
Abstract
Activation of nuclear factor-kappa B (NF- κ B) as a mechanism of host defense against infection and stress is the central mediator of inflammatory responses. A normal (acute) inflammatory response is activated on urgent basis and is auto-regulated. Chronic inflammation that results due to failure in the regulatory mechanism, however, is largely considered as a critical determinant in the initiation and progression of various forms of cancer. Mechanistically, NF- κ B favors this process by inducing various genes responsible for cell survival, proliferation, migration, invasion while at the same time antagonizing growth regulators including tumor suppressor p53. It has been shown by various independent investigations that a down regulation of NF- κ B activity directly, or indirectly through the activation of the p53 pathway reduces tumor growth substantially. Therefore, there is a huge effort driven by many laboratories to understand the NF- κ B signaling pathways to intervene the function of this crucial player in inflammation and tumorigenesis in order to find an effective inhibitor directly, or through the p53 tumor suppressor. We discuss here on the role of NF- κ B in chronic inflammation and cancer, highlighting mutual antagonism between NF- κ B and p53 pathways in the process. We also discuss prospective pharmacological modulators of these two pathways, including those that were already tested to affect this mutual antagonism.
Similar content being viewed by others

Introduction
Cancer is an extremely complex disease caused by cells that have lost their usual control over growth. The apparent cause of cancer formation may differ case by case, however the basic mechanism is thought to be the following. There are two classes of genes that can control cancer development. Oncogenes and tumor suppressor genes belong to one class, while the other class belongs to the caretaker genes. Healthy cells follow standard rules of growth and proliferation, and have a definitive life span. In contrast, cells with an oncogenic activation undergo much faster cell division with an indefinite life span. Tumor suppressor genes are evolved to inhibit deregulated cell growth. Usually cancer formation ensues when activation and inactivation of an oncogene and a tumor suppressor gene, respectively, occur in a cell at the same time. The caretaker genes control the rate of mutation in the genome. A defective caretaker gene would allow accumulation of mutation in the genome and thus leading to a higher rate of tumor formation. Therefore, cancer formation occurs due to functional defects in multiple genes.
Heredity plays important role in cancer formation. However, it appears to be a small causal factor compared to incidence attributed to the modern lifestyle and environment. Smoking, high calorie diet, obesity, alcohol consumption, chronic infection, exposure to radiation and environmental pollutants are considered to be the major risk factors for cancer formation [1]. In fact about 95% cancer can link modern life style and environment with inflammation as the basic underlying cause [2]. An acute inflammatory response is transient, self regulatory and protects our tissues from infection in a healthy cell. The level of pro-inflammatory cytokines that rises to the peak at the height of the response eventually leads to the production of anti-inflammatory cytokines [3]. Thus an acute inflammatory response is faded off to complete the process of healing. In contrast, chronic or persistent tissue inflammation or irritation is correlated with adverse effects and has long been linked with increasing rate of tumor formation by epidemiological studies [4–6]. Cancer promoted by chronic inflammation (called ‘arbuda’) has been cited in Ayurveda, a form of Indian traditional/alternative medicine ˜5000 years ago. Virchow (in1858) also had observed frequent cancer origination at the site of chronic irritation [4].
Inflammation as a natural host protective mechanism
As a default mechanism, a cell promptly responds to a tissue injury through activation of innate immunity. The first line of defense is launched by the resident immune cells such as macrophages, dendritic cells, neutrophils, eosinophils, mast cells, natural killer cells present in all tissues. Although neutrophils are the first effectors in acute inflammation, eosinophils are also recruited first sometimes. Monocytes then move to the site of injury and differentiate into macrophages which upon activation release soluble mediators such as interleukin 1 β (IL-1 β), tumor nicrosis factor α (TNF- α) interferon IFN- γ and chemokines as major effectors of local microenvironment. The mast cells as well affect the local microenvironment at the site of inflammation by secreting several pro-inflammatory mediators including histamines, matrix remodeling proteases to signal migration of different leukocytes (neutrophils, eosinophils, basophils) from adjacent blood vessels to the site of inflammation. Transendothelial migration of a leukocyte is accomplished through distinct sequential steps as follows: Attachment of circulating neutrophil on the vessel endothelium; Stretching and rolling of the attached leukocyte on the endothelium surface; Immobilisation and transendothelial migration of leukocytes to accumulate to the site of inflammation [5, 7]. The process of a leukocyte adhesion and rolling on the vascular endothelium surface is mediated by the interaction of the activated E-, and P-selectins on the leukocyte with the intercellular adhesion molecules ICAM1 and ICAM2 on the endothelium. Tight interaction of integrins such as CD11a/ α L β 2, α 4 β 1, α 4 β 7 α L β 2 with the adhesion molecules VCAM1, MadCAM1 help leukocytes immobilized on the vessel endothelium to migrate to the site of injury [8, 9]. Neutrophils are the first leukocytes to migrate to the site of injury followed by monocytes. Leukocytes, at the site of injury, release highly bioactive agents including reactive oxygen species (ROS), nitric oxide (NO), cationic peptides, eicosanoids, different matrix metalloproteases (MMPs) and elastases, clear the cell debris and invading agents through a phagocytosis like process [10, 11].
An innate immune reaction (inflammation) associates with appearance of correct profile of chemokines as well as cytokines in an appropriate order at the site of inflammation and is self limiting [12]. A normal inflammatory response is followed by its resolution, and is accompanied by down regulation of proinflammatory cytokines by expression of anti-inflammatory cytokines such as IL-10 [10, 13]. Regulatory T cells (Tregs) serve as important source of IL-10 [14]. Patients with mutation in IL-10 receptor develop aggressive diseases, and mice without an IL-10 receptor spontaneously develop inflammation associated colitis correlating with development of colorectal cancer [15]. This function of IL-10 is mediated through inhibition of NF- κ B activity involving STAT3 resulting the reduced expression of proinflammatory cytokines like TNF- α, IL-6 and IL-12 [16–19]. Many oncogenic factors are cooperatively regulated by STAT3 and NF- κ B [20]. Another mode of NF- κ B inhibition is mediated through activation of TNFAIP3 gene encoding A20. A20, a ubiquitin editing enzyme with E3 ubiquitin ligase as well as deubiquitinase activities, negatively regulates NF- κ B signaling by stepwise deubiquitination and ubiquitination of adaptor molecules associated with TNF- α and IL-1 receptors [21]. CYLD, another deubiquitinase negatively regulates NF- κ B signaling by targeting several key molecules including NEMO/IKK γ, and TNF receptors associated factors TRAF2 [21].
Correlation between chronic inflammation and cancer
Both epidemiologic as well as clinical studies strongly correlate chronic inflammation with tumor formation [5, 12, 22, 23]. Many individual malignancies are known to originate at the site of chronic infection where the source of infection could range from environmental agents to viral particles. It was estimated earlier that out of 2.2 million cancer cases diagnosed in the world on average more than 15% cases (22% in the developed and 7% in the developing world) can be rooted to infection [24]. There is about 1.2 million new cases of colorectal cancer each year which is majorly caused by chronic inflammation and takes about 60000 lives per year [25]. Persistent inflammation that may result from environmental factors such as exposure to asbestos, smoke, and UV irradiation has been underscored in lung and skin cancer [26, 27]. Long term alcohol consumption can cause chronic inflammation in the liver and thus cancer [28, 29]. Chronic infection by bacteria can be equally cancerous as well. Helicobacter pylori infection has been strongly associated with stomach cancer and MALT-lymphoma in the world [30, 31]. Gastric cancer is the second most prevalent cancer in the world [32, 33]. Chronic acid reflux is considered as a major reason for esophageal cancer [34].
Schistosomiasis, caused by infection with parasite genus Schistosoma predisposes individuals with increased risks of cancer in internal organs such as bladder and colon [5, 35, 36]. In fact, schistosomiasis is a socioeconomically devastating disease in developing countries like Asia and Africa [37]. The parasite Opisthorchis viverrini infection can lead to cancer in the bile duct, a rare kind of adenocarcinoma [38]. Inflammatory bowel disease such as Crohn’s disease and chronic ulcerative colitis are two good examples of intestinal diseases caused by chronic infection that affect millions of people in the world each year [35, 39, 40].
Persistent viral infection is thought to be a major cause of hepatocellular carcinoma (HCC). HCC is a third major cause of cancer related death worldwide which claims about 60000 lives each year. About 90% of HCC develops due to chronic infection caused by various agents such as hepatitis B and hepatitis C viruses and, long term alcohol consumption or non alcoholic fatty liver [28, 41–44]. Activation of oncogenes is caused by direct insertion of viral DNA such as human papilloma virus (HPV) and Epstein bar virus (EBV), although other mode of actions including degradation of tumor suppressor by viral protein could be critical player in the carcinogenesis process. In cervical cancer E6 protein of HPV degrades p53 tumor suppressor [45]. EBV, a common virus found in human, is conditionally responsible for several cancers such as Hodgkins lymphoma, Burkitt’s lymphoma, nasopharangial carcinoma and lymphoma in the central nervous system (CNS) [5, 46, 47]. Inflammation was thought to be an essential component in Rous sarcoma virus mediated tumor formation as well [48].
While chronic inflammation is a cause of various cancer as described above, prolong suppression of innate immune response pathway has also been attributed to increased risk for cancer [12, 49]. Long term use of antibiotics has been attributed to increased risk of breast cancer [50]. Use of antibiotics has been reportedly associated with increased prostagalandin E2 production catalysed by cyclooxygenases [51]. In fact, mice defective in producing interferon gamma and granulocyte stimulating factor, spontaneously carry low level of inflammation in various tissues that have been correlated with different types of cancer [22, 52].
Role of NF- κ B in chronic inflammation and cancer
Role of NF- κ B in inflammation was anticipated from the early phase of its discovery; it was activated by various cytokines to subsequently activate the same and other proinflammatory cytokines, chemokines, and adhesion molecules, acute phage proteins, inducible effector enzymes, regulators of cell proliferation and apoptosis. Based on the functional significance associated with innate and adaptive immunity and cell proliferation it is expected that the NF- κ B activity is tightly regulated in a cell such as macrophage, dendritic cell or lymphocyte [53].
NF- κ B was first discovered as an activity that binds to the κ B elements on the immunoglobulin kappa light chain enhancer in the B cells although it occurs, as discovered soon after, in all cell types [54, 55]. NF- κ B, refers to a group of five structurally related and conserved proteins in mammals i.e., RelA/p65, Rel/cRel, RelB, NF- κ B 1/p50, and NF- κ B 2/p52. The p50 and p52 are synthesised as larger precursors of p105 and p100, respectively [56]. The family members consist of well defined domain structure correlated with their distinct functions. The N-terminal rel homology domain (RHD) is required for formation of homodimers, or heterodimers between the members that is necessary to execute their transcriptional function [57]. The nuclear localization signal (NLS) resides towards the c-terminus of RHD. A major regulation in NF- κ B activity is executed through controlling the trafficking of NF- κ B between the nucleus and cytoplasm [53]. In unstimulated cells, the majority of cellular NF- κ B is sequestered in the cytoplasm by binding to the inhibitor I κ B. I κ B, specifically inhibits NF- κ B DNA binding function by trapping the NF- κ B in the cytoplasm. In I κ B-NF- κ B complex, the NLS of NF- κ B subunits is masked by ankyrin repeats [58]. Except p50/p50 and p52/p52 that are implicated in the repression of transcription, most NF- κ B heterodimers act as transcriptional activators [59]. p50/50 and p52/p52 homodimer do not have an activation domain but can activate gene expression by a nuclear mediator [57, 60, 61].
Activation of NF- κ B is mediated by various receptors located on the extra and intracellular membrane. Majority of the knowledge on NF- κ B activation pathway came from studying the activation of family of receptors in the class of IL-1 and TNF- α. Cytokines such as IL-1 β, TNF- α, can act in paracrine as well as autocrine manner to activate the NF- κ B activity through their cognate receptors on the cell surface. Toll like receptors (TLRs) belonging to the IL receptor family are a group of membrane anchored receptors activate NF- κ B in response to specific pathogen associated molecular patterns (PAMPs) including LPS, flageller protein like flagellin, viral double stranded RNA and many pro-inflammatory cytokines (Figure 1) [53, 62, 63]. Nod (nucleotide-binding oligomerization domain)-like receptors (NLRs) often considered cytoplasmic counter parts of TLRs are a group of 20 receptors activated by bacterial components and toxins to activate NF- κ B [64–67]. NLRs localized in the cytosol in mammals are also sensitive to signals created by the presence of dying and injured cells [68, 69]. C-type lectin receptors (CLRs) are another group of integral membrane bound receptors present predominantly on the myeloid cells i.e., monocyte, macrophage, granulocyte and dendrtic cells work in conjunction with the TLR and NLR [70]. Triggering receptors expressed on myeloid cells (TREM) proteins are yet another group of cell surface expressed receptors that are involved in the regulation of inflammatory response by leukocytes and differentiation of immune cells [71]. TREM-1 protein expressed on neutrophils and monocytes activates NF- κ B in response to bacterial products [72–74].

Activation of NF- κ B signaling through canonical and alternative pathways. Different cytokines and pathogen associated molecules (PAMs) interact with their specific receptors (cytokine receptors:TNF receptor, IL1 receptor; PAMs recognize TLRs present on outer cell membrane (TLR1,-2,-4,-5,-6, and -10) or on the endosomal membrane (TLR3, -7, -8 and -9) in the initial stage of NF- κ B activation pathway. The activated receptor recruit the adapter components (not shown) such as Myd88 and TRIF (TIR domain-containing adaptor inducing IFN- γ, except TLR3 which utilizes TRIF without Myd88 (Myeloid differentiation primary response protein 88) to transmit the signal through activation of several mediators components including IRAK4 (IL1 receptor associated kinase 4), TRAF6 (TNF receptor associated factor 6), to activate IKKK i.e., MEKK1 (mitogen activated protein (MAP) kinase/extracellular signa l regulated kinase (ERK) kinase kinase 1), MEKK3 and TAK1 (transforming growth factor β activated kinase) to act to phosphorylate IKK complex. The activated IKK modifies the inhibitor I κ B for its proteasomal degradation. NF- κ B is released free to enter into the nucleus for transcriptional activation of the target genes. The canonical pathway is active in innate immunity, inflammation and cell survival; the alternative pathway mediates the humoral immunity.
Upon interaction with a ligand, an activated receptor transmits the signal to I κ B kinase (IKK) complex through a mediator kinase [75]. Several kinases have been identified to act on IKK. Transforming growth factor β-activated kinase 1 (TAK1) mediated phosphorylation of IKK has been reported in response to several different stimuli such as IL-1 [76] and ubiquitin [77], TNF α[78] and LPS [79]. MEKK3 is another kinase that has been implicated in IKK activation in response to certain stimuli [80, 81]. Role of autophosphorylation in IKK activation has also been implicated in certain virus-induced NF- κ B activation [75, 82].
There are two distinct NF- κ B signaling pathways: canonical and non-canonical or alternate pathway. The canonical pathway involved in innate immunity is activated by pro-inflammatory stimuli including e.g., TNF- α, various interleukins, microbes, and virus related ligands. The alternative pathway on the other hand is associated with adaptive immunity such as lymphoid organogenesis and is stimulated by B cell activating factor (BAFF) [83–85], CD40 ligand, lymphotoxin β (LT β) [84] and receptor activator of NF- κ B ligand (RANKL) [86].
The IKK complex is a critical regulator of NF- κ B activation pathway. The I κ B kinase (IKK) involved in the canonical pathway is composed of IKK α IKK β and the regulatory subunit IKK γ or NEMO [87–89]. Although the three components of IKK complex is crucial for activation for NF- κ B, evidence of NF- κ B independent function of IKK β also exists [90]. In contrast, the IKK involved in the alternate/noncanonical pathway is composed of IKK α and IKK β[84]. The alternative pathway deals with the processing of p100 and translocation of p52-RelB dimer, and depends on the phosphorylation of IKK α not IKK β by NF- κ B inducing kinase (NIK) [53, 83, 84]. The activated IKK phosphorylates conserved residues on both I κ B α (at ser32 and ser36) and I κ B β (at ser19 and ser23) [82, 91, 92]. Phosphorylation induced conformation change tags I κ B for recognition by the receptor subunit β TrCP for polyubiquitination by specific E3 ubiquitin ligase of Skp1-Cull/F-box (SCF) family [93–95]. The polyubiquitinated I κ B undergoes rapid degradation by proteasome, allowing NF- κ B such as p50:p65 heterodimer to enter into the nucleus, and bind to the κ B motif on the target gene promoters for activation [53, 56, 96]. Because of the presence of activation domain (AD), only NF- κ B containing RelA/p65, RelB and cRel subunits can act as activator. Due to lack of AD, p50 and p52 homo and heterodimer associate with gene repression. The κ B bound NF- κ B mediates transcription activation function through recruitment of various coactivator including p300/CBP, or PCAF. Different post-transcriptional modifications (PTMs) including phosphorylation, acetylation, ubiquitination, nitrosylation of NF- κ B were shown to influence this process [54, 97]. For example, phosphorylation of p65 subunit by PKA and or MAPK at ser276 and ser311 by PKC ζ stabilizes RelA/p65-CBP interaction [97].
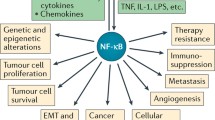
An innate immune response initiated by infection or injury recruits immune cells (such as neutrophils) at the site of injury as a protection mechanism. During this process neutrophils release several highly active antimicrobial agents such as reactive oxygen species, charged peptides, and proteases. Normally, these antimicrobial activities are required for a short period of time as the wound is repaired and self limiting. Secretion of these agents, however, for more than normal period may result in the induced genotoxicity complicated by the constant presence of inflammatory cells. A chronic inflammation associates with a constitutive activation of NF- κ B as a result of either an imbalance in the inflammatory signaling network such as inefficient anti-inflammatory mechanism, or persistent infection with pathogens. Furthermore, constant presence of pathogen proteins, activation of a kinase, over expression of a cytokine, and PAMP receptor can drive NF- κ B mediated gene expression to promote tumor initiation, progression, invasiveness and its persistence. In fact, the link between NF- κ B and cancer was first suspected by its close structural similarity with viral oncoprotein v-Rel [98, 99]. Identification of translocation of Bcl3, a member of I κ B family in chronic lymphoblastic leukemia (CLL) had at that time reaffirmed the connection of NF- κ B with cancer [100]. The significance strengthened when it was discovered that many cancer had constitutively active NF- κ B, and that down regulation of NF- κ B makes these cells more sensitive to treatments including chemo and radiation therapies [101, 102]. Directed inhibition of NF- κ B activity regresses tumor growth in mouse models of lung and colitis associated cancers in addition to tumor suppression in xenograft experiments [25, 103–105].
