
Abstract
Background
CETP is a plasma protein that modulates atherosclerosis risk through its HDL-cholesterol reducing action. The aim of this work was to examine the effect of the PPARα agonist, ciprofibrate, on the CETP gene expression, in the presence and absence of apolipoprotein (apo) CIII induced hypertriglyceridemia, and its impact on the HDL metabolism.
Results
Mice expressing apo CIII and/or CETP and non-transgenic littermates (CIII, CIII/CETP, CETP, non-Tg) were treated with ciprofibrate during 3 weeks. Drug treatment reduced plasma triglycerides (30-43%) and non-esterified fatty acids (19-47%) levels. Cholesterol (chol) distribution in plasma lipoprotein responses to ciprofibrate treatment was dependent on the genotypes. Treated CIII expressing mice presented elevation in VLDL-chol and reduction in HDL-chol. Treated CETP expressing mice responded with reduction in LDL-chol whereas in non-Tg mice the LDL-chol increased. In addition, ciprofibrate increased plasma post heparin lipoprotein lipase activity (1.3-2.1 fold) in all groups but hepatic lipase activity decreased in treated CETP and non-Tg mice. Plasma CETP activity and liver CETP mRNA levels were significantly increased in treated CIII/CETP and CETP mice (30-100%). Kinetic studies with 3H-cholesteryl ether (CEt) labelled HDL showed a 50% reduction in the 3H-CEt found in the LDL fraction in ciprofibrate treated compared to non-treated CETP mice. This means that 3H-CEt transferred from HDL to LDL was more efficiently removed from the plasma in the fibrate treated mice. Accordingly, the amount of 3H-CEt recovered in the liver 6 hours after HDL injection was increased by 35%.
Conclusion
Together these data showed that the PPARα agonist ciprofibrate stimulates CETP gene expression and changes the cholesterol flow through the reverse cholesterol transport, increasing plasma cholesterol removal through LDL.
Similar content being viewed by others

Background
Apolipoproteins (apo) CIII are small proteins mainly found on the surface of apoB containing lipoproteins (including VLDL, IDL and LDL), which strongly affect their metabolism. They inhibit catabolism of these lipoproteins by lipoprotein-lipase (LPL) and retard their clearance from plasma by decreasing their affinity for hepatic lipoprotein receptors [1–3]. The ability of apo CIII in inducing hypertriglyceridemia was directly demonstrated in transgenic mice over-expressing this protein [4]. Growing evidences have linked apo CIII concentration in VLDL and LDL to coronary heart disease (CHD) [5, 6]. Furthermore, Lee et al. [7] have reported that LDL containing apo CIII strongly predicts coronary events in diabetic patients independently of other lipid concentrations.
Cholesteryl ester transfer protein (CETP) promotes the transfer of cholesteryl esters (CE) from high density lipoprotein (HDL) to the apoB-containing lipoprotein particles, which are subsequently cleared from the circulation by the liver. Thus, CETP plays a critical role in the intravascular remodeling and recycling of HDL particles [8]. Inhibitors of CETP have been tested in human subjects and shown to markedly increase the concentration of HDL cholesterol while decreasing that of LDL cholesterol and apo B [9, 10], but the impact of CETP mediated lipid transfer reactions on atherogenesis remains controversial [11]. A large clinical trial with the CETP inhibitor torcetrapib has been prematurely interrupted due to elevation in mortality in the torcetrapib treated group [10]. Human subjects with genetic CETP deficiency and high levels of HDL-cholesterol (>60 mg/dL) showed reduced risk of CHD, whereas CETP-deficient subjects whose HDL levels were moderately increased (40-60 mg/dL) presented higher risk of CHD [12]. Studies in CETP transgenic mice have provided mixed results. The effects of CETP expression in this species can be neutral, pro- or antiatherogenic depending upon the metabolic context [13–17].
Fibrates are drugs widely used to correct hypertriglyceridemia. They exert their effect by activating specific transcription factors called peroxisome proliferator-activated receptors (PPARs), which in turn alter the transcription of several genes encoding for proteins that control lipoprotein metabolism. Thus, fibrates decrease plasma triglyceride (TG) concentrations by activating enzymes responsible for β-oxidation of long chain fatty acids, reducting TG synthesis, activating lipoprotein lipase and repressing the apo CIII expression [18, 19]. Fibrates can also alter the metabolism of HDL associated proteins. In humans, treatment with fibrates is associated with an increase in plasma HDL-cholesterol and apo AI concentrations [20]. However, in rodents, fibrates decrease apo AI mRNA and plasma concentrations [21] due to a polymorphism in the PPAR responsive elements in the apo AI gene [22]. Bouly et al. [23] showed that fenofibrate treatment decreased the activity of lecithin cholesterol acyl transferase (LCAT) and increased phospholipid transfer protein (PLTP), while no changes were observed hepatic lipase (HL) activity in mice. Regarding the plasma levels of CETP, varying results were obtained in fibrate treated hyperlipidemic subjects, as follow: CETP decreased with bezafibrate and fenofibrate [24–27], or did not change with gemfibrozil, fenofibrate and bezafibrate [25, 28–31], or increased with gemfibrozil [32]. In CETP transgenic mice, two studies reported conflicting results, estimulation [33] and repression [34] of CETP expression by fenofibrate. One type of fibrate largely employed in clinical interventions, ciprofibrate, has not been evaluated regarding its effect on CETP levels. Since CETP is a potent modulator of HDL levels, it is relevant to clarify whether fibrates affect its gene expression and impact HDL metabolism.
