) analyses. Therefore, efficacy data presented in this report are based on the primary efficacy responder population for the primary efficacy parameter, number of oocytes retrieved, and subsequent analyses. Baseline demographic characteristics and safety data comparisons are presented for all enrolled patients regardless of administration of Novarel™.
Demographic and baseline data, including endocrine status, primary infertility diagnosis and IVF history are presented in Table 1. The groups were generally well matched demographically, although women in the Bravelle® group had a significantly lower mean weight, and therefore lower BMI (23.3 vs. 24.5 kg/m2, respectively, P = 0.021). The modest difference in BMI was not considered to be clinically meaningful, in addition, for the ANCOVA analysis of efficacy parameters, BMI and age were used as covariates to account for the effect these differences may have had on outcomes.
During the course of treatment, there was no statistically significant differences between groups for the primary efficacy measure, mean number of oocytes retrieved per cycle, the numbers of oocytes that underwent normal fertilization or the number of embryos transferred (Figure 1). The implantation rate did not differ between groups, as it was 21.3% for the Bravelle® group and 20.3% for the Follistim® group.

Clinical response for the 113 patients that received Bravelle® (black bars) and the 115 patients that received Follistim® (gray bars). Mean value per patient of oocytes retrieved, oocytes fertilized and embryos transferred.
No significant differences were observed in the total dose of FSH administered, the duration of treatment or peak estradiol levels between groups (Table 2). Patients receiving Bravelle® had a numerical trend towards higher chemical, clinical, continuing pregnancy rates as well as live birth rates (Figure 2), but the results did not attain statistical significance.

Pregnancy and live birth results for the 113 patients that received Bravelle® (black bars) and the 115 patients that received Follistim® (gray bars).
There were no significant differences in the number of patients reporting adverse events (P = 0.680), severe adverse events (P = 0.307) or serious adverse events (P = 0.161) between the Bravelle® or Follistim® groups. The most frequently reported adverse events were headache, abdominal cramps, vaginal bleeding/spotting and nausea (Table 3). One woman taking Bravelle® experienced a 'severe' adverse event, compared with three patients in the Follistim® group. 'Serious' adverse events were reported by two patients in the Bravelle® group.
A two-way ANOVA model used to compare mean daily injection-site pain revealed a significantly lower pain score in Bravelle® patients on most treatment days and during the entire treatment period (P < 0.05). The difference had greater significance when a linear mixed model, which controls for intrasubject variation, was used to compare data across all time points [3.1 and 3.9, respectively (P < 0.001)] (Figure 3).

Comparison of injection site pain scores on a self-assessment scale of 0 (no pain) to 10 (severe pain) for the 113 patients that received Bravelle® (black bars) and the 115 patients that received Follistim® (gray bars).
Discussion
The data from these two randomized, controlled, clinical studies were used to evaluate the efficacy and safety of a new, highly purified, human derived FSH (Bravelle®) compared to follitropin-β (Follistim®) in patients undergoing controlled ovarian hyperstimulation for IVF-ET. Clinicians continually update their safety and efficacy data as patients are treated, in order to better understand their treatment protocols. This paper is a similar exercise in that it combines two regulatory grade IVF studies to confirm the efficacy and safety results from the previous study (8). The fact that the results and conclusions of the second study are similar to the first further highlights the consistency and reliability of Bravelle® in patients undergoing in vitro fertilization.
The baseline weight and BMI differences between Bravelle® and Follistim® patients were modestly different, but were unlikely to have affected trial outcomes since neither group could be categorized as obese (BMI ≥ 28 kg/m2) or underweight (BMI < 20 kg/m2), variables thought to correlate with reduced success during assisted conception [16–18]. Nonetheless, an ANCOVA analysis was performed using BMI and age as covariates, to ensure that the difference in BMI did not effect outcome measures.
Recombinant DNA engineering technology allows for the production of human FSH in Chinese hamster ovary cells [4]. The secreted protein is collected from the culture medium and purified. Efforts have been made in an attempt to identify differences in efficacy between recombinant and human derived gonadotropin preparations. It is recognized that differences exist in the glycosylation patterns of these preparations [5, 6]. However, collectively, the results from numerous, prospective, clinical studies [7–14], and recent meta-analyses [19, 20] of published data have failed to show any meaningful or reproducible differences.