Deregulation at any stage in the NF- κ B activation pathway can lead to persistent NF- κ B activation/chronic inflammation and eventually cancer [12, 23, 106]. Under this condition, resident immune cells such as macrophages and mast cells constantly monitor the tissue microenvironment, and sense the invading pathogen with pathogen associated molecular pattern (PAMP) through toll like receptors (TLRs) (Figure 1). Thus, TLRs are the first line of sensors for activation of innate immune response. Both epidemiological as well as genetic data link NF- κ B activating receptors with cancer [106]. Mutation in TLR cluster TLR1-6-10 in combination with interleukin receptor associated kinase (IRAK) 1 and 4 has been linked with greater risk of prostate cancer [107]. An elevated expression of several TLRs has been associated with different cancer types as well [108, 109]. In both mouse and human colorectal cancer TLR4 is over expressed, and mice deficient in TLR4 are insensitive to colon cancer [110]. Thus, TLRs are potentially important drug targets for cancer treatments. As well testing TLR expression profiling in patients is being considered as cancer diagnostic marker [111]. Linkage analysis detected association of 30 different genes including mutation in NOD2, a member of PAMP receptor with the increased incidence of Crohn’s disease [112, 113] and inflammatory bowel disease [114]. This has been linked with increased IL-1 β production in the inflammatory milieu [23, 115]. Several other findings associate DNA mutation with enhanced IL-1 β activity, particularly in gastric cancer [116, 117]. Notably, abundant IL-1 β level in cancer environment associated with increased cancer invasiveness and is considered a good therapeutic target [23, 118, 119]. Many cancers originate due to paracrine/autocrine expression of cytokines such as IL-1 β and TNF- α which constitutively activate NF- κ B by activation of their cognate receptors [53, 120, 121].
Abnormal activation of IKK has been implicated in many different cancer types including breast, prostate, brain and colon cancer, melanoma mantle cell lymphoma as recently reviewed elsewhere [75, 121]. Vlantis et al. [105] showed that a constitutive overexpression of IKK β in the intestinal epithelial cells (IEC) resulted in both inflammation and tumorigenesis. The IKK β overexpressing in IECs have elevated levels of pro inflammatory cytokines such as TNF- α, IL-1 β and various chemokines attracting increased level of infiltrated inflammatory immune cells. The elevated cytokine and chemokine levels modulate Wnt/ β catenin signaling leading to the activation of several IEC-associated stem cell factors providing a possible explanation for a switch from inflammation to transformation. Role of Wnt/ β -catenin pathway in intestinal cancers has already been implicated [122]. These studies noted IKK β as a potential therapeutic target in colorectal cancer. In MALT lymphoma resulting in AP12-MALT1 fusion leads to the constitutive activation of NF- κ B through aberrant IKK activity [16]. MUC1 over expressed in several cancers activate NF- κ B -p65 through direct interaction with IKK β and IKK γ[123]. In normal B lymphocytes CARD11 acts as a cytoplasmic scaffolding protein which coordinates the signal mediated- activation of IKK activity. Mutation in the coiled-coil domain of this protein resulting in the gain of function in the form of constitutive activation of IKK as observed in the diffuse B cell lymphoma [124]. In human T cell leukemia virus 1 (HTLV1) mediated transformation of host cell, the HTLV-1 protein Tax constitutively activate NF- κ B by direct interaction with the IKK complex [125].
Thus far a constitutively active mutation in the NF- κ B protein itself has not been found. Bcr-Abl, a tyrosine kinase, supports acute lymphoblastic leukemia (ALL) and chronic myelogenous leukemia (CML) by inducing NF- κ B function through enhancing nuclear translocation of NF- κ B. A defective I κ B leads to constitutively active NF- κ B in the Hodgkin cells [126]. An overexpression of tissue transglutaminase (TG) underlies a basis of many aggressive and drug resistant cancers including pancreatic ductal carcinoma [127]. Mann et al. have shown that elevated levels of TG drives constitutive NF- κ B overexpression in these cancers [128].
While NF- κ B promotes oncogenic potential of cells by driving expression of genes encoding prosurvival and proliferative functions, it is also antagonistic with tumor suppressor such as p53 [106, 129]. For example, NF- κ B target gene MDM2, an ubiquitin E3 ligase drive p53 for proteasomal degradation [130]. P53 can also antagonise NF- κ B by competing for cellular p300/CBP and vice versa [131]. Several other tumor suppressors including ARF (p14ARF) and PTEN may also antagonize transcriptional activation and function of NF- κ B [132–135]. Putative tumor suppressors LZAP (LXXLL/leucine-zipper-containing ARF-binding protein), transcription elongation factor A like 7 (TCEA7) and CHFR (check point with forkhead and ring finger domains) can also antagonize with NF- κ B activity by interfering with the transcriptional activities of RelA subunit [136–138]. While cross-talk activities of NF- κ B with the tumor suppressors is finely controlled during normal cellular homeostasis, a deregulation in any of the control points can result in its deregulation adding to tumor promoting capability of NF- κ B [133, 139].
Role of p53 in inflammation: p53 and NF- κ B antagonizes each other’s function
Tumor suppressor p53 is one of the most extensively studied proteins due to its functional association with the maintenance of genomic integrity. In fact, in more than 50% cancers the p53 protein is either absent or nonfunctional due to various other reasons. p53 is termed ‘the guardian of genome’ for the major function it plays in protecting cells from transformation and genomic mutation in response to a various stressors including DNA damage, oxidative stress, and oncogene activation through activation of cellular process like cell cycle arrest, apoptosis, or senescence [140, 141]. Activation of p53 also associates with induction of other important processes including autophagy, angiogenesis, cell migration, and differentiation [141].
A healthy cell constitutively maintains p53 at low level [142]. Usually, in an unstressed condition p53 is constantly ubiquitinated by MDM2, an E3 ubiquitin ligase to channel it to proteasomal degradation pathway [143]. MDM2, a p53 transcriptional target gene, is upregulated as p53 level goes up; an elevated MDM2 in its turn checks the normal low level of p53 in the cell by funneling extra level of p53 to proteasome for degradation. Thus p53 and MDM2 constitutively controls each others activity in a normal healthy cell. MDM2 executes this function as a heterodimer with a structurally related protein MDM4 (MDMX) [144, 145]. MDM2 controls the activity of MDM4, also a p53 target gene, through its E3-ubiquitin ligase activity. Thus p53 activity is tightly controlled by two autoregulatory loops; one through MDM2 and another through MDM4. The association of p53 with MDM2 and MDM4 is disrupted with phosphorylation by various enzymes such as DNA-PK, ABL, ATR, ATM and CHK -all activated by genotoxic stressors [146]. Phsophorylation of MDM2 at ser17 and MDM4 at Tyr99 by DNA-PK [147] and c-ABL [148], respectively, drives dissociation of MDM2, and activation of p53. MDM2 was reported to inhibit p53 transcription function by blocking p53 surface that interact with the basal transcription factors such as TFIIE [149]. ATM and CHK1 kinases induced by genotoxic stress signals activate p53 by driving dissociation of MDM2 and MDM4 through phophorylation [150]. In contrast, activation of AKT kinase function in cancer stabilizes MDM2 and MDM4 interactions resulting in inhibition of p53 activity [151, 152]. In addition to simple dissociation of p53 from its negative regulator MDM proteins, removal of ubiquitin moiety from p53 by ubiquitin proteases can lead to its stabilization as well [142].
In fact, the major part of p53 tumor suppressor activity is explained by its transcriptional activation function. The active p53 undergoes extensive post-translational modifications including phosphorylation, acetylation, monoubiquitination, and neddylation guided by specific stress signals. A particular posttranslational modification targets p53 to a subset of its target gene promoters [153, 154]. Furthermore, p53 executes its tumor suppressor activity in tissue specifc manner [155, 156]. A specific posttranslational modification may allow p53 to interact with a partner protein, or a promoter DNA sequence contributing to tissue specific gene activation [154, 157].
Depending on the nature of signal p53 induces a set of target genes resulting in a definite cell fate. For example, expression of p21/WAF1, 14-3-3 σ and Cdc25c genes links with reversible cell cycle arrest at different stages of cell cycle [158–161]. Elevated expression of genes such as Puma (p53-upregulated modulator of apoptosis), Noxa, Bax (Bcl-2 associated X protein), Apaf-1 (Apoptotic protease activating factor-1) results in cellular apoptosis [162]. Expression of Pai-1 (plasminogen activator inhibitor type 1) and p21 associates with a state of an irreversible cell cycle arrest or replicative senescence [163, 164]. Induction of DRAM (Damage-regulated autophagy modulator) leads to autophagy [165]. In addition to its role in transcriptional activation, p53 is also implicated in transcriptional repression of many genes involving various mechanisms [166].
In principle p53 should be induced in an inflammatory condition, however, this is not the case normally. In fact most cells have mechanisms to suppress p53 when NF- κ B is activated to tilt the situation in favor of the cellular transformation process. Through various mechanisms, NF- κ B may cripple cellular p53 activities (Figure 2) [129, 167]. MDM2-p53 network discussed earlier can be interrupted by NF- κ B. First, MDM2 is a NF- κ B target gene. NF- κ B can suppress p53 levels by upregulating the MDM2 expression mediated through Bcl3 [168], or IKK β[169]. It has been shown that DNA damage induced upregulation of Bcl3 can inhibit p53 through upregulation of Hdm2 gene transcription [168]. Bcl3, a member of I κ B family, helps activate NF- κ B in a cooperative manner in cellular proliferation [168]. It has been shown that the presence of an active IKK β can block doxorubicin induced p53 mediated apoptosis and this event requires IKK β kinase function which is mediated by NF- κ B through MDM2 [169].

NF- κ B and p53 antagonises each others activity. Various mediators involved in the pathways are indicated. In addition, p53 and NF- κ B can inhibit each other by direct physical interaction through their multimerization domain. Eventually, the effect of activation NF- κ B pathway prevents the activation of p53 pathway and vice versa. The detail is described in the text. The black colored upward arrows adjacent to NF- κ B and p53 indicate activation of these transcription factors. MDM2: mouse double mute 2; β-TrCP1: beta transducing repeat containing protein1; ARF: alternate reading frame of INK4/ARF locus; ATR: ATM-Rad3 related; CHK: check point kinase; I κ B: inhibitior of kB; IKK: inhibitor of kappaB kinase.
Activation of AKT (a serine threonin kinase) by PI3 kinase in response to growth factor, plays important role in cancer cell growth and proliferation (mediated by induction of anti-apoptotic mechanism). AKT favors prosurvival pathway by inhibiting p53 and supporting NF- κ B activities at the same time. It inactivates proapoptotic Bad protein through phosphorylation at three positions (at ser112, -136 and -155) which prevents its binding with anti-apoptotic protein Bcl-xL[170, 171]. Bad, a member of Bcl2 family proteins, is p53 target gene [171]. AKT can block p53 accumulation and apoptosis by augmenting ubiquitin ligase activity of MDM2 activity through phosphorylation [172]. AKT mediated phosphorylation of MDM2 at ser166 and ser186 facilitates nuclear entry of MDM2 and faster degradation of p53. AKT also activates NF- κ B through phosphorylation of both IKK α and IKK β subunits of IKK that facilitates nuclear translocation by I κ B phosphorylation and degradation [173]. Furthermore, AKT was reported to activate IKK α in response to PDGF [174], and IKK β in response to TNF α[175].
IKK β can also block cellular p53 stability through direct post translational modification while activating NF- κ B through phosphorylation of I κ B. It was shown that p53 can be phosphorylated at ser366 and potentially at ser362 by IKK β facilitating its ubiquitination by β-TrCP, a member of SCFb-TrCP E3 ligase complex and proteosomal degradation [176].
ARF tumor suppressor known as p14ARF and p19ARF in human and mice, respectively, induced upon oncogene such as E2F activation, has been shown to facilitate p53 activation [134]. ARF induces p53 accumulation by inhibiting p53 association with MDM2/Hdm2 [177]. In addition, ARF was shown to block NF- κ B function. ARF inhibits NF- κ B activation by inducing association with histone deacetylase, HDAC1 [134]. RelA/p65 subunit when phosphorylated at thr505 by Chk1 kinase was shown to associate with HDAC1. ARF induction activates Chk1 kinase via induction of ATR kinase [178].
An effect of p53 products on cell fate can be neutralized by the products of the NF- κ B target genes; NF- κ B induced antiapoptotic genes including Bcl2 can abrogate the pro-apoptotic functions of PUMA, and Noxa induced by p53 [179].
In addition, Ikeda et al. showed that NF- κ B/RelA and p53 can inhibit each other’s activity through direct physical interaction with multimerisation domains of the transcription factors [180].
A major interference of p53 activity by NF- κ B is mediated by their use of a common coactivator p300 and CBP for their optimal activity in response to a stress signal [181, 182]. It has been shown that through the depletion of limited pool of cellular p300/CBP, NF- κ B competes out and thus represses p53 function [183]. IKK α mediated phosphorylation of a specific amino acid residue on CBP makes it preferentially utilized by NF- κ B instead of p53 [131, 184].
It was shown that ectopic expression of p53 inhibits NF- κ B function in a unique way; expression of p53 enhanced NF- κ B DNA binding but blocked it transactivation function [185]. NF- κ B and p53 cross action has been associated with the regulation of actin cytoskeleton function and integrin signaling both of which play important role in tumor progression. It has been shown that in the absence of p53, STAT3 is constitutively activated in an NF- κ B-dependent manner which regulate lamellipodia formation via integrin signaling [186].
Glucose metabolism is another aspect which is inversely controlled by p53 and NF-k B. Absence of p53 enhances NF- κ B activity through activation of IKK kinase and thereby increase the rate of glycolysis. It was shown that absence of p53 upregulates expression of high affinity glucose transporter GLUT3 as observed in tumor microenvironment [187]. In contrast, activation of p53 has been linked with reducing the expression of several glycolysis regulating factor including glucose transporters [188]. In addition to slowing down glucose uptake, p53 induction was shown to promote oxidative phosphorylation [189, 190].
Finally, an inverse correlation between NF- κ B and p53 has been demonstrated in model animals. First, it was shown that mice bearing p53 null homozygous mutation was born normal from embryonic stem cell but prone to tumor formation during development starting at 6 month of age [191]. Later it was shown that p53-/- mice had relatively higher level of proinflammatory cytokines and chemokines and reduced level of oxidation products, while those mice ectopically expressed p53 carried lower level of those cytokines compared to their respective controls. Komarova et al. also shown that LNCaP cells transduced with p53 suppressor element expressed much lower level of proinflammatory markers [192]. Furthermore, treatement of cells with inducer of p53 caused inhibition of NF- κ B target genes expression [103, 104]. Recently, Natarajan et al. shows that transduction of cells with NF- κ B activating polypeptides isolated by genetic selection called as NF- κ B activating selectable peptides (NASP), reduced p53 accumulation [106].
Small molecule NF- κ B inhibitors as chemopreventive agents
The presence of constitutively active NF- κ B apears to be a common underlying factor in inflammation related cancer (responsible for constitutive induction of different prosurvival genes such as antiapoptotic genes: Bcl2, Bcl-x; proangiogenic gene: VEGF; genes encoding metastatic and invasion activities: MMPs). Consistently, both clinical and epidemiological data suggest strong chemopreventive potential for NF- κ B inhibitors. Chemoprevention refers to use of chemicals, including even food supplements to prevent the development and progression of cancer.
Given the significance of NF- κ B function, many laboratories have undertaken prime interests to deliver a specific and potent inhibitor of NF- κ B pathway (Table 1). A series of small molecules acting on single as well as multiple steps in the NF- κ B signaling pathway have been reported, some of which are in various stages of clinical trials [58, 129, 193, 194]. The nature of these molecules includes protein/antibodies, small peptide, small RNA/DNA, and small molecular weight chemicals. Development of inhibitors targeting basically every possible step in the NF- κ B signaling pathway is underway including molecules interfering with the receptor-ligand interaction, interference of the adapter with the activated receptor, activation of IKK and subsequent phosphorylation leading to proteasomal degradation of I κ B, nuclear translocation of NF- κ B, binding of NF- κ B with the target gene promoter, and interaction of the activator with the co-activator (for detail list of inhibitors see [194]). Normally the outcome of activation of NF- κ B is determined by the cross-talk among the parallel signaling pathways such as p53, PTEN, and p38 MAPK [133].
A major point of regulation in the NF- κ B activation cascade involves signal induced release of NF- κ B from I κ B in the cytoplasm (Table 1). A signal activated IKK phosporylates I κ B α which is targeted by β-TrCP mediated ubiquitination and subsequent proteosomal degradation. β-TrCP, the F-box component of SCF-E3 ubiquitin ligase, through its WD40 domain recognizes the destruction motif of I κ B α in phosphorylation dependent manner [93–95]. β-TrCP inhibitors which target I κ B α degradation in the NF- κ B activation pathway blocking the release of NF- κ B for entry into the nucleus have shown a great promise in the treatment of disease such as multiple myeloma (MM) [58, 195]. MM cells are very sensitive to a proteasome inhibitor like bortezomib due to two reasons: a constitutively activated NF κ B and an over dependence on proteasome activity [196]. Bortezomib (or Velcade) was the first drug as proteasome inhibitor (reversible inhibitor) approved by US food and drug administration for treatment of MM [197–199] (Table 2). By definition, however, proteasome inhibitors will be nonspecific because cellular proteins recognized by the proteasome regulating various signaling pathways will also be sensitive to this treatment. For example, many proteasome substrates not related to NF- κ B pathway having pro-apoptotic and cell cycle regulatory activities will be stabilized and thus enhance bortezomib activity [200]. These players include program cell death regulator (PDC4) [201], myeloid cell leukemia 1 (Mcl-1) [202] and cell cycle division cycle homologue 25A (CDC25A) [203, 204]. Furthermore, bortezomib treatment stabilizes a factor like β-catenin, also a β-TrCP substrate. In fact, an elevated level of β-catenin has been observed in several cancers including colorectal and liver cancer [58, 205–207]. However, GS143, a proteasome inhibitor was reported to degrade β-catenin. It was reported to block I κ B α degration, not its phosphorylation [208]. MG-132 was reported as another specific, potent, reversible, and cell-permeable proteasome inhibitor with Ki= 4 nM. It was shown to reduce the degradation of ubiquitin-conjugated proteins in mammalian cells and in strains of yeast (with membrane permeable ability) by the 26S complex without affecting its ATPase or isopeptidase activities. MG132 can also inhibit NF- κ B activation with an IC50 of 3 μ M. MG-132 by blocking I κ B α degradation has been shown to potently inhibit TNF- α-induced NF- κ B activation, interleukin-8 (IL-8) gene transcription, and IL-8 protein release in A549 cells [209].
Although 35% MM patients respond to bortezomib, it was a great success for treating MM and mantle cell lymphoma patients. Nevertheless, acquisition of resistance to bortezomib in MM patients is common with reported over expression of or mutation in the β 5 subunit of the proteasome catalytic core complex. This has led to the development of second generation proteasome inhibitor carfilzomib. Carfilzomib, reported to be a potent and an irreversible inhibitor of the proteasome, that drives higher rate of apoptosis has completed phase III clinical trial [193, 266–268]. Marizomib is another proteasome inhibitor with increased potency and sustained inhibitory activity compared to bortezomib [269–271]. Marizomib, isolated from marine actinomycete Salinspora tropica is the first irreversible proteasome inhibitor from natural sources, had been in phase I clinical trial to assess its efficacy against solid tumors and treatment of refractory multiple myeloma [272, 273]. Marizomib blocks all three catalytic sites of 26S proteasome. In addition, several newly developed proteasome inhibitors including CEP-18770, MLN-9709, ONX-0912, NPI-0052 are currently in various phases of clinical trials for their efficacy against myeloma and solid tumors [199, 274–277].
Thalidomide and its analogues known as immunomodulatory drugs (IMiDs), have anticancer as well as anti-inflammatory effects. Recently, these agents, including IMiD CC-5013 and IMiD CC-4047 [278], have shown promise in clinical trials for the treatment of different cancers. Among several different hypotheses, the inhibition of NF- κ B activation has been proposed to explain the therapeutic activity of thalidomide and related drugs [279]. In endothelial cells, thalidomide blocks the degradation of I κ B- α by inhibiting IKK- β, which is consistent with its role in inhibiting cytokine-induced NF- κ B activation [279]. The inhibitory effect of thalidomide on TNF- α and H2O2-induced NF- κ B activation is also seen in other cell types, including T lymphocytes, and myeloid and epithelial cells [280]. IMiD-induced apoptosis in multiple myeloma cells is associated with downregulation of NF- κ B DNA-binding activity, as well as the reduced expression of NF- κ B-dependent proteins [281]. Hence a major part of the immunosuppressive effects of thalidomide might be due to inhibition of NF- κ B activation.
Cyclopentenone prostaglandins (cyPGs) are naturally occurring prostaglandin metabolites which inhibit NF- κ B activation or activity [282]. This effect could be partly due to the ability of cyPGs to activate the peroxisome proliferation-activated receptor- γ (PPAR- γ), which was shown to antagonize NF- κ B transcriptional activity [283]. The treatment of peritoneal macrophages with the cyPG 15-deoxy- Δ 12, 14-prostaglandin J2 (15d-PGJ2) inhibits the expression of inducible nitric oxide synthase (iNOS), as well as NF- κ B activity in a PPAR- γ-dependent manner. However, cyPGs can directly inhibit activation of NF- κ B pathway by blocking IKK- β activity [284].
Nonsteroidal anti-inflammatory drugs (NSAIDs) are known to act as protective agents in cancer treatments, in particular inflammation associated cancer in model animals and humans. Although NSAIDs are popularly known as the blocker of prostaglandin (PGE) synthesis by COX-2, they have considerable NF- κ B inhibitory activities unrelated to COX inhibition. COX-2, the rate limiting enzyme for production of PGE from arachidonic acid is NF- κ B inducible [285–287]. Indomethacin and related compounds sulindac, sulindac sulfide and sulindac sulfone, were shown to block IKK β kinase activity in colon cancer and other cell lines [288]. Aspirin and salicylate act as competitive inhibitors of IKK- β for its ATP binding pocket and thus prevent I κ B phosphorylation and NF- κ B activation [289]. These NSAIDs are also shown to block NF- κ B mediated expression of VCAM-I and ICAM-I induced by TNF- α[288]. Salicylate is also reported to prevent expression of endothelial leukocyte adhesion molecule and leukocyte transmigration through endothelial monolayer [290]. Sulphasalazine is another NSAID that is widely used to treat inflammatory bowel disease. This compound is cleaved following oral administration to 5-amino-salicylic acid (5-ASA) and sulphapyridine. The treatment of human colonic epithelial cells with sulphasalazine, but not 5-ASA or sulphapyridine, inhibits NF- κ B activation through blocking I κ B phosphorylation and degradation in response to TNF- α, LPS or phorbol esters [291]. However, in a more recent study, 5-ASA was shown to block NF- κ B activation by inhibiting both IKK- α and IKK- β kinase activity in mouse colonic cells [292]. Mesalamine, a related aminosalicylate, can block phosphorylation of p65 without affecting I κ B degradation [293]. These results indicate that these agents can block the NF- κ B activation pathway at multiple steps.
‘The development of selective IKK or NF- κ B inhibitors has been undertaken by several pharmaceutical industries (Table 1). Yet, no potent IKK- α-specific inhibitors have been described so far. Several compounds, which are under development, can inhibit IKK- α kinase activity in the low micromolar range, although these agents were initially identified as IKK- β inhibitors. The unique role of IKK- α in the alternative pathway, important for B-cell mediated responses, and the recent demonstration of the auxiliary role of IKK- α in the classical pathway, indicate that IKK- α might be an important target for therapeutic intervention in autoimmune diseases and cancer [84, 294–296]. By comparison, the development of specific IKK- β inhibitors has progressed rather rapidly. Although most IKK- β inhibitors reported so far are still in preclinical stages of development, a number of novel small-molecule inhibitors of IKK- β have been described. For example, SPC-839, a member of a series of quinazoline analogues developed by Celgene [297–299], is one of the more extensively studied IKK- β inhibitors. SPC-839 inhibits IKK- β with an IC50 of 62 nM, and has a 200-fold selectivity for IKK- β over IKK- α (IC50 = 13 μ M). Several groups have indicated the inhibition of IKK- β activity by β-carboline derivatives [222, 300, 301]. PS-1145, which was developed from a β-carboline natural product inhibits the IKK complex with an IC50 of 150 nM, blocks TNF- α-induced I κ B phosphorylation and degradation in HeLa cells [222]. Another molecule that inhibits IKK- β is BMS-345541, which is an imidazoquinoxaline derivative [210, 302]. BMS-345541 shows greater than tenfold selectivity for IKK- β (IC50 =0.3μ M) over IKK- α (IC50 =4μ M). In addition, several other compounds like ureidocarboxamido thiophenes [303–305], 2-amino-3-cyano-4,6,-diarylpyridines [306–308], anilinopyrimidine derivatives [309], a group of optically active pyridine analogues [310], and a group of related pyridyl cyanoguanidines [311, 312] have been reported as nanomolar-range selective inhibitors of IKK- β kinase activity. Bay 11-7082 (BAY) is an inhibitor of κ B kinase (IKK) that has pharmacological activities that include anticancer, neuroprotective, and anti-inflammatory effects. BAY-11-7082 selectively and irreversibly inhibits NF- κ B activation by blocking TNF- α-induced phosphorylation of I κ B- α without affecting constitutive I κ B- α phosphorylation [313].
Dehydroxymethylepoxyquinomicin (DHMEQ), derived from the structure of an antibiotic epoxyquinomicin C is a novel NF- κ B inhibitor. DHMEQ could be qualified as a candidate for a new chemotherapeutic agent against human hepatoma [314]. It can also enhance antitumor activities of taxanes in anaplastic thyroid cancer (ATC) cells. DHMEQ blocks the nuclear translocation of NF- κ B. Inhibition of NF- κ B by DHMEQ creates a chemosensitive environment and greatly enhances apoptosis in taxanes-treated ATC cells in vitro and in vivo [315].
I3C/DIM Indole-3-carbinol (I3C) is a glucosinolate when given orally is converted to diindolylmethane (DIM) and other oligomers catalyzed by stomach acid. DIM is the predominant active agent and that I3C is a precursor. Combinatorial treatment of I3C/DIM with N-acetyl-S-(N-2-phenethylthiocarbamoyl)-l-cysteine (PEITC-NAC) and myo-inositol (MI) caused marked reductions in the activation of Akt, ERK and NF-kB in lung tumor tissues and thereby demonstrated the promise of combination therapy using I3C/DIM for the chemoprevention of lung carcinogenesis in smokers [316].
Small molecule inhibitors of NF- κ B from natural sources
From the beginning of civilization, herbal medicines, fruit and vegetables have been used in the disease treatment and well being of humans. It is also believed that plant based treatments are without side effects. Interestingly, many phytochemicals with NF- κ B inhibitory activity with cancer cell type specificity have been isolated (for a detail review on inhibitors see [194]) (Table 3). These compounds can make cancer cells sensitive to apoptosis or inhibit the expression of genes responsible for growth, proliferation and the metastasization of cancer cells. Different groups isolated battery of compounds from different traditional and ethnic medicinal plant sources against cancer cells of different tissue origins such as multiple myeloma cells (U266 line and MM.1 line), prostate cancer cells. Mode of action of these compounds is increasingly becoming clear. Many of these compounds inhibit both inducible as well as constitutively active NF- κ B activities. Compounds with diverse specificity have been isolated with specificity towards the IKK or IKKK (IKK kinase), I κ B α stability, p65 translocation or DNA binding in the NF- κ B activation pathway. Celastrol isolated from Celastrus orbiculatus inhibit various stimuli -induced phophorylation of I κ B α and its degradation. Importantly, celastrol is found to block IKK function and IKK β activity in dose dependent manner [225]. Epicatechin present in green tea, for example, prevents constitutive NF- κ B activity facilitating apoptosis by preventing p65 translocation to nucleus [317]. Apigenin, a flavonoid present in most fruits and vegetables blocks p65 phosphorylation by inhibiting IKK function [318, 319].
An administration of antioxidant N-acetyl-L-cysteine (NAC) suppresses LPS-induced NF- κ B activity [333]. In another study it is shown that vitamin C inhibits TNF- α- and IL-1 β-induced IKK phosphorylation of I κ B- α and subsequent NF- κ B DNA binding in endothelial cell lines [334]. Further studies reveal that dehydroascorbic acid (DHA), which is the oxidized form of ascorbic acid, suppresses TNF- α-induced NF- κ B activation by the direct inhibition of IKK- β kinase activity independent of p38 MAPK [335]. It should be noted that antioxidants can also inhibit the activity of other components of NF- κ B signaling pathways, including TNF receptors and the proteasome, without any direct effect on IKK [336].
Various molecules have been isolated that target IKK include xantholhumol from hops [255], epigallocatechin-3-gallate from green tea [337], genistein from soy [338], capsaisin from chilli pepper [339], boswellin from Boswellia serrata[340], and sulforaphane from cruciferous vegetables [341] (Table 3). Aggarwal and colleagues have isolated and functionally characterized a group of compounds targeting the NF- κ B activation pathways [121]. Some of these include a pentacyclic triterpenoid escin isolated from horse chestnut whose extract is used as traditional medicine in China. Escin inhibits activation of IKK activity [328]; Sesamin isolated from sesame seed inhibits IKK kinase [329]; Bharangin, a diterpenoid quinonemethide isolated from Premna herbacea , an Indian medicinal plant also has similar activity [342].
Other compounds from natural/dietary sources, with NF- κ B inhibitory activity and are currently in preclinical or clinical trials include Luteolin and parthenolide. Luteolin, a polyphenol flavonoid, has been reported to sensitize colorectal cancer cells to TNF-induced apoptosis through suppression of NF- κ B. Accumulation of ROS induced by luteolin plays a pivotal role in suppression of NF- κ B and potentiation of JNK to sensitize lung cancer cells to undergo TNF-induced apoptosis [331]. Zerumbone, isolated from subtropical ginger has been shown to inhibit angiogenesis through inhibition of NF- κ B in gastric cancer cell line [343]. Parthenolide is a major active component of the herbal medicine feverfew (Tanacetum parthenium), which is conventionally used in Europe to treat inflammatory diseases such as fever, migraine, and arthritis [344]. Parthenolide has been shown to inhibit growth or induce apoptosis in a number of tumor cell lines [345–348]. Many mechanisms are postulated to be involved in the antitumor effect of parthenolide, including inhibition of NF- κ B [347]. It has also been shown that parthenolide sensitizes cancer cells to various apoptosis-inducing agents mainly through inhibition of NF- κ B [349].
Several compounds have been isolated from traditionally used plants that have inhibitory activities against multiple components of NF- κ B activation pathway [121]. Curcumin from common Indian spice turmeric and resveratrol from grape have drawn a lot of attention because of their activity against multiple signaling pathways. Curcumin inhibits IKK- mediated phosphorylation of I κ B as demonstrated in Burkitt lymphoma cells. Curcumin prevents NF- κ B/p65 phospohorylation at serine 536 and its acetylation through p300 inhibition (Table 2) [350]. Burkitt lymphoma cells expressing wild type Bax protein undergo apoptosis by curcumin treatment. Curcumin sensitizes Bax negative Burkitt lymphoma cells to TNF-related apoptosis inducing ligand (TRAIL) which activates apoptosis through the extrinsic pathway [351]. As a transcriptional regulator, it inhibits production of eicosanoids prostaglandin E (PGE2) and 5-hydroxyeicosatetraenoic acid (5-HETE) and is in phase II clinical trial for different malignancies including pancreatic colorectal cancers [352, 353]. Recently, curcumin has been described to block proteasome function in cell based assays [354]. Curcumin was described to block the chymotrypsin like activity of the catalytic core of rabbit 20S and cellular 26S proteasome. In a recent study curcumin was described to target different components of ubiquitin proteasome pathway [355]. Resveratrol, a plant polyphenol found in different fruits (grapes, berries etc) came to lime light in 1997 for its anti tumor activity tested against various tumors such as myeloid, breast, and prostate [356]. Resveratrol has been shown to down regulate the expression of many antiapoptotic genes. Resveratrol inhibits constitutive activation of NF- κ B by inhibiting both IKK as well as p65 phosphorylation [357, 358]. Preparation of resveratrol deratives for better afficacy is underway [259].
Small molecule activators of p53 pathway
A significant cancer population carries a non-functional p53 tumor suppressor gene. The p53 functional defect can result due to the overexpression of its negative regulator MDM2 and or MDMX, or mutation in the gene body producing p53 non-functional for not properly folding. An elevated level of MDM2/MDMX prevents proper accumulation of p53. As conceivable, a mis-folded p53 can be dysfunctional in DNA binding or interaction with itself or another protein. Some population of p53 associated cancer is known to carry a nonsense mutation in their p53 gene. Given the strong mechanistic link between p53 and cancer, many small molecule activators of p53 with potential pharmacological values have been reported with some targeting the p53 defects discussed above. See [157] for review on p53 activators.
The first non-peptide small molecule that demonstrated the possibility of inhibiting the p53–MDM2 interaction was 4,5 dihydroimidazoline (nutlin; Roche) [252]. The crystal structure of nutlin 3a when bound to MDM2, provided the template for the design of better inhibitors such as the benzodiazepinedione family of compounds [359], chromenotriazolopyrimidine [360], terphenyls [361, 362] and chalcones [363, 364]. At the same time, structure-based screening of compounds combined with molecular modelling enabled the development of a new class of inhibitors like the spiro-oxindole based molecules. This led to the identification of MI219, which has a subnanomolar affinity for MDM2, can taken orally. MI-219 has good pharmacokinetic and pharmacodynamic (PK/PD) properties and increases the level of p53 as well as the p53 target genes CDKN1A and MDM2 [365]. The same group also developed a series of diastereomeric spiro-oxindoles such as MI888 which is a potent MDM2 inhibitor (Ki =0.44 nM) with a superior pharmacokinetic profile and enhanced in vivo efficacy [366]. Simultaneously, other drugs that have been reported so far include pyrazole and imidazole compounds [367], imidazole-indoles [368], isoindolinones [369] pyrrolidinones such as PXN822 [370, 371], piperidines [372], spirooxindoles [373] and the sulphonamide NSC279287 [374].
Restoration of p53 activity by inhibition of the p53-MDM2 interaction has been considered an attractive approach for cancer treatment. Currently, the most advanced MDM2 inhibitors include RG7112 (Roche), RO5503781 (Roche), MI773 (Sanofi), DS3032b (Daiichi Sankyo), which are at various stages of Phase I clinical trials [157]. After the discovery of RG7112, which was the first small-molecule p53-MDM2 inhibitor in clinical development, many groups reported the discovery and characterization of a second generation clinical MDM2 inhibitor, with superior potency and selectivity like RG7388 [375], RO5353 and RO2468 (two new potent, selective, and orally active p53-MDM2 antagonists [376], AM-8553 (a potent and selective piperidinone inhibitor of the MDM2-p53 interaction.with promising potential for clinical development) [377] and AMG 232 (an extremely potent MDM2 inhibitor with remarkable pharmacokinetic properties and in vivo antitumor activity) [378]. Recently a putative small-molecule inhibitor of p53-MDM2 interaction, pyranoxanthone, was discovered using a yeast p53 transactivation assay based approach [379]. Pyranoxanthone mimicked the activity of known p53 activators, leading to p53 stabilization and activation of p53-dependent transcriptional activity. A novel and promising lead structure for the development of anticancer drugs as MDM2-p53 interaction disruptor, 3-benzylideneindolin-2-one derivative, was also identified recently using both pharmacophore- and structure-based approaches [380].
A few preclinical lead compounds which restore the activity of mutant p53 also recently completed Phase I trials. PRIMA1 (a 2,2bis(hydroxymethyl)-3quinuclidinone and its structural analogue PRIMA-1 Met (APR246, developed by Aprea) have been shown to restore mutant p53 activity in vitro and in vivo[381–383]. PRIMA-1 Met seems to lead to the formation of covalent adducts on mutant p53-R175H and p53-R273H proteins, but its exact mechanism of action has yet to be fully understood [384] and needs further investigation. PRIMA-1 Met also seems to be able to restore the function of mutant p63 (a p53 homologue) [385, 386]. Other therapeutics that target mutant p53 via various mechanisms have been described; for example, NSC176327 and RETRA disrupt the binding interactions between mutant p53 and p73, whereas MIRA1 eliminates mutant p53 [371, 387].
Two nonsense mutations at position R196X and R213X in p53 gene have been linked with large number of cancer patients. Thus, small molecules and drugs that promote the read-through of nonsense codons in p53 could provide a novel approach for treating tumours carrying this type of mutation [388, 389]. A recent study showed that treatment of the human tumour cell line HDQP1, which contains a homozygous nonsense mutation at codon 213 (CGA-TGA), with the read-through-promoting aminoglycoside antibiotic G418 led to a dramatic increase in the level of TP53 mRNA and full-length p53 protein [390]. This restored full-length protein might re-establish the p53-MDM2 feedback loop, so a combination of G418 and nutlin may be especially effective. There are a number of potential read-through drugs in development because of high demand due to the association of p53-readthrough mutation with genetic diseases like Duchenne muscular dystrophy [157].
Cell-based screens for activators of the p53 pathway have identified large numbers of compounds which have unknown targets and incompletely defined mechanisms of action. In a few cases, the mechanisms by which these compounds activate p53 have been determined. These include the small molecule RITA (to target p53 itself) [391] (cyclin-dependent kinase (CDK) inhibitors such as roscovitine [246, 392], RNA polymerase inhibitors such as actinomycin D [393], exportin 1-binding compounds such as leptomycin B and KPT-330 [394], the NEDD1 (neural precursor cell expressed developmentally downregulated protein 1) ligase inhibitor MLN4924 [395] and sirtuin inhibitors such as the tenovins [396]. All of the compounds described above (with the possible exception of RITA) act on targets that will affect other pathways in addition to the p53 pathway and will therefore require careful evaluation in preclinical models.
Several MDM2 inhibitors derived from natural products have also been identified, such as the prenylated xanthones α-mangostin (from the fruit of Garcinia mangostana L.) and gambogic acid (from the resin of Garcinia hanburyi) [379, 397]. These molecules are thought to bind in a manner similar to nutlins and open up future prospect for the development of new classes of non-toxic inhibitors. As natural products, they also offer potential as chemopreventive anticancer agents without side effects. Marine organisms are now also being considered as a potential resource for a variety of lead compounds. For example, the siladenoserinols, derived from the marine invertebrate family Didemnidae, were recently reported to inhibit the p53–MDM2 interactions [398].