Nonetheless, it is well known that the success of a controlled ovarian hyperstimulation cycle is in part dependent on the presence of LH. Bravelle® provides a low level of LH activity which might benefit certain patients by enhancing intrafollicular estradiol levels, resulting in the excellent single cycle pregnancy rates observed in this analysis. The presence of small amounts of LH activity may be of specific benefit in patients with low endogenous LH levels that undergo pituitary down regulation. Overall, Bravelle® provides a safe and highly effective alternative to gonadotropins devoid of LH activity.
The adverse event data from this study demonstrate that Bravelle® is at least as well tolerated as Follistim®, but causes significantly less injection-related pain. This finding, reported in a previous study [8] may be related to the purity of Bravelle® and the differences in excipients used in the manufacturing processes for the two compounds. For example, Follistim® contains sodium citrate which has previously been implicated with injection site pain by some [22] while Bravelle® does not.
In conclusion, this analysis further supports the clinical data indicating the comparable efficacy and overall safety of Bravelle® as compared to Follistim® in controlled ovarian hyperstimulation for IVF-ET, and highlights the potential benefit of Bravelle® which produced less injection site pain.
References
Gemzell C, Diczfalusy E, Tillinger K: Clinical effects of human pituitary follicle stimulating hormone. J Clin Endocrinol Metab. 1958, 18: 1333-1348.
Lunenfeld B, Sulimovici S, Rabau E, Eshkol A: L'induction de l'ovulation dans les amenorrhees hypophysaies par un traitement combiné de gonadotropines urinaires ménopausiques et de gonadotropines chorioniques. Soc Fr Gynecol. 1962, 5: 346-351.
Donini P, Puzzuoli D, D'Alessio I, Lunenfeld B, Eshkol A, Parlow AF: Purification and separation of follicle stimulating hormone (FSH) and luteinizing hormone (LH) from human postmenopausal gonadotropin (HMG). II. Preparation of biological apparently pure FSH by selective binding of the LH with an anti-HCG serum and subsequent chromatography. Acta Endocrinol (Copenh). 1966, 52: 186-198.
Sairam M, Sebok K: Pharmacokinetics of natural/recombinant FSH (follitropin) and some analogs. In: Ovulation Induction: Basic Science and Clinical Advances. Edited by: Filicori M, Flamigni C. 1994, Amsterdam: Excerpta Medica, 199-208.
Matikainen T, De Leeuw R, Mannaerts B, Huhtaniemi I: Circulating bioactive and immunoreactive recombinant human follicle stimulating hormone (Org 32489) after administration to gonadotropin-deficient subjects. Fertil Steril. 1994, 61: 62-69.
Fauser B, Rutherford A, Strauss J, Van Steirteghem A, Eds: In: Gonadotrophin receptors: Molecular biology in reproductive medicine. 1999, New York, NY: Parthenon Publishing Group, 165-200.
Selman H, De Santo M, Sterzik K, Coccia E, El-Danasouri I: Effect of highly purified urinary follicle-stimulating hormone on oocyte and embryo quality. Fertil Steril. 2002, 78: 1061-1067. 10.1016/S0015-0282(02)04202-4.
Dickey R, Thornton M, Nichols J, Marshall D, Fein S, Nardi R, for the Bravelle™ IVF Study Group: Comparison of the efficacy and safety of a highly purified human follicle-stimulating hormone (Bravelle™) and recombinant follitropin-β for in vitro fertilization: a prospective, randomized study. Fertil Steril. 2002, 77: 1202-1208. 10.1016/S0015-0282(02)03131-X.
Ravhon A, Lavery S, Aurell R, Trew G, Margara R, Winston R: Clinical experience with recombinant follicle-stimulating hormone (FSH) and urinary FSH: a retrospective case-controlled analysis. Fertil Steril. 2001, 75: 920-925. 10.1016/S0015-0282(01)01684-3.
Franco J, Baruffi R, Coelho J, Mauri A, Petersen C, Garbellini E: A prospective and randomized study of ovarian stimulation for ICSI with recombinant FSH versus highly purified urinary FSH. Gynecol Endocrinol. 2000, 14: 5-10.
Lenton E, Soltan A, Hewitt J, Thomson A, Davies W, Ashraf N, Sharma V, Jenna L, Ledger W, McVeigh E: Induction of ovulation in women undergoing assisted reproductive techniques: recombinant human FSH (follitropin alpha) versus highly purified urinary FSH (urofollitropin HP). Hum Reprod. 2000, 15: 1021-1027. 10.1093/humrep/15.5.1021.