Small molecule modulators of both p53 and NF- κ B pathways
Cancer is a complex disease and results due to defect in multiple signaling pathways [399]. Thus drug targeting multiple pathways is thought to be a better option in cancer therapy than a single component [102, 129]. In fact, combinatorial therapy or drugs that affect multiple signaling pathways or multiple steps in one pathway are expected to be more effective in cancer treatment. Critical roles of NF- κ B in cancer include induction of genes that prevent cell death and promote cell proliferation, and antagonize tumor suppressor p53. Several compounds simulteneously targeting more than one pathways such as inhibition of NF- κ B while activating p53 have been considered very promising chemopreventive agents with some already are in the various phases of clinical trials (Table 2). Anti-malaria drug quinacrine was identified to have dual activities of inhibiting NF- κ B and activating inactive cellular p53 by cell based assays. Quinacrine as well as other derivatives of aminoacridines are DNA-damage mimetic, non genotoxic with excellent anticancer therapeutic potential as tested in mouse xenograph models. This is notable because anticancer agents like cisplatin induces p53 by formation of covalent-DNA adducts. Quinacrine inhibits both constitutive and inducible form of NF- κ B [104]. This NF- κ B inhibitory activity has been shown to be independent of the p53 status of a cell because quinacrine strongly inhibits NF- κ B irrespective of their p53 status [104]. The p53 inducing activity of quinacrine which acts in micromolar range is thought to be based on its ability the planar molecule to intercalate between the adjacent DNA base pairs with its side chain interacting with the minor groove [103]. Quinacrine can also target several signaling pathways such as PI3K/AKT/mTOR in addition to NF- κ B and p53 [257]. Curaxins are newly discovered prospective anticancer drugs that at nano molar concentration simultaneously down regulate NF- κ B while activating the p53 [400]. Like quinacrine, curaxins activate p53 without detectable damage in the cellular DNA. These carbazol structure based compounds intercalates between the DNA base pairs while its side chain binds the minor group of nucleic acids. Mechanistically, the anticancer property of this group of compounds may rely on the trapping of the transcription elongation factor FACT (facilitates chromatin transcription) on the chromatin making it unavailable for transcription of the NF- κ B target genes that are dependent on FACT [401, 402]. FACT upon binding to DNA-curaxin complex induces casein kinase 2 (CK2). Activated CK2 phosphorylates p53 (at serine 392) to drive p53 for interaction with FACT. Curaxins apparently does not interfere with other aspects in the NF- κ B activation pathway such as nuclear translocation of p65/p50 heterodimer. FACT, originally implicated as histone H2A/2B chaperone and a heterodimer of SSRP1 and Spt16 is utilized by RNA polymerase II for transcription of nucleosomal DNA [403]. Lippard and colleagues earlier identified FACT complex by its ability to bind cisplatin modified DNA [404].
R-Roscovitine (seliciclib, CYC202), a 2, 6, 9 substituted purine analogue is another small molecule that prevent tumor growth by targeting multiple signaling pathways simultaneously. It blocks constitutively active NF- κ B activity while activating the p53 tumor suppressor and in phase II clinical trial for its anticancer property. It was shown that R-Roscovitine abrogated TNF- α and IL-1 mediated induction of NF- κ B by preventing I κ B kinase (IKK) kinase activity [245]. The purine analogue also blocks p65 phosphorylation at ser536 by IKK that is required for nuclear translocation and chromatin remodeling function. At the level of transcription, R-Roscovitine represses transcription of NF- κ B target genes MCP-1, ICAM-1, COX2 and IL-8 [245, 392]. R-Roscovitine originally isolated as cyclin dependent kinase inhibitor (CDK), induces p53 by disrupting p53-MDM2 interaction. R-Roscovitine was shown to downregulate MDM2 both at the level of protein and mRNA [247]. Retinoblastoma protein phosphorylation and activation of MAP kinase are other identified functions of R-Roscovitine [405].
Flavoridol (Table 2), an inhibitor of multiple CDKs (CDK1, -2, -4 and -7) as well simultaneously inhibits NF- κ B and activates p53 is considered as an agent in combinational chemotherapy. A semisynthetic flavonoid, flavoperidol blocks TNF α induced NF- κ B induction by blocking steps in the NF- κ B activation pathway such as I κ B α kinase and p65 nuclear translocation, and cyclin D1 transcription [250]. Flavoperidol inhibits cellular transcription through targeting positive transcription elongation factor (pTEFb), a kinase targeting the serine 2 residue of largest subunit of RNA polymerase II [249, 406]. Flavoperidol was shown to reversibly activate p53 by inhibiting MDM2, and sensitize cells towards apoptosis [406, 407].
Nutlin was isolated as the first specific interrupter of p53-MDM2 interaction and is considered as a potent inducer of p53. It was also shown to inhibit activation of NF- κ B target genes such as ICAM-1 and MCP-1 through p53 dependent manner in lung cancer cells [251, 252]. While p53 can induce MDM2 expression; MDM2 on the other hand can influence p53 activity in multiple ways including inhibition of transactivation function, removing the tumor suppressor out of nucleus, and directing it to the proteasomal degradation pathway through its own E3 ubiquitin ligase activity [408]. Grasberger and colleagues also isolated benzodiazepinedione based MDM2 inhibitors with similar functional properties [359, 409]. A number of compounds (peptide/small molecules) are being studied that is thought to stabilize p53 activity inducing proper folding correcting readthrough error in the p53 gene to activate p53 functions that include for example, PRIMA-1 [383] or RTC13 [410], respectively. Compounds that are doubly inhibitory against MDM2 and MDM4 have also been under investigation [146]. These later group of compounds should be used along with MDM2 inhibtors for effective p53 accumulation. In principle these compounds would be able to work like nutlins mechanistically for their anticancer properties.
Synthetic derivative of benzofuran lignan (Benfur) has a reported antitumor activity. Benfur-mediated cell death is partially regulated by the inhibition of NF- κ B activity but it is predominantly dependent on p53-mediated pathway [258]. Benfur potentially inhibits NF- κ B DNA binding activity by inhibiting I κ B α and thereby blocking the nuclear translocation of p65 in both Jurkat and U937 cells. Benfur treatment was shown to increase p53 level by inhibiting Sp1 binding on the MDM2 promoter [258].
Conclusions
NF- κ B, a central regulator of innate immune response, normally is activated in a time dependent manner as a host protection mechanism. It has been now established by numerous independent studies that a persistent long term activation of this factor is tumorigenic and blockade of the activities of an inflammatory mediator regress tumor progression as well as its aggressiveness. Studies from various independent laboratories have already pinpointed some mediators in the NF- κ B activation pathway that undergo aberrant regulation in various cancers. Studies from many laboratories have firmly established a reverse correlation of activation between the NF- κ B and p53 pathways highlighting a prospective avenue in cancer chemotherapy. Several small molecules of natural or synthetic origin many of which target multiple signaling pathways including NF- κ B and p53 apparently hold a great promise to move the cancer treatment and management in the desired direction. In this direction, various potential pharmacological agents have been isolated that activate p53 in tumor cells with high potency at very low concentration. Most of these small molecules appears to have at least part their mode action through inhibition of NF- κ B. Conversely, it is conceivable that the anticancer activity of many NF- κ B inhibitors is partly due to their ability to induce p53 in cancer cells. Efforts are already underway to find small molecules that will with higher specificity rectify the defect in the pathways to suppress NF- κ B activity. Clearly, more studies are needed to provide us with more insightful understanding of the mechanism of deregulation and the underlying cause. Given the role of NF- κ B in innate immunity and cancer, a desired objective would be to achieve the ability to turn off and on the function of NF- κ B with high precision and as needed.
References
Stein CJ, Colditz GA:Modifiable risk factors for cancer. Br J Cancer. 2004, 90: 299-303.
Anand P, Kunnumakkara AB, Sundaram C, Harikumar KB, Tharakan ST, Lai OS, Sung B, Aggarwal BB:Cancer is a preventable disease that requires major lifestyle changes. Pharm Res. 2008, 25: 2097-2116.
Rossi AG, Sawatzky DA: The Resolution of Inflammation. 2008, Birkhauser: Basel
Balkwill F, Mantovani A:Inflammation and cancer: back to Virchow?. Lancet. 2001, 357: 539-545.
Coussens LM, Werb Z:Inflammation and cancer. Nature. 2002, 420: 860-867.
Philip M, Rowley DA, Schreiber H:Inflammation as a tumor promoter in cancer induction. Semin Cancer Biol. 2004, 14: 433-439.
Beekhuizen H, van Furth R:Monocyte adherence to human vascular endothelium. J Leukoc Biol. 1993, 54: 363-378.
Kulidjian AA, Inman R, Issekutz TB:Rodent models of lymphocyte migration. Semin Immunol. 1999, 11: 85-93.
van Gils JM, Zwaginga JJ, Hordijk PL:Molecular and functional interactions among monocytes, platelets, and endothelial cells and their relevance for cardiovascular diseases. J Leukoc Biol. 2009, 85: 195-204.
Eming SA, Krieg T, Davidson JM:Inflammation in wound repair: molecular and cellular mechanisms. J Invest Dermatol. 2007, 127: 514-525.
Weiss SJ:Tissue destruction by neutrophils. N Engl J Med. 1989, 320: 365-376.
Balkwill F, Coussens LM:Cancer: an inflammatory link. Nature. 2004, 431: 405-406.
Sato Y, Ohshima T, Kondo T:Regulatory role of endogenous interleukin-10 in cutaneous inflammatory response of murine wound healing. Biochem Biophys Res Commun. 1999, 265: 194-199.
Erdman SE, Poutahidis T:Roles for inflammation and regulatory T cells in colon cancer. Toxicol Pathol. 2010, 38: 76-87.
Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W:Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993, 75: 263-274.
Ho L, Davis RE, Conne B, Chappuis R, Berczy M, Mhawech P, Staudt LM, Schwaller J:MALT1 and the API2-MALT1 fusion act between CD40 and IKK and confer NF-kappa B-dependent proliferative advantage and resistance against FAS-induced cell death in B cells. Blood. 2005, 105: 2891-2899.
Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A:Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001, 19: 683-765.
Rizzo A, Pallone F, Monteleone G, Fantini MC:Intestinal inflammation and colorectal cancer: a double-edged sword?. World J Gastroenterol. 2011, 17: 3092-3100.
Schottelius AJ, Mayo MW, Sartor RB, Baldwin AS:Interleukin-10 signaling blocks inhibitor of kappaB kinase activity and nuclear factor kappaB DNA binding. J Biol Chem. 1999, 274: 31868-31874.
Mantovani A:Molecular pathways linking inflammation and cancer. Curr Mol Med. 2010, 10: 369-373.
Harhaj EW, Dixit VM:Deubiquitinases in the regulation of NF-kappaB signaling. Cell Res. 2012, 21: 22-39.
de Visser KE, Eichten A, Coussens LM:Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006, 6: 24-37.
Lin WW, Karin M:A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007, 117: 1175-1183.
Kuper H, Adami HO, Trichopoulos D:Infections as a major preventable cause of human cancer. J Intern Med. 2000, 248: 171-183.
Sunami Y, Wirth T:Intestinal carcinogenesis: IKK can go all the way. J Clin Invest. 2011, 121: 2551-2553.
Mossman BT, Lounsbury KM, Reddy SP:Oxidants and signaling by mitogen-activated protein kinases in lung epithelium. Am J Respir Cell Mol Biol. 2006, 34: 666-669.
Vaid M, Katiyar SK:Molecular mechanisms of inhibition of photocarcinogenesis by silymarin, a phytochemical from milk thistle (Silybum marianum L. Gaertn.) (Review). Int J Oncol. 2010, 36: 1053-1060.
Schutte K, Bornschein J, Malfertheiner P:Hepatocellular carcinoma–epidemiological trends and risk factors. Dig Dis. 2009, 27: 80-92.
Stauffer JK, Scarzello AJ, Jiang Q, Wiltrout RH:Chronic inflammation, immune escape and oncogenesis in the liver: a unique neighborhood for novel intersections. Hepatology. 2012, 56 (4): 1567-1574.
Loffeld RJ, Willems I, Flendrig JA, Arends JW:Helicobacter pylori and gastric carcinoma. Histopathology. 1990, 17: 537-541.
Stolte M:Helicobacter pylori gastritis and gastric MALT-lymphoma. Lancet. 1992, 339: 745-746.
Ernst PB, Gold BD:The disease spectrum of Helicobacter pylori: the immunopathogenesis of gastroduodenal ulcer and gastric cancer. Annu Rev Microbiol. 2000, 54: 615-640.
Shacter E, Weitzman SA:Chronic inflammation and cancer. Oncology (Williston Park). 2002, 16: 217-226, 229; discussion 230–212.
Nicholson A, Jankowski J:Acid reflux and oesophageal cancer. Recent Results Cancer Res. 2011, 185: 65-82.
Gryseels B:Schistosomiasis. Infect Dis Clin North Am. 2012, 26: 383-397.
Parkin DM:The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006, 118: 3030-3044.
King CH:Parasites and poverty: the case of schistosomiasis. Acta Trop. 2010, 113: 95-104.
Schwartz DA:Helminths in the induction of cancer: Opisthorchis viverrini, Clonorchis sinensis and cholangiocarcinoma. Trop Geogr Med. 1980, 32: 95-100.
Abraham C, Medzhitov R:Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology. 2011, 140: 1729-1737.
Clapper ML, Gary MA, Coudry RA, Litwin S, Chang WC, Devarajan K, Lubet RA, Cooper HS:5-aminosalicylic acid inhibits colitis-associated colorectal dysplasias in the mouse model of azoxymethane/dextran sulfate sodium-induced colitis. Inflamm Bowel Dis. 2008, 14: 1341-1347.
de Martel C, Franceschi S:Infections and cancer: established associations and new hypotheses. Crit Rev Oncol Hematol. 2009, 70: 183-194.
Grewal P, Viswanathen VA:Liver cancer and alcohol. Clin Liver Dis. 2012, 16: 839-850.
Rook GA, Dalgleish A:Infection, immunoregulation, and cancer. Immunol Rev. 240: 141-159.
Wang HJ, Gao B, Zakhari S, Nagy LE:Inflammation in alcoholic liver disease. Annu Rev Nutr. 2012, 32: 343-368.
Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM:The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990, 63: 1129-1136.
James JA, Kaufman KM, Farris AD, Taylor-Albert E, Lehman TJ, Harley JB:An increased prevalence of Epstein-Barr virus infection in young patients suggests a possible etiology for systemic lupus erythematosus. J Clin Invest. 1997, 100: 3019-3026.
Kutok JL, Wang F:Spectrum of Epstein-Barr virus-associated diseases. Annu Rev Pathol. 2006, 1: 375-404.
Mackenzie I, Rous P:The experimental disclosure of latent neoplastic changes in tarred skin. J Exp Med. 1941, 73: 391-416.
Balkwill F, Charles KA, Mantovani A:Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005, 7: 211-217.
Velicer CM, Heckbert SR, Lampe JW, Potter JD, Robertson CA, Taplin SH:Antibiotic use in relation to the risk of breast cancer. JAMA. 2004, 291: 827-835.
Attur MG, Patel RN, Patel PD, Abramson SB, Amin AR:Tetracycline up-regulates COX-2 expression and prostaglandin E2 production independent of its effect on nitric oxide. J Immunol. 1999, 162: 3160-3167.
Enzler T, Gillessen S, Manis JP, Ferguson D, Fleming J, Alt FW, Mihm M, Dranoff G:Deficiencies of GM-CSF and interferon gamma link inflammation and cancer. J Exp Med. 2003, 197: 1213-1219.
Ghosh S, Karin M:Missing pieces in the NF-kappaB puzzle. Cell. 2002, 109 Suppl: S81-S96.
Ghosh S, Hayden MS:Celebrating 25 years of NF-kappaB research. Immunol Rev. 2012, 246: 5-13.
Sen R, Baltimore D:Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell. 1986, 47: 921-928.
Karin M, Ben-Neriah Y:Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000, 18: 621-663.
Vallabhapurapu S, Karin M:Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009, 27: 693-733.
Karin M, Yamamoto Y, Wang QM:The IKK NF-kappa B system: a treasure trove for drug development. Nat Rev Drug Discov. 2004, 3: 17-26.
Zhong H, May MJ, Jimi E, Ghosh S:The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol Cell. 2002, 9: 625-636.
Cha-Molstad H, Agrawal A, Zhang D, Samols D, Kushner I:The Rel family member P50 mediates cytokine-induced C-reactive protein expression by a novel mechanism. J Immunol. 2000, 165: 4592-4597.
Perkins ND:Oncogenes, tumor suppressors and p52 NF-kappaB. Oncogene. 2003, 22: 7553-7556.
Akira S:Pathogen recognition by innate immunity and its signaling. Proc Jpn Acad Ser B Phys Biol Sci. 2009, 85: 143-156.
Medzhitov R:Toll-like receptors and innate immunity. Nat Rev Immunol. 2001, 1: 135-145.
Girardin SE, Tournebize R, Mavris M, Page AL, Li X, Stark GR, Bertin J, DiStefano PS, Yaniv M, Sansonetti PJ, Philpott DJ:CARD4/Nod1 mediates NF-kappaB and JNK activation by invasive Shigella flexneri. EMBO Rep. 2001, 2: 736-742.
Newton K, Dixit VM:Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012, 4: 1-19.
Philpott DJ, Yamaoka S, Israel A, Sansonetti PJ:Invasive Shigella flexneri activates NF-kappa B through a lipopolysaccharide-dependent innate intracellular response and leads to IL-8 expression in epithelial cells. J Immunol. 2000, 165: 903-914.
Robertson SJ, Girardin SE:Nod-like receptors in intestinal host defense: controlling pathogens, the microbiota, or both?. Curr Opin Gastroenterol. 2012, 29: 15-22.
Barnich N, Aguirre JE, Reinecker HC, Xavier R, Podolsky DK:Membrane recruitment of NOD2 in intestinal epithelial cells is essential for nuclear factor-{kappa}B activation in muramyl dipeptide recognition. J Cell Biol. 2005, 170: 21-26.
Fritz JH, Ferrero RL, Philpott DJ, Girardin SE:Nod-like proteins in immunity, inflammation and disease. Nat Immunol. 2006, 7: 1250-1257.
Sancho D, Reis ESC:Signaling by myeloid C-type lectin receptors in immunity and homeostasis. Annu Rev Immunol. 2012, 30: 491-529.
Klesney-Tait J, Turnbull IR, Colonna M:The TREM receptor family and signal integration. Nat Immunol. 2006, 7: 1266-1273.
Arts RJ, Joosten LA, van der Meer JW, Netea MG:TREM-1: intracellular signaling pathways and interaction with pattern recognition receptors. J Leukoc Biol. 2012, 93: 209-215.
Bouchon A, Dietrich J, Colonna M:Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J Immunol. 2000, 164: 4991-4995.
Ford JW, McVicar DW:TREM and TREM-like receptors in inflammation and disease. Curr Opin Immunol. 2009, 21: 38-46.
Israel A:The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. 2010, 2: a.000158-
Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K:The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature. 1999, 398: 252-256.
Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ:TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001, 412: 346-351.
Takaesu G, Surabhi RM, Park KJ, Ninomiya-Tsuji J, Matsumoto K, Gaynor RB:TAK1 is critical for IkappaB kinase-mediated activation of the NF-kappaB pathway. J Mol Biol. 2003, 326: 105-115.
Irie T, Muta T, Takeshige K:TAK1 mediates an activation signal from toll-like receptor(s) to nuclear factor-kappaB in lipopolysaccharide-stimulated macrophages. FEBS Lett. 2000, 467: 160-164.
Huang Q, Yang J, Lin Y, Walker C, Cheng J, Liu ZG, Su B:Differential regulation of interleukin 1 receptor and Toll-like receptor signaling by MEKK3. Nat Immunol. 2004, 5: 98-103.
Yang J, Lin Y, Guo Z, Cheng J, Huang J, Deng L, Liao W, Chen Z, Liu Z, Su B:The essential role of MEKK3 in TNF-induced NF-kappaB activation. Nat Immunol. 2001, 2: 620-624.
Delhase M, Hayakawa M, Chen Y, Karin M:Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science. 1999, 284: 309-313.
Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, Li ZW, Karin M, Ware CF, Green DR:The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity. 2002, 17: 525-535.
Senftleben U, Cao Y, Xiao G, Greten FR, Krähn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M:Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001, 293: 1495-1499.
Sil AK, Maeda S, Sano Y, Roop DR, Karin M:IkappaB kinase-alpha acts in the epidermis to control skeletal and craniofacial morphogenesis. Nature. 2004, 428: 660-664.
Novack DV, Yin L, Hagen-Stapleton A, Schreiber RD, Goeddel DV, Ross FP, Teitelbaum SL:The IkappaB function of NF-kappaB2 p100 controls stimulated osteoclastogenesis. J Exp Med. 2003, 198: 771-781.
DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M:A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997, 388: 548-554.
Rothwarf DM, Zandi E, Natoli G, Karin M:IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature. 1998, 395: 297-300.
Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M:The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. 1997, 91: 243-252.
Liu F, Xia Y, Parker AS, Verma IM:IKK biology. Immunol Rev. 2012, 246: 239-253.
Chen Z, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D, Maniatis T:Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995, 9: 1586-1597.
Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A:IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science. 1997, 278: 860-866.
Kanarek N, Ben-Neriah Y:Regulation of NF-kappaB by ubiquitination and degradation of the IkappaBs. Immunol Rev. 2012, 246: 77-94.
Yaron A, Gonen H, Alkalay I, Hatzubai A, Jung S, Beyth S, Mercurio F, Manning AM, Ciechanover A, Ben-Neriah Y:Inhibition of NF-kappa-B cellular function via specific targeting of the I-kappa-B-ubiquitin ligase. EMBO J. 1997, 16: 6486-6494.
Yaron A, Hatzubai A, Davis M, Lavon I, Amit S, Manning AM, Andersen JS, Mann M, Mercurio F, Ben-Neriah Y:Identification of the receptor component of the IkappaBalpha-ubiquitin ligase. Nature. 1998, 396: 590-594.
Ghosh G, Wang VY, Huang DB, Fusco A:NF-kappaB regulation: lessons from structures. Immunol Rev. 2012, 246: 36-58.
Diaz-Meco MT, Moscat J:The atypical PKCs in inflammation: NF-kappaB and beyond. Immunol Rev. 2012, 246: 154-167.
Ghosh S, Gifford AM, Riviere LR, Tempst P, Nolan GP, Baltimore D:Cloning of the p50 DNA binding subunit of NF-kappa B: homology to rel and dorsal. Cell. 1990, 62: 1019-1029.
Gilmore TD:NF-kappa B, KBF1, dorsal, and related matters. Cell. 1990, 62: 841-843.
Karin M, Greten FR:NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005, 5: 749-759.
Aggarwal BB, Gehlot P:Inflammation and cancer: how friendly is the relationship for cancer patients?. Curr Opin Pharmacol. 2009, 9: 351-369.
Staudt LM:Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol. 2010, 2: a.000109-
Gasparian AV, Burkhart CA, Purmal AA, Brodsky L, Pal M, Saranadasa M, Bosykh DA, Commane M, Guryanova OA, Pal S, Safina A, Sviridov S, Koman IE, Veith J, Komar AA, Gudkov AV, Gurova KV:Curaxins: anticancer compounds that simultaneously suppress NF-kappaB and activate p53 by targeting FACT. Sci Transl Med. 2011, 3: 95ra74-
Gurova KV, Hill JE, Guo C, Prokvolit A, Burdelya LG, Samoylova E, Khodyakova AV, Ganapathi R, Ganapathi M, Tararova ND, Bosykh D, Lvovskiy D, Webb TR, Stark GR, Gudkov AV:Small molecules that reactivate p53 in renal cell carcinoma reveal a NF-kappaB-dependent mechanism of p53 suppression in tumors. Proc Natl Acad Sci U S A. 2005, 102: 17448-17453.
Vlantis K, Wullaert A, Sasaki Y, Schmidt-Supprian M, Rajewsky K, Roskams T, Pasparakis M:Constitutive IKK2 activation in intestinal epithelial cells induces intestinal tumors in mice. J Clin Invest. 2011, 121: 2781-2793.
Natarajan V, Komarov AP, Ippolito T, Bonneau K, Chenchik AA, Gudkov AV:Peptides genetically selected for NF-kappaB activation cooperate with oncogene Ras and model carcinogenic role of inflammation. Proc Natl Acad Sci U S A. 2014, 111: E474-E483.
Sun J, Wiklund F, Hsu FC, Bälter K, Zheng SL, Johansson JE, Chang B, Liu W, Li T, Turner AR, Li L, Li G, Adami HO, Isaacs WB, Xu J, Grönberg H:Interactions of sequence variants in interleukin-1 receptor-associated kinase4 and the toll-like receptor 6-1-10 gene cluster increase prostate cancer risk. Cancer Epidemiol Biomarkers Prev. 2006, 15: 480-485.
So EY, Ouchi T:The application of Toll like receptors for cancer therapy. Int J Biol Sci. 2010, 6: 675-681.
Yu L, Chen S:Toll-like receptors expressed in tumor cells: targets for therapy. Cancer Immunol Immunother. 2008, 57: 1271-1278.
Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S, Hsu D, Xu R, Harpaz N, Dannenberg AJ, Subbaramaiah K, Cooper HS, Itzkowitz SH, Abreu MT:Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology. 2007, 133: 1869-1881.
Wolska A, Lech-Maranda E, Robak T:Toll-like receptors and their role in carcinogenesis and anti-tumor treatment. Cell Mol Biol Lett. 2009, 14: 248-272.
Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH, Targan SR, Xavier RJ, Libioulle C, Sandor C, Lathrop M, Belaiche J, Dewit O, Gut I, Heath S, et al:Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008, 40: 955-962.
Hugot JP:[Role of NOD2 gene in Crohn’s disease]. Gastroenterol Clin Biol. 2002, 26: 13-15.
Xavier RJ, Podolsky DK:Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007, 448: 427-434.
Eckmann L, Karin M:NOD2 and Crohn’s disease: loss or gain of function?. Immunity. 2005, 22: 661-667.
Sehouli J, Mustea A, Konsgen D, Katsares I, Lichtenegger W:Polymorphism of IL-1 receptor antagonist gene: role in cancer. Anticancer Res. 2002, 22: 3421-3424.
Troost E, Hold GL, Smith MG, Chow WH, Rabkin CS, McColl KE, El-Omar EM:The role of interleukin-1beta and other potential genetic markers as indicators of gastric cancer risk. Can J Gastroenterol. 2003, 17 Suppl B: 8B-12B.
Apte RN, Dotan S, Elkabets M, White MR, Reich E, Carmi Y, Song X, Dvozkin T, Krelin Y, Voronov E:The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 2006, 25: 387-408.
Lewis AM, Varghese S, Xu H, Alexander HR:Interleukin-1 and cancer progression: the emerging role of interleukin-1 receptor antagonist as a novel therapeutic agent in cancer treatment. J Transl Med. 2006, 4: 48-
Lawrence T:The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009, 1: a.001651-
Prasad S, Ravindran J, Aggarwal BB:NF-kappaB and cancer: how intimate is this relationship. Mol Cell Biochem. 2010, 336: 25-37.
Fodde R, Smits R, Clevers H:APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer. 2001, 1: 55-67.
Ahmad R, Raina D, Trivedi V, Ren J, Rajabi H, Kharbanda S, Kufe D:MUC1 oncoprotein activates the IkappaB kinase beta complex and constitutive NF-kappaB signalling. Nat Cell Biol. 2007, 9: 1419-1427.
Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, Dave SS, Zhao H, Xu W, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Fisher RI, Chan WC, Staudt LM:Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008, 319: 1676-1679.
Yamaoka S, Inoue H, Sakurai M, Sugiyama T, Hazama M, Yamada T, Hatanaka M:Constitutive activation of NF-kappa B is essential for transformation of rat fibroblasts by the human T-cell leukemia virus type I Tax protein. EMBO J. 1996, 15: 873-887.
Wood KM, Roff M, Hay RT:Defective IkappaBalpha in Hodgkin cell lines with constitutively active NF-kappaB. Oncogene. 1998, 16: 2131-2139.
Mehta K:Biological and therapeutic significance of tissue transglutaminase in pancreatic cancer. Amino Acids. 2009, 36: 709-716.
Mann AP, Verma A, Sethi G, Manavathi B, Wang H, Fok JY, Kunnumakkara AB, Kumar R, Aggarwal BB, Mehta K:Overexpression of tissue transglutaminase leads to constitutive activation of nuclear factor-kappaB in cancer cells: delineation of a novel pathway. Cancer Res. 2006, 66: 8788-8795.
Gudkov AV, Gurova KV, Komarova EA:Inflammation and p53: a tale of two stresses. Genes Cancer. 2011, 2: 503-516.
Vucic D, Dixit VM, Wertz IE:Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat Rev Mol Cell Biol. 2011, 12: 439-452.
Huang WC, Ju TK, Hung MC, Chen CC:Phosphorylation of CBP by IKKalpha promotes cell growth by switching the binding preference of CBP from p53 to NF-kappaB. Mol Cell. 2007, 26: 75-87.
Mayo MW, Madrid LV, Westerheide SD, Jones DR, Yuan XJ, Baldwin ASJr., Whang YE:PTEN blocks tumor necrosis factor-induced NF-kappa B-dependent transcription by inhibiting the transactivation potential of the p65 subunit. J Biol Chem. 2002, 277: 11116-11125.
Perkins ND:The diverse and complex roles of NF-kappaB subunits in cancer. Nat Rev Cancer. 2012, 12: 121-132.
Rocha S, Campbell KJ, Perkins ND:p53- and Mdm2-independent repression of NF-kappa B transactivation by the ARF tumor suppressor. Mol Cell. 2003, 12: 15-25.
Wolff B, Naumann M:INK4 cell cycle inhibitors direct transcriptional inactivation of NF-kappaB. Oncogene. 1999, 18: 2663-2666.
Kashima L, Toyota M, Mita H, Suzuki H, Idogawa M, Ogi K, Sasaki Y, Tokino T:CHFR, a potential tumor suppressor, downregulates interleukin-8 through the inhibition of NF-kappaB. Oncogene. 2009, 28: 2643-2653.
Rattan R, Narita K, Chien J, Maguire JL, Shridhar R, Giri S, Shridhar V:TCEAL7, a putative tumor suppressor gene, negatively regulates NF-kappaB pathway. Oncogene. 2010, 29: 1362-1373.
Wang J, An H, Mayo MW, Baldwin AS, Yarbrough WG:LZAP, a putative tumor suppressor, selectively inhibits NF-kappaB. Cancer Cell. 2007, 12: 239-251.
Dell’Orso S, Fontemaggi G, Stambolsky P, Goeman F, Voellenkle C, Levrero M, Strano S, Rotter V, Oren M, Blandino G:ChIP-on-chip analysis of in vivo mutant p53 binding to selected gene promoters. OMICS. 2011, 15: 305-312.
Gudkov AV, Komarova EA:Pathologies associated with the p53 response. Cold Spring Harb Perspect Biol. 2010, 2: a.001180-
Murray-Zmijewski F, Slee EA, Lu X:A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol. 2008, 9: 702-712.
Ak P, Levine AJ:p53 and NF-kappaB: different strategies for responding to stress lead to a functional antagonism. FASEB J. 2010, 24: 3643-3652.
Hu W, Feng Z, Levine AJ:The regulation of multiple p53 stress responses is mediated through MDM2. Genes Cancer. 2012, 3: 199-208.
Huang L, Yan Z, Liao X, Li Y, Yang J, Wang ZG, Zuo Y, Kawai H, Shadfan M, Ganapathy S, Yuan ZM:The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proc Natl Acad Sci U S A. 2011, 108: 12001-12006.
Pant V, Xiong S, Iwakuma T, Quintas-Cardama A, Lozano G:Heterodimerization of Mdm2 and Mdm4 is critical for regulating p53 activity during embryogenesis but dispensable for p53 and Mdm2 stability. Proc Natl Acad Sci U S A. 2011, 108: 11995-12000.
Wade M, Li YC, Wahl GM:MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013, 13: 83-96.
Mayo LD, Turchi JJ, Berberich SJ:Mdm-2 phosphorylation by DNA-dependent protein kinase prevents interaction with p53. Cancer Res. 1997, 57: 5013-5016.
Zuckerman V, Lenos K, Popowicz GM, Silberman I, Grossman T, Marine JC, Holak TA, Jochemsen AG, Haupt Y:c-Abl phosphorylates Hdmx and regulates its interaction with p53. J Biol Chem. 2009, 284: 4031-4039.
Thut CJ, Goodrich JA, Tjian R:Repression of p53-mediated transcription by MDM2: a dual mechanism. Genes Dev. 1997, 11: 1974-1986.
Meek DW, Hupp TR:The regulation of MDM2 by multisite phosphorylation–opportunities for molecular-based intervention to target tumours?. Semin Cancer Biol. 2010, 20: 19-28.
Lopez-Pajares V, Kim MM, Yuan ZM:Phosphorylation of MDMX mediated by Akt leads to stabilization and induces 14-3-3 binding. J Biol Chem. 2008, 283: 13707-13713.
Mayo LD, Donner DB:A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A. 2001, 98: 11598-11603.
Goh AM, Lim CY, Chiam PC, Li L, Mann MB, Mann KM, Menendez S, Lane DP:Using targeted transgenic reporter mice to study promoter-specific p53 transcriptional activity. Proc Natl Acad Sci U S A. 2012, 109: 1685-1690.
Toledo F, Wahl GM:Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006, 6: 909-923.
Fei P, Bernhard EJ, El-Deiry WS:Tissue-specific induction of p53 targets in vivo. Cancer Res. 2002, 62: 7316-7327.
Goh AM, Lane DP:How p53 wields the scales of fate: arrest or death?. Transcription. 2012, 3: 240-244.
Khoo KH, Verma CS, Lane DP:Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014, 13: 217-236.
el-Deiry WS:Regulation of p53 downstream genes. Semin Cancer Biol. 1998, 8: 345-357.
el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B:Definition of a consensus binding site for p53. Nat Genet. 1992, 1: 45-49.
Piwnica-Worms H:Cell cycle. Fools Rush Nature. 1999, 401: 535, 537-
St Clair S, Giono L, Varmeh-Ziaie S, Resnick-Silverman L, Liu WJ, Padi A, Dastidar J, DaCosta A, Mattia M, Manfredi JJ:DNA damage-induced downregulation of Cdc25C is mediated by p53 via two independent mechanisms: one involves direct binding to the cdc25C promoter. Mol Cell. 2004, 16: 725-736.
Shaw P, Bovey R, Tardy S, Sahli R, Sordat B, Costa J:Induction of apoptosis by wild-type p53 in a human colon tumor-derived cell line. Proc Natl Acad Sci U S A. 1992, 89: 4495-4499.
Kortlever RM, Higgins PJ, Bernards R:Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006, 8: 877-884.
Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi A, Oren M:Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature. 1991, 352: 345-347.
Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM:DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006, 126: 121-134.
Beckerman R, Prives C:Transcriptional regulation by p53. Cold Spring Harb Perspect Biol. 2010, 2: a.000935-
Gurova K:New hopes from old drugs: revisiting DNA-binding small molecules as anticancer agents. Future Oncol. 2009, 5: 1685-1704.
Kashatus D, Cogswell P, Baldwin AS:Expression of the Bcl-3 proto-oncogene suppresses p53 activation. Genes Dev. 2006, 20: 225-235.
Tergaonkar V, Pando M, Vafa O, Wahl G, Verma I:p53 stabilization is decreased upon NFkappaB activation: a role for NFkappaB in acquisition of resistance to chemotherapy. Cancer Cell. 2002, 1: 493-503.
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME:Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997, 91: 231-241.
Jiang P, Du W, Heese K, Wu M:The Bad guy cooperates with good cop p53: Bad is transcriptionally up-regulated by p53 and forms a Bad/p53 complex at the mitochondria to induce apoptosis. Mol Cell Biol. 2006, 26: 9071-9082.
Vivanco I, Sawyers CL:The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002, 2: 489-501.
Khwaja A:Akt is more than just a Bad kinase. Nature. 1999, 401: 33-34.
Romashkova JA, Makarov SS:NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999, 401: 86-90.
Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB:NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999, 401: 82-85.
Xia Y, Padre RC, De Mendoza TH, Bottero V, Tergaonkar VB, Verma IM:Phosphorylation of p53 by IkappaB kinase 2 promotes its degradation by beta-TrCP. Proc Natl Acad Sci U S A. 2009, 106: 2629-2634.
Lowe SW, Sherr CJ:Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 2003, 13: 77-83.
Rocha S, Perkins ND:ARF the integrator: linking NF-kappaB, p53 and checkpoint kinases. Cell Cycle. 2005, 4: 756-759.
Perkins ND:Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007, 8: 49-62.
Ikeda A, Sun X, Li Y, Zhang Y, Eckner R, Doi TS, Takahashi T, Obata Y, Yoshioka K, Yamamoto K:p300/CBP-dependent and -independent transcriptional interference between NF-kappaB RelA and p53. Biochem Biophys Res Commun. 2000, 272: 375-379.
Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine AS, Kelly K:Recruitment of p300/CBP in p53-dependent signal pathways. Cell. 1997, 89: 1175-1184.
Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T:CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci U S A. 1997, 94: 2927-2932.
Webster GA, Perkins ND:Transcriptional cross talk between NF-kappaB and p53. Mol Cell Biol. 1999, 19: 3485-3495.
Tergaonkar V, Perkins ND:p53 and NF-kappaB crosstalk: IKKalpha tips the balance. Mol Cell. 2007, 26: 158-159.
Kawauchi K, Araki K, Tobiume K, Tanaka N:Activated p53 induces NF-kappaB DNA binding but suppresses its transcriptional activation. Biochem Biophys Res Commun. 2008, 372: 137-141.
Guo AK, Hou YY, Hirata H, Yamauchi S, Yip AK, Chiam KH, Tanaka N, Sawada Y, Kawauchi K:Loss of p53 enhances NF-kappaB-dependent lamellipodia formation. J Cell Physiol. 2014, 229: 696-704.
Kawauchi K, Araki K, Tobiume K, Tanaka N:p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol. 2008, 10: 611-618.
Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E:The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64: 2627-2633.
Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH:TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006, 126: 107-120.
Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM:p53 regulates mitochondrial respiration. Science. 2006, 312: 1650-1653.
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CAJr., Butel JS, Bradley A:Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992, 356: 215-221.
Komarova EA, Krivokrysenko V, Wang K, Neznanov N, Chernov MV, Komarov PG, Brennan ML, Golovkina TV, Rokhlin OW, Kuprash DV, Nedospasov SA, Hazen SL, Feinstein E, Gudkov AV:p53 is a suppressor of inflammatory response in mice. FASEB J. 2005, 19: 1030-1032.
Appel A:Drugs: more shots on target. Nature. 2011, 480: S40-S42.
Gilmore TD, Herscovitch M:Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene. 2006, 25: 6887-6899.
Alkalay I, Yaron A, Hatzubai A, Orian A, Ciechanover A, Ben-Neriah Y:Stimulation-dependent I kappa B alpha phosphorylation marks the NF-kappa B inhibitor for degradation via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1995, 92: 10599-10603.
Meister S, Schubert U, Neubert K, Herrmann K, Burger R, Gramatzki M, Hahn S, Schreiber S, Wilhelm S, Herrmann M, Jäck HM, Voll RE:Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer Res. 2007, 67: 1783-1792.