Bergh C, Howles C, Borg K, Hamberger L, Josefsson B, Nilsson L, Wikland M: Recombinant human follicle stimulating hormone (r-hFSH ; Gonal F) versus highly purified urinary FSH (Metrodin HP): results of a randomized comparative study in women undergoing assisted reproductive techniques. Hum Reprod. 1997, 12: 2133-2139. 10.1093/humrep/12.10.2133.
Recombinant Human FSH Study Group: Clinical assessment of recombinant human follicle-stimulating hormone in stimulating ovarian follicular development before in vitro fertilization. Fertil Steril. 1995, 63: 77-86.
Feigenbaum S, Miller P, Kaufmann R, Elkind-Hirsch K, Fein S, Marshall D: A new highly purified human-derived FSH, Bravelle™, is as effective and well-tolerated as recombinant follitropin beta in ovulation induction in infertile women with ovulatory dysfunction. Today's Therapeutic Trends. 2001, 19: 297-313.
World Health Organisation: . Laboratory manual for the examination of human semen and sperm cervical mucus interaction. 1999, Cambridge: Cambridge University Press, 4
Wittemer C, Ohl J, Bailly M, Bettahar-Lebugle K, Nisand I: Does body mass index of infertile women have an impact on IVF procedure and outcome?. J Assist Reprod Genet. 2000, 17: 547-552. 10.1023/A:1026477628723.
Loveland JB, McClamrock H, Malinow A, Sharara F: Increased body mass index has a deleterious effect on in vitro fertilization outcome. J Assist Reprod Genet. 2000, 18: 382-386. 10.1023/A:1016622506479.
Salha O, Dada T, Sharma V: Influence of body mass index and self-administration of hCG on the outcome of IVF cycles: a prospective cohort study. Hum Fertil(camb). 2001, 4: 37-42.
Van Wely M, Bayram N, van der Veen F: Recombinant FSH in alternative doses or versus uinary gonadotrophins for ovulation induction in subfertility associted with polycystic ovary syndrome: a systematic review based on a Cochrane review. Hum Reprod. 2003, 18: 1143-1149. 10.1093/humrep/deg229.
Al-Inany H, Aboulghar M, Mansour R, Serour G: Meta-analysis of recombinant versus urinary-derived FSH: an update. Hum Reprod. 2003, 18: 305-313. 10.1093/humrep/deg088.
Fevold HL: Synergism of the follicle stimulating and luteinizing hormones in producing estrogen secretion. Endocrinology. 1941, 28: 33-36.
Yu AW, Leung CB, Li PK, Lui SF, Lai KN: Pain perception following subcutaneous injections of citrate-buffered and phosphate-buffered epoetin alpha. Int J Artif Organs. 1998, 21: 341-343.
Acknowledgements
Other members of the Bravelle® IVF study group are: Frankfurter, D, Women's and Infants Hospital, Providence, RI; Gorrill, M, Oregon Health Science University, Women's Health Research Unit, Portland, OR; Katayama, P, Advanced Institute of Fertility, Milwaukee, WI; Kettel, M, San Diego Fertility Center, San Diego, CA; Kort, H, Reproductive Biology Associates, Atlanta, GA; Kutteh, W, University of Tennessee, Memphis, TN; Magarelli, P, Reproductive Medicine and Fertility Center, Colorado Springs, CO; Najmabadi, S, Center for Repoductive Health and Gynecology, Valencia, CA; Patton, G, Southeastern Fertility Center, PA, Mount Pleasant, SC; Somkuti, S, Abington Reproductive Medicine, PC, Abington, PA; Yeko, T, Verkauf, Bernhisel, Tarantino, Goodman and Yeko, MDs, PA, Tampa, FL.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Author contributions
Dr.'s Dickey, Nichols, Steinkampf, Gocial, Thornton, Webster, Bello and Crain contributed to the treatment of patients and collection of data.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Dickey, R.P., Nichols, J.E., Steinkampf, M.P. et al. Highly purified human-derived follicle-stimulating hormone (Bravelle®) has equivalent efficacy to follitropin-beta (Follistim ®) in infertile women undergoing in vitro fertilization. Reprod Biol Endocrinol 1, 63 (2003). https://doi.org/10.1186/1477-7827-1-63
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1477-7827-1-63