Kane RC, Bross PF, Farrell AT, Pazdur R:Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist. 2003, 8: 508-513.
Kane RC, Farrell AT, Sridhara R, Pazdur R:United States Food and Drug Administration approval summary: bortezomib for the treatment of progressive multiple myeloma after one prior therapy. Clin Cancer Res. 2006, 12: 2955-2960.
Ruschak AM, Slassi M, Kay LE, Schimmer AD:Novel proteasome inhibitors to overcome bortezomib resistance. J Natl Cancer Inst. 2011, 103: 1007-1017.
DiDonato JA, Mercurio F, Karin M:NF-kappaB and the link between inflammation and cancer. Immunol Rev. 2012, 246: 379-400.
Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M:S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006, 314: 467-471.
Ding Q, He X, Hsu JM, Xia W, Chen CT, Li LY, Lee DF, Liu JC, Zhong Q, Wang X, Hung MC:Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol. 2007, 27: 4006-4017.
Busino L, Donzelli M, Chiesa M, Guardavaccaro D, Ganoth D, Dorrello NV, Hershko A, Pagano M, Draetta GF:Degradation of Cdc25A by beta-TrCP during S phase and in response to DNA damage. Nature. 2003, 426: 87-91.
Kanemori Y, Uto K, Sagata N:Beta-TrCP recognizes a previously undescribed nonphosphorylated destruction motif in Cdc25A and Cdc25B phosphatases. Proc Natl Acad Sci U S A. 2005, 102: 6279-6284.
Fuchs SY, Chen A, Xiong Y, Pan ZQ, Ronai Z:HOS, a human homolog of Slimb, forms an SCF complex with Skp1 and Cullin1 and targets the phosphorylation-dependent degradation of IkappaB and beta-catenin. Oncogene. 1999, 18: 2039-2046.
Kitagawa M, Hatakeyama S, Shirane M, Matsumoto M, Ishida N, Hattori K, Nakamichi I, Kikuchi A, Nakayama K:An F-box protein, FWD1, mediates ubiquitin-dependent proteolysis of beta-catenin. EMBO J. 1999, 18: 2401-2410.
Mudduluru G, Medved F, Grobholz R, Jost C, Gruber A, Leupold JH, Post S, Jansen A, Colburn NH, Allgayer H:Loss of programmed cell death 4 expression marks adenoma-carcinoma transition, correlates inversely with phosphorylated protein kinase B, and is an independent prognostic factor in resected colorectal cancer. Cancer. 2007, 110: 1697-1707.
Nakajima H, Fujiwara H, Furuichi Y, Tanaka K, Shimbara N:A novel small-molecule inhibitor of NF-kappaB signaling. Biochem Biophys Res Commun. 2008, 368: 1007-1013.
Fiedler MA, Wernke-Dollries K, Stark JM:Inhibition of TNF-alpha-induced NF-kappaB activation and IL-8 release in A549 cells with the proteasome inhibitor MG-132. Am J Respir Cell Mol Biol. 1998, 9: 259-268.
Burke JR, Pattoli MA, Gregor KR, Brassil PJ, MacMaster JF, McIntyre KW, Yang X, Iotzova VS, Clarke W, Strnad J, Qiu Y, Zusi FC:BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J Biol Chem. 2003, 278: 1450-1456.
Murata T, Shimada M, Sakakibara S, Yoshino T, Masuda T, Shintani T, Sato H, Koriyama Y, Fukushima K, Nunami N, Yamauchi M, Fuchikami K, Komura H, Watanabe A, Ziegelbauer KB, Bacon KB, Lowinger TB:Synthesis and structure-activity relationships of novel IKK-beta inhibitors. Part 3: Orally active anti-inflammatory agents. Bioorg Med Chem Lett. 2004, 14: 4019-4022.
Lambert C, Li J, Jonscher K, Yang TC, Reigan P, Quintana M, Harvey J, Freed BM:Acrolein inhibits cytokine gene expression by alkylating cysteine and arginine residues in the NF-kappaB1 DNA binding domain. J Biol Chem. 2007, 282: 19666-19675.
Yore MM, Liby KT, Honda T, Gribble GW, Sporn MB:The synthetic triterpenoid 1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole blocks nuclear factor-kappaB activation through direct inhibition of IkappaB kinase beta. Mol Cancer Ther. 2006, 5: 3232-3239.
Guichard C, Pedruzzi E, Fay M, Marie JC, Braut-Boucher F, Daniel F, Grodet A, Gougerot-Pocidalo MA, Chastre E, Kotelevets L, Lizard G, Vandewalle A, Driss F, Ogier-Denis E:Dihydroxyphenylethanol induces apoptosis by activating serine/threonine protein phosphatase PP2A and promotes the endoplasmic reticulum stress response in human colon carcinoma cells. Carcinogenesis. 2006, 27: 1812-1827.
Nagashima K, Sasseville VG, Wen D, Bielecki A, Yang H, Simpson C, Grant E, Hepperle M, Harriman G, Jaffee B, Ocain T, Xu Y, Fraser CC:Rapid TNFR1-dependent lymphocyte depletion in vivo with a selective chemical inhibitor of IKKbeta. Blood. 2006, 107: 4266-4273.
Kishore N, Sommers C, Mathialagan S, Guzova J, Yao M, Hauser S, Huynh K, Bonar S, Mielke C, Albee L, Weier R, Graneto M, Hanau C, Perry T, Tripp CS:A selective IKK-2 inhibitor blocks NF-kappa B-dependent gene expression in interleukin-1 beta-stimulated synovial fibroblasts. J Biol Chem. 2003, 278: 32861-32871.
Morwick T, Berry A, Brickwood J, Cardozo M, Catron K, DeTuri M, Emeigh J, Homon C, Hrapchak M, Jacober S, Jakes S, Kaplita P, Kelly TA, Ksiazek J, Liuzzi M, Magolda R, Mao C, Marshall D, McNeil D, Prokopowicz A3rd, Sarko C, Scouten E, Sledziona C, Sun S, Watrous J, Wu JP, Cywin CL:Evolution of the thienopyridine class of inhibitors of IkappaB kinase-beta: part I: hit-to-lead strategies. J Med Chem. 2006, 49: 2898-2908.
Pandey MK, Sandur SK, Sung B, Sethi G, Kunnumakkara AB, Aggarwal BB:Butein, a tetrahydroxychalcone, inhibits nuclear factor (NF)-kappaB and NF-kappaB-regulated gene expression through direct inhibition of IkappaBalpha kinase beta on cysteine 179 residue. J Biol Chem. 2007, 282: 17340-17350.
Yoon JW, Kang JK, Lee KR, Lee HW, Han JW, Seo DW, Kim YK:beta-Carboline alkaloid suppresses NF-kappaB transcriptional activity through inhibition of IKK signaling pathway. J Toxicol Environ Health A. 2005, 68: 2005-2017.
Pandey MK, Sung B, Kunnumakkara AB, Sethi G, Chaturvedi MM, Aggarwal BB:Berberine modifies cysteine 179 of IkappaBalpha kinase, suppresses nuclear factor-kappaB-regulated antiapoptotic gene products, and potentiates apoptosis. Cancer Res. 2008, 68: 5370-5379.
Inayama M, Nishioka Y, Azuma M, Muto S, Aono Y, Makino H, Tani K, Uehara H, Izumi K, Itai A, Sone S:A novel IkappaB kinase-beta inhibitor ameliorates bleomycin-induced pulmonary fibrosis in mice. Am J Respir Crit Care Med. 2006, 173: 1016-1022.
Castro AC, Dang LC, Soucy F, Grenier L, Mazdiyasni H, Hottelet M, Parent L, Pien C, Palombella V, Adams J:Novel IKK inhibitors: beta-carbolines. Bioorg Med Chem Lett. 2003, 13: 2419-2422.
Yan SS, Li Y, Wang Y, Shen SS, Gu Y, Wang HB, Qin GW, Yu Q:17-Acetoxyjolkinolide B irreversibly inhibits IkappaB kinase and induces apoptosis of tumor cells. Mol Cancer Ther. 2008, 7: 1523-1532.
Mo SJ, Son EW, Lee SR, Lee SM, Shin DH, Pyo S:CML-1 inhibits TNF-alpha-induced NF-kappaB activation and adhesion molecule expression in endothelial cells through inhibition of IkBalpha kinase. J Ethnopharmacol. 2007, 109: 78-86.
Lee JH, Koo TH, Yoon H, Jung HS, Jin HZ, Lee K, Hong YS, Lee JJ:Inhibition of NF-kappa B activation through targeting I kappa B kinase by celastrol, a quinone methide triterpenoid. Biochem Pharmacol. 2006, 72: 1311-1321.
Shin HM, Lee YR, Chang YS, Lee JY, Kim BH, Min KR, Kim Y:Suppression of interleukin-6 production in macrophages by furonaphthoquinone NFD-37. Int Immunopharmacol. 2006, 6: 916-923.
Yadav VR, Prasad S, Gupta SC, Sung B, Phatak SS, Zhang S, Aggarwal BB:3-Formylchromone interacts with cysteine 38 in p65 protein and with cysteine 179 in IkappaBalpha kinase, leading to down-regulation of nuclear factor-kappaB (NF-kappaB)-regulated gene products and sensitization of tumor cells. J Biol Chem. 2011, 287: 245-256.
Catley MC, Sukkar MB, Chung KF, Jaffee B, Liao SM, Coyle AJ, Haddad el B, Barnes PJ, Newton R:Validation of the anti-inflammatory properties of small-molecule IkappaB Kinase (IKK)-2 inhibitors by comparison with adenoviral-mediated delivery of dominant-negative IKK1 and IKK2 in human airways smooth muscle. Mol Pharmacol. 2006, 70: 697-705.
Sethi G, Ahn KS, Sung B, Aggarwal BB:Pinitol targets nuclear factor-kappaB activation pathway leading to inhibition of gene products associated with proliferation, apoptosis, invasion, and angiogenesis. Mol Cancer Ther. 2008, 7: 1604-1614.
Callister ME, Pinhu L, Catley MC, Westwell AD, Newton R, Leaver SK, Quinlan GJ, Evans TW, Griffiths MJ, Burke-Gaffney A:PMX464, a thiol-reactive quinol and putative thioredoxin inhibitor, inhibits NF-kappaB-dependent proinflammatory activation of alveolar epithelial cells. Br J Pharmacol. 2008, 155: 661-672.
Kuefer R, Genze F, Zugmaier W, Hautmann RE, Rinnab L, Gschwend JE, Angelmeier M, Estrada A, Buechele B:Antagonistic effects of sodium butyrate and N-(4-hydroxyphenyl)-retinamide on prostate cancer. Neoplasia. 2007, 9: 246-253.
de-Blanco EJ, Pandit B, Hu Z, Shi J, Lewis A, Li PK:Inhibitors of NF-kappaB derived from thalidomide. Bioorg Med Chem Lett. 2007, 17: 6031-6035.
Huang X, Chen Y, Zhang H, Ma Q, Zhang YW, Xu H:Salubrinal attenuates beta-amyloid-induced neuronal death and microglial activation by inhibition of the NF-kappaB pathway. Neurobiol Aging. 2011. 1007, 33: e1009-1017.
Syed DN, Afaq F, Sarfaraz S, Khan N, Kedlaya R, Setaluri V, Mukhtar H:Delphinidin inhibits cell proliferation and invasion via modulation of Met receptor phosphorylation. Toxicol Appl Pharmacol. 2008, 231: 52-60.
Jagielska J, Salguero G, Schieffer B, Bavendiek U:Digitoxin elicits anti-inflammatory and vasoprotective properties in endothelial cells: therapeutic implications for the treatment of atherosclerosis?. Atherosclerosis. 2009, 206: 390-396.
Xu J, Itoh Y, Hayashi H, Takii T, Miyazawa K, Onozaki K:Dihydrotestosterone inhibits interleukin-1alpha or tumor necrosis factor alpha-induced proinflammatory cytokine production via androgen receptor-dependent inhibition of nuclear factor-kappaB activation in rheumatoid fibroblast-like synovial cell line. Biol Pharm Bull. 2011, 34: 1724-1730.
Kim HK, Park HR, Lee JS, Chung TS, Chung HY, Chung J:Down-regulation of iNOS and TNF-alpha expression by kaempferol via NF-kappaB inactivation in aged rat gingival tissues. Biogerontology. 2007, 8: 399-408.
Chiu FL, Lin JK:Tomatidine inhibits iNOS and COX-2 through suppression of NF-kappaB and JNK pathways in LPS-stimulated mouse macrophages. FEBS Lett. 2008, 582: 2407-2412.
Sarkar D, Saha P, Gamre S, Bhattacharjee S, Hariharan C, Ganguly S, Sen R, Mandal G, Chattopadhyay S, Majumdar S, Chatterjee M:Anti-inflammatory effect of allylpyrocatechol in LPS-induced macrophages is mediated by suppression of iNOS and COX-2 via the NF-kappaB pathway. Int Immunopharmacol. 2008, 8: 1264-1271.
Hwang J, Zheng LT, Ock J, Lee MG, Kim SH, Lee HW, Lee WH, Park HC, Suk K:Inhibition of glial inflammatory activation and neurotoxicity by tricyclic antidepressants. Neuropharmacology. 2008, 55: 826-834.
Rajapakse N, Kim MM, Mendis E, Kim SK:Inhibition of inducible nitric oxide synthase and cyclooxygenase-2 in lipopolysaccharide-stimulated RAW264.7 cells by carboxybutyrylated glucosamine takes place via down-regulation of mitogen-activated protein kinase-mediated nuclear factor-kappaB signaling. Immunology. 2008, 123: 348-357.
Zhu WW, Liu XP, Wu N, Zhao TT, Zhao Y, Zhang J, Shao JH:Beneficial effects of losartan on vascular injury induced by advanced glycosylation end products and their receptors in spontaneous hypertension rats. Mol Cell Biochem. 2007, 304: 35-43.
Kim HG, Hien TT, Han EH, Hwang YP, Choi JH, Kang KW, Kwon KI, Kim BH, Kim SK, Song GY, Jeong TC, Jeong HG:Metformin inhibits P-glycoprotein expression via the NF-kappaB pathway and CRE transcriptional activity through AMPK activation. Br J Pharmacol. 2008, 162: 1096-1108.
Boost KA, Leipold T, Scheiermann P, Hoegl S, Sadik CD, Hofstetter C, Zwissler B:Sevoflurane and isoflurane decrease TNF-alpha-induced gene expression in human monocytic THP-1 cells: potential role of intracellular IkappaBalpha regulation. Int J Mol Med. 2009, 23: 665-671.
Dey A, Wong ET, Cheok CF, Tergaonkar V, Lane DP:R-Roscovitine simultaneously targets both the p53 and NF-kappaB pathways and causes potentiation of apoptosis: implications in cancer therapy. Cell Death Differ. 2008, 15: 263-273.
Doleckova I, Cesnek M, Dracinsky M, Brynda J, Voller J, Janeba Z, Krystof V:Synthesis and biological evaluation of guanidino analogues of roscovitine. Eur J Med Chem. 2013, 62: 443-452.
Lu W, Chen L, Peng Y, Chen J:Activation of p53 by roscovitine-mediated suppression of MDM2 expression. Oncogene. 2001, 20: 3206-3216.
Lew QJ, Chia YL, Chu KL, Lam YT, Gurumurthy M, Xu S, Lam KP, Cheong N, Chao SH:Identification of HEXIM1 as a positive regulator of p53. J Biol Chem. 2012, 287: 36443-36454.
Peterlin BM, Price DH:Controlling the elongation phase of transcription with P-TEFb. Mol Cell. 2006, 23: 297-305.
Takada Y, Aggarwal BB:Flavopiridol inhibits NF-kappaB activation induced by various carcinogens and inflammatory agents through inhibition of IkappaBalpha kinase and p65 phosphorylation: abrogation of cyclin D1, cyclooxygenase-2, and matrix metalloprotease-9. J Biol Chem. 2004, 279: 4750-4759.
Dey A, Wong ET, Bist P, Tergaonkar V, Lane DP:Nutlin-3 inhibits the NFkappaB pathway in a p53-dependent manner: implications in lung cancer therapy. Cell Cycle. 2007, 6: 2178-2185.
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA:In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004, 303: 844-848.
Anand P, Sung B, Kunnumakkara AB, Rajasekharan KN, Aggarwal BB:Suppression of pro-inflammatory and proliferative pathways by diferuloylmethane (curcumin) and its analogues dibenzoylmethane, dibenzoylpropane, and dibenzylideneacetone: role of Michael acceptors and Michael donors. Biochem Pharmacol. 2011, 82: 1901-1909.
Gupta SC, Prasad S, Kim JH, Patchva S, Webb LJ, Priyadarsini IK, Aggarwal BB:Multitargeting by curcumin as revealed by molecular interaction studies. Nat Prod Rep. 2011, 28: 1937-1955.
Harikumar KB, Kunnumakkara AB, Ahn KS, Anand P, Krishnan S, Guha S, Aggarwal BB:Modification of the cysteine residues in IkappaBalpha kinase and NF-kappaB (p65) by xanthohumol leads to suppression of NF-kappaB-regulated gene products and potentiation of apoptosis in leukemia cells. Blood. 2009, 113: 2003-2013.
Shehzad A, Lee YS:Molecular mechanisms of curcumin action: signal transduction. Biofactors. 2013, 39: 27-36.
Guo C, Gasparian AV, Zhuang Z, Bosykh DA, Komar AA, Gudkov AV, Gurova KV:9-Aminoacridine-based anticancer drugs target the PI3K/AKT/mTOR, NF-kappaB and p53 pathways. Oncogene. 2009, 28: 1151-1161.
Manna SK, Bose JS, Gangan V, Raviprakash N, Navaneetha T, Raghavendra PB, Babajan B, Kumar CS, Jain SK:Novel derivative of benzofuran induces cell death mostly by G2/M cell cycle arrest through p53-dependent pathway but partially by inhibition of NF-kappaB. J Biol Chem. 2010, 285: 22318-22327.
Lu C, Guo Y, Yan J, Luo Z, Luo HB, Yan M, Huang L, Li X:Design, synthesis, and evaluation of multitarget-directed resveratrol derivatives for the treatment of Alzheimer’s disease. J Med Chem. 2013, 56: 5843-5859.
Michan S, Sinclair D:Sirtuins in mammals: insights into their biological function. Biochem J. 2007, 404: 1-13.
Saunders LR, Verdin E:Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene. 2007, 26: 5489-5504.
Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW:Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23: 2369-2380.
Culmsee C, Zhu X, Yu QS, Chan SL, Camandola S, Guo Z, Greig NH, Mattson MP:A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J Neurochem. 2001, 77: 220-228.
Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV:A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999, 285: 1733-1737.
Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP:Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets. 2011, 11: 239-253.
Ahn SH, Keogh MC, Buratowski S:Ctk1 promotes dissociation of basal transcription factors from elongating RNA polymerase II. EMBO J. 2009, 28: 205-212.
Kuhn DJ, Chen Q, Voorhees PM, Strader JS, Shenk KD, Sun CM, Demo SD, Bennett MK, van Leeuwen FW, Chanan-Khan AA, Orlowski RZ:Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood. 2007, 110: 3281-3290.
Mujtaba T, Dou QP:Advances in the understanding of mechanisms and therapeutic use of bortezomib. Discov Med. 2011, 12: 471-480.
Chauhan D, Catley L, Li G, Podar K, Hideshima T, Velankar M, Mitsiades C, Mitsiades N, Yasui H, Letai A, Ovaa H, Berkers C, Nicholson B, Chao TH, Neuteboom ST, Richardson P, Palladino MA, Anderson KC:A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell. 2005, 8: 407-419.
Groll M, Potts BC:Proteasome structure, function, and lessons learned from beta-lactone inhibitors. Curr Top Med Chem. 2011, 11: 2850-2878.
Obaidat A, Weiss J, Wahlgren B, Manam RR, Macherla VR, McArthur K, Chao TH, Palladino MA, Lloyd GK, Potts BC, Enna SJ, Neuteboom ST, Hagenbuch B:Proteasome regulator marizomib (NPI-0052) exhibits prolonged inhibition, attenuated efflux, and greater cytotoxicity than its reversible analogs. J Pharmacol Exp Ther. 2011, 337: 479-486.
Potts BC, Albitar MX, Anderson KC, Baritaki S, Berkers C, Bonavida B, Chandra J, Chauhan D, Cusack JCJr, Fenical W, Ghobrial IM, Groll M, Jensen PR, Lam KS, Lloyd GK, McBride W:McConkey: Marizomib, a proteasome inhibitor for all seasons: preclinical profile and a framework for clinical trials. Curr Cancer Drug Targets. 2011, 11: 254-284.
Yang H, Zonder JA, Dou QP:Clinical development of novel proteasome inhibitors for cancer treatment. Expert Opin Investig Drugs. 2009, 18: 957-971.
Chauhan D, Singh AV, Aujay M, Kirk CJ, Bandi M, Ciccarelli B, Raje N, Richardson P, Anderson KC:A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood. 2010, 116: 4906-4915.
Dick LR, Fleming PE:Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov Today. 2010, 15: 243-249.
Kisselev AF, van der Linden WA, Overkleeft HS:Proteasome inhibitors: an expanding army attacking a unique target. Chem Biol. 2012, 19: 99-115.
Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, Yu J, Yang Y, Hales P, Bruzzese F, Liu J, Blank J, Garcia K, Tsu C, Dick L, Fleming P, Yu L, Manfredi M, Rolfe M, Bolen J:Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010, 70: 1970-1980.
Dredge K, Dalgleish AG, Marriott JB:Thalidomide analogs as emerging anti-cancer drugs. Anticancer Drugs. 2003, 14: 331-335.
Keifer JA, Guttridge DC, Ashburner BP, Baldwin AS:Inhibition of NF-B activity by thalidomide through suppression of IB kinase activity. J Biol Chem. 2001, 276: 22382-22387.
Majumdar S, Lamothe B, Aggarwal BB:Thalidomide suppresses NF-κB activation induced by TNF and H2O2, but not that activated by ceramide, lipopolysaccharides, or phorbol ester. J Immunol. 2002, 168: 2644-2651.
Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Richardson PG, Hideshima T, Munshi NC, Treon SP, Anderson KC:Apoptotic signaling induced by immunomodulatory thalidomide analogs in human multiple myeloma cells: therapeutic implications. Blood. 2002, 99: 4525-4530.
Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA:Inducible cyclooxygenase may have antiinflammatory properties. Nat Med. 1999, 5: 698-701.
Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK:The peroxisome proliferator-activated receptor-γis a negative regulator of macrophage activation. Nature. 1998, 391: 79-82.
Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG:Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IκB kinase. Nature. 2000, 403: 103-108.
Chen J, Zhao M, Rao R, Inoue H, Hao CM:C/EBP{beta} and its binding element are required for NF{kappa}B-induced COX2 expression following hypertonic stress. J Biol Chem. 2005, 280: 16354-16359.
Okada Y, Voznesensky O, Herschman H, Harrison J, Pilbeam C:Identification of multiple cis-acting elements mediating the induction of prostaglandin G/H synthase-2 by phorbol ester in murine osteoblastic cells. J Cell Biochem. 2000, 78: 197-209.
Tazawa R, Xu XM, Wu KK, Wang LH:Characterization of the genomic structure, chromosomal location and promoter of human prostaglandin H synthase-2 gene. Biochem Biophys Res Commun. 1994, 203: 190-199.
Yamamoto Y, Yin MJ, Lin KM, Gaynor RB:Sulindac inhibits activation of the NF-kappaB pathway. J Biol Chem. 1999, 274: 27307-27314.
Yin MJ, Yamamoto Y, Gaynor RB:The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. 1998, 396: 77-80.
Pierce JW, Read MA, Ding H, Luscinskas FW, Collins T:Salicylates inhibit I kappa B-alpha phosphorylation, endothelial-leukocyte adhesion molecule expression, and neutrophil transmigration. J Immunol. 1996, 156: 3961-3969.
Wahl C, Liptay S, Adler G, Schmid RM:Sulfasalazine: A potent and specific inhibitor of NF-B. J Clin Invest. 1997, 101: 1163-1174.
Yan F:Polk DB Aminosalicylic acid inhibits IκB kinase-αphosphorylation of Iκ B αin mouse intestinal epithelial cells. J Biol Chem. 1999, 274: 36631-36636.
Egan LJ, Mays DC, Huntoon CJ, Bell MP, Pike MG, Sandborn WJ, Lipsky JJ, McKean DJ:Inhibition of interleukin-1-stimulated NF-κB RelA/p65 phosphorylation by mesalamine is accompanied by decreased transcriptional activity. J Biol Chem. 1999, 274: 26448-26453.
Yamamoto Y, Verma UN, Prajapati S, Kwak YT, Gaynor RB:Histone H3 phosphorylation by IKK-αis critical for cytokine-induced gene expression. Nature. 2003, 423: 655-659.
Cao Y, Bonizzi G, Seagroves TN, Greten FR, Johnson R, Schmidt EV, Karin M:IKKαprovides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell. 2001, 107: 763-775.
Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS:A nucleosomal function for IκB kinase-αin NF-κB-dependent gene expression. Nature. 2003, 423: 659-663.
Signal Pharmaceuticals, Inc:Quinazoline analogs and related compounds and methods for treating inflammatory conditions. 1999, WO 199901441-
Leisten JC, Grimshaw CE, Bennett B, Satoh Y, Xu W, O’Leary EC, Firestein GS, Boyle DL, Dreano M, Bhagwat SS, Ramon HK:Identification of a disease modifying IKK2 inhibitor in rat adjuvant arthritis. Inflamm Res. 2002, 51 (Suppl 2): A25-
Palanki MS, Gayo-Fung LM, Shevlin GI, Erdman P, Sato M, Goldman M, Ransone LJ, Spooner C:Structure–activity relationship studies of ethyl 2-[(3-methyl-2,5-dioxo(3-pyrrolinyl))amino]-4-(trifluoromethyl)pyrimidine-5-carboxylate: an inhibitor of AP-1 and NF-κB mediated gene expression. Bioorg Med Chem Lett. 2002, 12: 2573-2577.
Adams J, Castro A, Grenier L, Hancock WW, Mazdiyasni H, Palombella V, Ritzeler O, Soucy F:Preparation of substituted carbolines as potential therapeutics in diseases associated with increased IB kinase activity. Aventis Pharma Gmbh. 2001, WO 2001068648- .,
Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, Munshi N, Dang L, Castro A, Palombella V, Adams J, Anderson KC:NF-κB as a therapeutic target in multiple myeloma. J Biol Chem. 2003, 277: 16639-16647.
Burke JR, Nadler S, Qiu Y, Townsend RM, Zusi FC:Method of treating inflammatory and immune diseases using 4-amino substituted imidazoquinoxaline, benzopyrazoloquinazoline, benzoimidazoquinoxaline and benzoimidazoquinoline inhibitors of IB kinase (IKK). Bristol-Myers Squibb Co. 2002, WO 2002060386-
Burgess JL, Callahan JF, Wan Z:NF-kB inhibitors. SmithKline Beecham Corp. 2003, WO 2003029242-
Griffiths D, Johnstone C:Preparation of ureido–carboxamido thiophene as inhibitors of IKK2 kinase. AstraZeneca. 2003, WO 2003010163-
Baxter A, Brough S, Faull A, Johnstone C, Mcinally T, AstraZeneca:Preparation of thiophenecarboxamides as inhibitors of the enzyme IKK-2. AstraZeneca. 2001, WO 2001058890-
Fuchikami K, Ikegami Y, Komura H, Lowinger TB, Masuda T, Murata T, Sakakibara S, Shimada M, Shimazaki M, Shintani T, Umeda M, Yoshida N, Yoshino T, Ziegelbauer KB:Preparation of 2,4-diarylpyridines as IB kinase inhibitors useful as antiinflammatories. Bayer Ag. 2002, WO 2002044153-
Fuchikami K, Hiroshi K, Komura H, Koriyama Y, Lowinger TB, Masuda T, Murata T, Sakakibara S, Sato H, Shimada M, Shintani T, Umeda M, Yoshino T, Ziegelbauer KB:Preparation of hydroxyarylpyridines with IB kinase (IKK) inhibiting activity. Bayer Ag. 2002, WO 2002024679-
Murata T, Shimada M, Sakakibara S, Yoshino T, Kadono H, Masuda T, Shimazaki M, Shintani T, Fuchikami K, Sakai K, Inbe H, Takeshita K, Niki T, Umeda M, Bacon KB, Ziegelbauer KB, Lowinger TB:Discovery of novel and selective IKK-βserine-threonine protein kinase inhibitors. Part 1. Bioorg Med Chem Lett. 2003, 13: 913-918.
Bhagwat SS, Erdman PE, Kois A, Macfarlane KJ, Palanki MSS, Parnes JS, Satoh Y:Preparation of anilinopyrimidines as IKK inhibitors. Signal Pharmaceuticals, Inc. 2002, WO 2002046171-
Bacon K, Fuchikami K, Fukushima K, Grosser R, Koriyama Y, Lowinger T, Murata T, Nunami N, Sasaki S, Sato H, Yamauchi M, Yoshino T:Preparation of optically active pyridooxazinones as antiinflammatory agents. Bayer Ag. 2003, WO 2003076447-
Madsen MW, Olsen LS:A method using cyanoguanidine compounds for modulating NFkB activity and use for the treatment of cancer. Pharma L. 2002, WO 2002094265-
Binderup L, Bramm E, Hamberg KJ, Hjarnaa P-JV:Antitumor drug–cyanoguanidine IKK inhibitor combination. Pharma L. 2002, WO 2002094322-
Pierce JW, Schoenleber R, Jesmok G, Best J, Moore SA, Collins T, Gerritsen ME:Novel inhibitors of cytokine-induced Iκ B αphosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem. 1997, 272: 21096-21103.
Nishimura D, Ishikawa H, Matsumoto K, Shibata H, Motoyoshi Y, Fukuta M, Kawashimo H, Goto T, Taura N, Ichikawa T, Hamasaki K, Nakao K, Umezawa K, Eguchi K:DHMEQ, a novel NF-kappaB inhibitor, induces apoptosis and cell-cycle arrest in human hepatoma cells. Int J Oncol. 2006, 29: 713-719.
Meng Z, Mitsutake N, Nakashima M, Starenki D, Matsuse M, Takakura S, Namba H, Saenko V, Umezawa K, Ohtsuru A, Yamashita S:DHMEQ, a novel NF-{kappa}B inhibitor, enhances anti-tumor activity of taxanes in anaplastic thyroid cancer cells. Endocrinology. 2008, 149: 5357-5365.
Kassie F, Melkamu T, Endalew A, Upadhyaya P, Luo X, Hecht SS:Inhibition of lung carcinogenesis and critical cancer-related signaling pathways by N-acetyl-S-(N-2-phenethylthiocarbamoyl)-l-cysteine, indole-3-carbinol and myo-inositol, alone and in combination. Carcinogenesis. 2010, 12: 1634-1641.
Mackenzie GG, Oteiza PI:Modulation of transcription factor NF-kappaB in Hodgkin’s lymphoma cell lines: effect of (-)-epicatechin. Free Radic Res. 2006, 40: 1086-1094.
Nicholas C, Batra S, Vargo MA, Voss OH, Gavrilin MA, Wewers MD, Guttridge DC, Grotewold E, Doseff AI:Apigenin blocks lipopolysaccharide-induced lethality in vivo and proinflammatory cytokines expression by inactivating NF-kappaB through the suppression of p65 phosphorylation. J Immunol. 2007, 179: 7121-7127.
Shukla S, Gupta S:Suppression of constitutive and tumor necrosis factor alpha-induced nuclear factor (NF)-kappaB activation and induction of apoptosis by apigenin in human prostate carcinoma PC-3 cells: correlation with down-regulation of NF-kappaB-responsive genes. Clin Cancer Res. 2004, 10: 3169-3178.
Dai Y, Desano J, Tang W, Meng X, Meng Y, Burstein E, Lawrence TS, Xu L:Natural proteasome inhibitor celastrol suppresses androgen-independent prostate cancer progression by modulating apoptotic proteins and NF-kappaB. PLoS One. 2010, 5: e14153-
Wheeler DS, Catravas JD, Odoms K, Denenberg A, Malhotra V, Wong HR:Epigallocatechin-3-gallate, a green tea-derived polyphenol, inhibits IL-1 beta-dependent proinflammatory signal transduction in cultured respiratory epithelial cells. J Nutr. 2004, 134: 1039-1044.
Kundu JK, Surh YJ:Epigallocatechin gallate inhibits phorbol ester-induced activation of NF-kappa B and CREB in mouse skin: role of p38 MAPK. Ann N Y Acad Sci. 2007, 1095: 504-512.
Xu L, Zhang L, Bertucci AM, Pope RM, Datta SK:Apigenin, a dietary flavonoid, sensitizes human T cells for activation-induced cell death by inhibiting PKB/Akt and NF-kappaB activation pathway. Immunol Lett. 2008, 121: 74-83.
Gao X, Deeb D, Liu Y, Gautam S, Dulchavsky SA, Gautam SC:Immunomodulatory activity of xanthohumol: inhibition of T cell proliferation, cell-mediated cytotoxicity and Th1 cytokine production through suppression of NF-kappaB. Immunopharmacol Immunotoxicol. 2009, 31: 477-484.
Natarajan K, Manna SK, Chaturvedi MM, Aggarwal BB:Protein tyrosine kinase inhibitors block tumor necrosis factor-induced activation of nuclear factor-kappaB, degradation of IkappaBalpha, nuclear translocation of p65, and subsequent gene expression. Arch Biochem Biophys. 1998, 352: 59-70.
Sarkar FH, Li Y:Mechanisms of cancer chemoprevention by soy isoflavone genistein. Cancer Metastasis Rev. 2002, 21: 265-280.
Wang H, Syrovets T, Kess D, Buchele B, Hainzl H, Lunov O, Weiss JM, Scharffetter-Kochanek K, Simmet T:Targeting NF-kappa B with a natural triterpenoid alleviates skin inflammation in a mouse model of psoriasis. J Immunol. 2009, 183: 4755-4763.
Harikumar KB, Sung B, Pandey MK, Guha S, Krishnan S, Aggarwal BB:Escin, a pentacyclic triterpene, chemosensitizes human tumor cells through inhibition of nuclear factor-kappaB signaling pathway. Mol Pharmacol. 2010, 77: 818-827.
Harikumar KB, Sung B, Tharakan ST, Pandey MK, Joy B, Guha S, Krishnan S, Aggarwal BB:Sesamin manifests chemopreventive effects through the suppression of NF-kappa B-regulated cell survival, proliferation, invasion, and angiogenic gene products. Mol Cancer Res. 2010, 8: 751-761.
Jeng KC, Hou RC, Wang JC, Ping LI:Sesamin inhibits lipopolysaccharide-induced cytokine production by suppression of p38 mitogen-activated protein kinase and nuclear factor-kappaB. Immunol Lett. 2005, 97: 101-106.
Ju W, Wang X, Shi H, Chen W, Belinsky SA, Lin Y:A critical role of luteolin-induced reactive oxygen species in blockage of tumor necrosis factor-activated nuclear factor-kappaB pathway and sensitization of apoptosis in lung cancer cells. Mol Pharmacol. 2007, 71: 1381-1388.
Kwok BH, Koh B, Ndubuisi MI, Elofsson M, Crews CM:The anti-inflammatory natural product parthenolide from the medicinal herb Feverfew directly binds to and inhibits IkappaB kinase. Chem Biol. 2001, 8: 759-766.
Blackwell TS, Blackwell TR, Holden EP, Christman BW, Christman JW:In vivo antioxidant treatment suppresses nuclear factor-κB activation and neutrophilic lung inflammation. J Immunol. 1996, 157: 1630-1637.
Bowie AG, O’Neill LA:Vitamin C inhibits NF-κB activation by TNF via the activation of p38 mitogen-activated protein kinase. J Immunol. 2000, 165: 7180-7188.
Carcamo JM, Pedraza A, Borquez-Ojeda O, Golde DW:Vitamin C suppresses TNFα-induced NF-κB activation by inhibiting Iκ B αphosphorylation. Biochemistry. 2002, 41: 12995-13002.
Hayakawa M, Miyashita H, Sakamoto I, Kitagawa M, Tanaka H, Yasuda H, Karin M, Kikugawa K:Evidence that reactive oxygen species do not mediate NF-κB activation. EMBO J. 2003, 22: 3356-3366.
Rasheed Z, Anbazhagan AN, Akhtar N, Ramamurthy S, Voss FR, Haqqi TM:Green tea polyphenol epigallocatechin-3-gallate inhibits advanced glycation end product-induced expression of tumor necrosis factor-alpha and matrix metalloproteinase-13 in human chondrocytes. Arthritis Res Ther. 2009, 11: R71-
Tabary O, Escotte S, Couetil JP, Hubert D, Dusser D, Puchelle E, Jacquot J:Genistein inhibits constitutive and inducible NFkappaB activation and decreases IL-8 production by human cystic fibrosis bronchial gland cells. Am J Pathol. 1999, 155: 473-481.
Patel PS, Varney ML, Dave BJ, Singh RK:Regulation of constitutive and induced NF-kappaB activation in malignant melanoma cells by capsaicin modulates interleukin-8 production and cell proliferation. J Interferon Cytokine Res. 2002, 22: 427-435.
Syrovets T, Gschwend JE, Buchele B, Laumonnier Y, Zugmaier W, Genze F, Simmet T:Inhibition of IkappaB kinase activity by acetyl-boswellic acids promotes apoptosis in androgen-independent PC-3 prostate cancer cells in vitro and in vivo. J Biol Chem. 2005, 280: 6170-6180.
Moon DO, Kim MO, Kang SH, Choi YH, Kim GY:Sulforaphane suppresses TNF-alpha-mediated activation of NF-kappaB and induces apoptosis through activation of reactive oxygen species-dependent caspase-3. Cancer Lett. 2009, 274: 132-142.
Gupta SC, Kannapan R, Hye Kim J, Rahman GM, Francis SK, Raveendran R, Nair MS, Das J, Aggarwal BB:Bharangin, a diterpenoid quinonemethide, abolishes constitutive and inducible NF-kB activation by modifying p65 on cysteine 38 residue and inhibiting IkBalpha kinase activation, leading to suppression of NF-kB-regulated gene expression and sensitization of tumor cells to chemotherapeutic agents. Mol Pharmacol. 2011, 80: 769-781.
Tsuboi K, Matsuo Y, Shamoto T, Shibata T, Koide S, Morimoto M, Guha S, Sung B, Aggarwal BB, Takahashi H, Takeyama H:Zerumbone inhibits tumor angiogenesis via NF-kappaB in gastric cancer. Oncol Rep. 2014, 31: 57-64.
Knight DW:Feverfew: chemistry and biological activity. Nat Prod Rep. 1995, 12: 271-276.
Wen J, You KR, Lee SY, Song CH, Kim DG:Oxidative stress-mediated apoptosis. The anticancer effect of the sesquiterpene lactone parthenolide. J Biol Chem. 2002, 277: 38954-38964.
Guzman ML, Rossi RM, Karnischky L, Li X, Peterson DR, Howard DS, Jordan CT:The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood. 2005, 105: 4163-4169.
Zhang S, Ong CN, Shen HM:Critical roles of intracellular thiols and calcium in parthenolide-induced apoptosis in human colorectal cancer cells. Cancer Lett. 2004, 208: 143-153.
Kim JH, Liu L, Lee SO, Kim YT, You KR, Kim DG:Susceptibility of cholangiocarcinoma cells to parthenolide-induced apoptosis. Cancer Res. 2005, 65: 6312-6320.
Patel NM, Nozaki S, Shortle NH, Bhat-Nakshatri P, Newton TR, Rice S, Gelfanov V, Boswell SH, Goulet RJ, Sledge GW, Nakshatri H:Paclitaxel sensitivity of breast cancer cells with constitutively active NF-κB is enhanced by Iκ B αsuper-repressor and parthenolide. Oncogene. 2000, 19: 4159-4169.
Aggarwal S, Ichikawa H, Takada Y, Sandur SK, Shishodia S, Aggarwal BB:Curcumin (diferuloylmethane) down-regulates expression of cell proliferation and antiapoptotic and metastatic gene products through suppression of IkappaBalpha kinase and Akt activation. Mol Pharmacol. 2006, 69: 195-206.
Hussain AR, Ahmed M, Al-Jomah NA, Khan AS, Manogaran P, Sultana M, Abubaker J, Platanias LC, Al-Kuraya KS, Uddin S:Curcumin suppresses constitutive activation of nuclear factor-kappa B and requires functional Bax to induce apoptosis in Burkitt’s lymphoma cell lines. Mol Cancer Ther. 2008, 7: 3318-3329.
Carroll RE, Benya RV, Turgeon DK, Vareed S, Neuman M, Rodriguez L, Kakarala M, Carpenter PM, McLaren C, Meyskens FL, Brenner DE:Phase IIa clinical trial of curcumin for the prevention of colorectal neoplasia. Cancer Prev Res (Phila). 2011, 4: 354-364.
Saif MW:Is there a role for herbal medicine in the treatment of pancreatic cancer? Highlights from the “44th ASCO Annual Meeting”. Chicago IL, USA. May 30–June 3, 2008. JOP. 2008, 9: 403-407.
Milacic V, Banerjee S, Landis-Piwowar KR, Sarkar FH, Majumdar AP, Dou QP:Curcumin inhibits the proteasome activity in human colon cancer cells in vitro and in vivo. Cancer Res. 2008, 68: 7283-7292.
Hasima N, Aggarwal BB:Targeting proteasomal pathways by dietary curcumin for cancer prevention and treatment. Curr Med Chem. 2014, 21: 1583-1594.
Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta RG, Moon RC, Pezzuto JM:Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997, 275: 218-220.
Bhardwaj A, Sethi G, Vadhan-Raj S, Bueso-Ramos C, Takada Y, Gaur U, Nair AS, Shishodia S, Aggarwal BB:Resveratrol inhibits proliferation, induces apoptosis, and overcomes chemoresistance through down-regulation of STAT3 and nuclear factor-kappaB-regulated antiapoptotic and cell survival gene products in human multiple myeloma cells. Blood. 2007, 109: 2293-2302.
Harikumar KB, Kunnumakkara AB, Sethi G, Diagaradjane P, Anand P, Pandey MK, Gelovani J, Krishnan S, Guha S, Aggarwal BB:Resveratrol, a multitargeted agent, can enhance antitumor activity of gemcitabine in vitro and in orthotopic mouse model of human pancreatic cancer. Int J Cancer. 2010, 127: 257-268.
Grasberger BL, Lu T, Schubert C, Parks DJ, Carver TE, Koblish HK, Cummings MD, LaFrance LV, Milkiewicz KL, Calvo RR, Maguire D, Lattanze J, Franks CF, Zhao S, Ramachandren K, Bylebyl GR, Zhang M, Manthey CL, Petrella EC, Pantoliano MW, Deckman IC, Spurlino JC, Maroney AC, Tomczuk BE, Molloy CJ, Bone RF:Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J Med Chem. 2005, 48: 909-912.
Allen JG, Bourbeau MP, Wohlhieter GE, Bartberger MD, Michelsen K, Hungate R, Gadwood RC, Gaston RD, Evans B, Mann LW, Matison ME, Schneider S, Huang X, Yu D, Andrews PS, Reichelt A, Long AM, Yakowec P, Yang EY, Lee TA, Oliner JD:Discovery and optimization of chromenotriazolopyrimidines as potent inhibitors of the mouse double minute 2 −tumor protein 53 protein −protein interaction. J Med Chem. 2009, 52: 7044-7053.
Orner BP, Ernst JT, Hamilton AD:Toward proteomimetics: terphenyl derivatives as structural and functional mimics of extended regions of anαhelix. J Am Chem Soc. 2001, 123: 5382-5383.
Yin H, Lee GI, Park HS, Payne GA, Rodriguez JM, Sebti SM, Hamilton AD:Terphenyl-based helical mimetics that disrupt the p53/HDM2 interaction. Angew Chem Int Ed Engl. 2005, 44: 2704-2707.
Go ML, Wu X, Liu XL:Chalcones: an update on cytotoxic and chemoprotective properties. Curr Med Chem. 2005, 12: 483-499.
Stoll R, Renner C, Hansen S, Palme S, Klein C, Belling A, Zeslawski W, Kamionka M, Rehm T, Mühlhahn P, Schumacher R, Hesse F, Kaluza B, Voelter W, Engh RA, Holak TA:Chalcone derivatives antagonize interactions between the human oncoprotein MDM2 and p53. Biochemistry. 2001, 40: 336-344.
Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, Nikolovska-Coleska Z, Ding K, Wang G, Chen J, Bernard D, Zhang J, Lu Y, Gu Q, Shah RB, Pienta KJ, Ling X, Kang S, Guo M, Sun Y, Yang D, Wang S:Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci USA. 2008, 105: 3933-3938.
Zhao Y, Yu S, Sun W, Liu L, Lu J, McEachern D, Shargary S, Bernard D, Li X, Zhao T, Zou P, Sun D, Wang S:A potent small-molecule inhibitor of the MDM2–p53 interaction (MI888) achieved complete and durable tumor regression in mice. J Med Chem. 2013, 56: 5553-5561.
Czarna A, Beck B, Srivastava S, Popowicz GM, Wolf S, Huang Y, Bista M, Holak TA, Dömling A:Robust generation of lead compounds for protein–protein interactions by computational and MCR chemistry: 53/Hdm2 antagonists. Angew Chem Int Ed. 2010, 49: 5352-5356.
Boettcher A, Buschmann N, Furet P, Groell J, Kallen J, Hergovich LJ, Masuya K, Mayr A, Vaupel A:3imidazolyl-indoles for the treatment of proliferative diseases. 2008, WO Patent2008119741-
Hardcastle IR, Liu J, Valeur E, Watson A, Ahmed SU, Blackburn TJ, Bennaceur K, Clegg W, Drummond C, Endicott JA, Golding BT, Griffin RJ, Gruber J, Haggerty K, Harrington RW, Hutton C, Kemp S, Lu X, McDonnell JM, Newell DR, Noble ME, Payne SL, Revill CH, Riedinger C, Xu Q, Lunec J:Isoindolinone inhibitors of the murine double minute 2 (MDM2)-p53 protein-protein interaction: structure-activity studies leading to improved potency. J Med Chem. 2011, 54: 1233-1243.
Burdack C, Kalinsky C, Ross G, Weber L, Khazak V:Pyrrolidin-2-ones as hdm2 ligands. Priaxon Ag. 2010, WO 2010028862-
Essmann F, Schulze-Osthoff K:Translational approaches targeting the p53 pathway for anti-cancer therapy. Br J Pharmacol. 2012, 165: 328-344.
Bogen SL, Ma Y, Wang Y, Lahue BR, Nair LG, Shizuka M, Voss ME, Kirova-Snover M, Pan W, Tian Y, Kulkarni BA, Gibeau CR, Liu Y, Scapin G, Rindgen D, Doll RJ, Guzi TJ, Hicklin DJ, Nomeir A, Seide Dugan C, Shipps GW, Maccoss M:Substituted piperidines that increase p53 activity and the uses thereof. 2011, WO 2011046771A1-
Bertamino A, Soprano M, Musella S, Rusciano MR, Sala M, Vernieri E, Di Sarno V, Limatola A, Carotenuto A, Cosconati S, Grieco P, Novellino E, Illario M, Campiglia P, Gomez-Monterrey I:Synthesis, in vitro: and in cell studies of a new series of [indoline3,2thiazolidine]-based p53 modulators. J Med Chem. 2013, 56: 5407-5421.
Galatin PS, Abraham DJ:A nonpeptidic sulfonamide inhibits the p53mdm2 interaction and activates p53dependent transcription in mdm2overexpressing cells. J Med Chem. 2004, 47: 4163-4165.
Ding Q, Zhang Z, Liu JJ, Jiang N, Zhang J, Ross TM, Chu XJ, Bartkovitz D, Podlaski F, Janson C, Tovar C, Filipovic ZM, Higgins B, Glenn K, Packman K, Vassilev LT, Graves B:Discovery of RG7388, a Potent and Selective p53-MDM2 Inhibitor in Clinical Development. J Med Chem. 2013, 56: 5979-5983.
Zhang Z, Chu XJ, Liu JJ, Ding Q, Zhang J, Bartkovitz D, Jiang N, Karnachi P, So SS, Tovar C, Filipovic ZM, Higgins B, Glenn K, Packman K, Vassilev L, Graves B:Discovery of Potent and Orally Active p53-MDM2 Inhibitors RO5353 and RO2468 for Potential Clinical Development. ACS Med Chem Lett. 2013, 5: 124-127.
Lucas BS, Fisher B, McGee LR, Olson SH, Medina JC, Cheung E:An expeditious synthesis of the MDM2-p53 inhibitor AM-8553. J Am Chem Soc. 2012, 134: 12855-12860.
Sun D, Li Z, Rew Y, Gribble M, Bartberger MD, Beck HP, Canon J, Chen A, Chen X, Chow D, Deignan J, Duquette J, Eksterowicz J, Fisher B, Fox BM, Fu J, Gonzalez AZ, Gonzalez-Lopez De Turiso F, Houze JB, Huang X, Jiang M, Jin L, Kayser F, Liu JJ, Lo MC, Long AM, Lucas B, McGee LR, McIntosh J, Mihalic J, et al:Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J Med Chem. 2014, 57: 1454-1472.
Leão M, Pereira C, Bisio A, Ciribilli Y, Paiva AM, Machado N, Palmeira A, Fernandes MX, Sousa E, Pinto M, Inga A, Saraiva L:Discovery of a new small-molecule inhibitor of p53-MDM2 interaction using a yeast-based approach. Biochem Pharmacol. 2013, 85: 1234-1245.
Zheng GH, Shen JJ, Zhan YC, Yi H, Xue ST, Wang Z, Ji XY, Li ZR:Design, synthesis and in vitro and in vivo antitumour activity of 3-benzylideneindolin-2-one derivatives, a novel class of small-molecule inhibitors of the MDM2-p53 interaction. Eur J Med Chem. 2014, 81C: 277-288.
Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, Bergman J, Wiman KG, Selivanova G:Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002, 8: 282-288.
Zache N, Lambert JM, Wiman KG, Bykov VJ:PRIMA-1MET inhibits growth of mouse tumors carrying mutant p53. Cell Oncol. 2008, 30: 411-418.
Zandi R, Selivanova G, Christensen CL, Gerds TA, Willumsen BM, Poulsen HS:PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin Cancer Res. 2011, 17: 2830-2841.
Lambert JM, Gorzov P, Veprintsev DB, Soderqvist M, Segerback D, Bergman J, Fersht AR, Hainaut P, Wiman KG, Bykov VJ:PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. 2009, 15: 376-388.
Shalom-Feuerstein R, Serror L, Aberdam E, Muller FJ, van Bokhoven H, Wiman KG, Zhou H, Aberdam D, Petit I:Impaired epithelial differentiation of induced pluripotent stem cells from ectodermal dysplasia-related patients is rescued by the small compound APR-246/PRIMA-1MET. Proc Natl Acad Sci U S A. 2013, 110: 2152-2156.
Shen J, van den Bogaard EH, Kouwenhoven EN, Bykov VJ, Rinne T, Zhang Q, Tjabringa GS, Gilissen C, van Heeringen SJ, Schalkwijk J, van Bokhoven H, Wiman KG, Zhou H:APR-246/PRIMA-1(MET) rescues epidermal differentiation in skin keratinocytes derived from EEC syndrome patients with p63 mutations. Proc Natl Acad Sci U S A. 2013, 110: 2157-2162.
Stegh AH:Targeting the p53 signaling pathway in cancer therapy - the promises, challenges and perils. Expert Opin Ther Targets. 2012, 16: 67-83.
Linde L, Kerem B:Introducing sense into nonsense in treatments of human genetic diseases. Trends Genet. 2008, 24: 552-563.
Rowe SM, Clancy JP:Pharmaceuticals targeting nonsense mutations in genetic diseases: progress in development. BioDrugs. 2009, 23: 165-174.
Floquet C, Deforges J, Rousset JP, Bidou L:Rescue of non-sense mutated p53 tumor suppressor gene by aminoglycosides. Nucleic Acids Res. 2011, 39: 3350-3362.
Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, Pramanik A, Selivanova G:Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med. 2004, 10: 1321-1328.
MacCallum DE, Melville J, Frame S, Watt K, Anderson S, Gianella-Borradori A, Lane DP, Green SR:Seliciclib (CYC202, R-Roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res. 2005, 65: 5399-5407.
Choong ML, Yang H, Lee MA, Lane DP:Specific activation of the p53 pathway by low dose actinomycin D: a new route to p53 based cyclotherapy. Cell Cycle. 2009, 8: 2810-2818.
Smart P, Lane EB, Lane DP, Midgley C, Vojtesek B, Lain S:Effects on normal fibroblasts and neuroblastoma cells of the activation of the p53 response by the nuclear export inhibitor leptomycin B. Oncogene. 1999, 18: 7378-7386.
Blank JL, Liu XJ, Cosmopoulos K, Bouck DC, Garcia K, Bernard H, Tayber O, Hather G, Liu R, Narayanan U, Milhollen MA, Lightcap ES:Novel DNA damage checkpoints mediating cell death induced by the NEDD8-activating enzyme inhibitor MLN4924. Cancer Res. 2013, 73: 225-234.
Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, McCarthy A, Appleyard V, Murray KE, Baker L, Thompson A, Mathers J, Holland SJ, Stark MJ, Pass G, Woods J, Lane DP, Westwood NJ:Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. 2008, 13: 454-463.
Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, Xie X, Byrne EH, McCarroll SA, Gaudet R, Schaffner SF, Lander ES, Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, Pasternak S, Wheeler DA, Willis TD, Yu F, Yang H, Zeng C, Gao Y, Hu H, et al:Genome-wide detection and characterization of positive selection in human populations. Nature. 2007, 449: 913-918.
Nakamura Y, Kato H, Nishikawa T, Iwasaki N, Suwa Y, Rotinsulu H, Losung F, Maarisit W, Mangindaan RE, Morioka H, Yokosawa H, Tsukamoto S:Siladenoserinols A-L: new sulfonated serinol derivatives from a tunicate as inhibitors of p53-Hdm2 interaction. Org Lett. 2012, 15: 322-325.
Vogelstein B, Kinzler KW:Cancer genes and the pathways they control. Nat Med. 2004, 10: 789-799.
Gasparian AV, Neznanov N, Jha S, Galkin O, Moran JJ, Gudkov AV, Gurova KV, Komar AA:Inhibition of encephalomyocarditis virus and poliovirus replication by quinacrine: implications for the design and discovery of novel antiviral drugs. J Virol. 2010, 84: 9390-9397.
Draetta GF, Depinho RA:Cancer drug discovery faces the FACT. Sci Transl Med. 2011, 3: 95ps34-
Reinberg D, Sims RJ:de FACTo nucleosome dynamics. J Biol Chem. 2006, 281: 23297-23301.
Orphanides G, LeRoy G, Chang CH, Luse DS, Reinberg D:FACT, a factor that facilitates transcript elongation through nucleosomes. Cell. 1998, 92: 105-116.
Bruhn SL, Pil PM, Essigmann JM, Housman DE, Lippard SJ:Isolation and characterization of human cDNA clones encoding a high mobility group box protein that recognizes structural distortions to DNA caused by binding of the anticancer agent cisplatin. Proc Natl Acad Sci U S A. 1992, 89: 2307-2311.
Whittaker SR, Walton MI, Garrett MD, Workman P:The Cyclin-dependent kinase inhibitor CYC202 (R-roscovitine) inhibits retinoblastoma protein phosphorylation, causes loss of Cyclin D1, and activates the mitogen-activated protein kinase pathway. Cancer Res. 2004, 64: 262-272.
Marshall NF, Price DH:Control of formation of two distinct classes of RNA polymerase II elongation complexes. Mol Cell Biol. 1992, 12: 2078-2090.
Demidenko ZN, Blagosklonny MV:Flavopiridol induces p53 via initial inhibition of Mdm2 and p21 and, independently of p53, sensitizes apoptosis-reluctant cells to tumor necrosis factor. Cancer Res. 2004, 64: 3653-3660.
Wu X, Bayle JH, Olson D, Levine AJ:The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993, 7: 1126-1132.
Parks DJ, LaFrance LV, Calvo RR, Milkiewicz KL, Marugan JJ, Raboisson P, Schubert C, Koblish HK, Zhao S, Franks CF, Lattanze J, Carver TE, Cummings MD, Maguire D, Grasberger BL, Maroney AC, Lu T:Enhanced pharmacokinetic properties of 1,4-benzodiazepine-2,5-dione antagonists of the HDM2-p53 protein-protein interaction through structure-based drug design. Bioorg Med Chem Lett. 2006, 16: 3310-3314.
Kayali R, Ku JM, Khitrov G, Jung ME, Prikhodko O, Bertoni C:Read-through compound 13 restores dystrophin expression and improves muscle function in the mdx mouse model for Duchenne muscular dystrophy. Hum Mol Genet. 2012, 21: 4007-4020.
Acknowledgments
This work was supported by grants from Bose Institute, Department of Science and Technology, and Department of Biotechnology, Government of India (MP). AB is supported by Ramalingaswami fellowship, Government of India. AA is a CSIR (ad-hoc) Research Fellow. We thank Katerina Gurova and Andrei Gudkov for help.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SP, NCM, SCM, and MP conceived of- and designed the review. SP, AB, NCM, SCM, and MP wrote the manuscript. AA contributed to critical reading and comments of the manuscript. All authors read and approved the final manuscript.
Srabani Pal, Ashish Bhattacharjee contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Pal, S., Bhattacharjee, A., Ali, A. et al. Chronic inflammation and cancer: potential chemoprevention through nuclear factor kappa B and p53 mutual antagonism. J Inflamm 11, 23 (2014). https://doi.org/10.1186/1476-9255-11-23
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-9255-11-23