Mouse models genetically modified to express specific human genes provide a good opportunity to re-examine the effect of established drugs and to characterize new sites and mode of action. Thus, the aims of the present work were to examine the effect of ciprofibrate treatment on the CETP gene expression, in the presence and absence of apo CIII induced hypertriglyceridemia, and its impact on the HDL metabolism. For this purpose we used transgenic mice over expressing human apo CIII and CETP genes driven by their natural promoters.
Materials and methods
Animal procedure
All animal protocols were approved by the university's Committee for Ethics in Animal Experimentation (CEEA/UNICAMP). The mice were housed in a temperature-controlled room on a 12 h light-dark cycle and had free access to food (standard rodent chow; Nuvital, Colombo, Brazil) and water. Hemizygous human CETP transgenic mice, line 5203, C57BL6/J background, expressing a human CETP minigene under the control of its natural flanking sequences [35] and mice overexpressing the human apolipoprotein CIII, line 3707 [36] were derived from Dr. Alan R. Tall's colony (Columbia University, New York, NY). The pups' tail tips were utilized for screening for the presence of the CETP gene promoter by polymerase chain reaction (PCR) DNA amplification of the -538 to -222 CETP promoter region (GeneBank U71187). Tail blood was also drawn for determining plasma CETP activity [37]. Apo CIII transgenic mice were screened by the level of plasma triglycerides: transgenic > 300 mg/dL and non-transgenic < 100 mg/dL. CETP and apoCIII transgenic mice were crossbred to generate 4 genotypic groups: CETP, CIII, CETP/CIII and non-transgenic. Male and female littermates, 10-14 weeks of age, were studied after 3 weeks of treatment (v.o.) with vehicle (2% arabic gum) or ciprofibrate (10 mg/Kg). Mice fasted overnight were anesthetized using ketamine (50 mg/kg, i.p., Parke-Davis, São Paulo, Brazil) and xylazine (16 mg/kg, i.p., Bayer S.A., São Paulo, Brazil) and killed by exsanguination through the retroorbital plexus. Blood samples were centrifuged at 2,500 × g at 4°C for 10 min. Aliquots of plasma were stored at -80°C until analyses.
Biochemical analyses in plasma
Lipoproteins from pooled or individual plasmas of mice were separated by fast protein liquid chromatography (FPLC) using a HR10/30 Superose 6 column (Amersham-Pharmacia Biotech., Uppsala, Sweden) as described previously [38]. Total cholesterol and triacylglycerols (Chod-Pap, Roche Diagnostic GmbH., Mannheim, Germany) and plasma free fatty acids (Wako Chemical, Neuss, Germany) were determined by enzymatic-colorimetric methods according to the manufacturer's instructions. Plasma glucose concentrations were determined by the glucose oxidase method using the Merck Diagnostic-Biotrol® Kit (Chennevières-les-Louvres, France) according to the instructions. Insulin concentrations were determined by radioimmunoassay as described previously [39] using rat insulin as standard.
Intravascular lipases activities
Total lipase activity was determined according to Ehnholm & Kuusi [40]. Briefly, overnight fasted mice plasmas, obtained before (basal) and 10 minutes after heparin I.V. injection (100 U/Kg body weight), were incubated with a 3H-triolein/arabic gum substrate (9,10 3H (N)-triolein, New England Nuclear, Boston, MA) in 0.2 M Tris-HCl buffer, pH 8.5, 37°C, during 1 hour. Hepatic lipase (HL) activity was determined in paralell tubes where the lipoprotein lipase (LPL) was inhibited by 2 M NaCl. The hydrolyzed labeled free fatty acids were extracted with methanol/chloroform/heptane (1.4 : 1.25 : 1), 0.14 M K2CO3/H3BO3, pH 10.5, dried under N2, and their radioactivity was determined in a liquid scintillation solution in a LS6000 Beckman Beta Counter. The LPL activity was calculated as the difference between the total and the hepatic lipase activities.
Cholesteryl ester transfer protein activity (exogenous assay)
CETP activity which reflects the plasma CETP concentration [41] was measured using an exogenous substrates assay as previously described [37]. Briefly, a mixture of human VLDL and LDL protein (100 μg) were incubated with 10,000 dpm of human HDL3 labeled with [14C]-cholesteryl ester (CE) [42] and 5 μl of mice plasma as the source of CETP in a final volume of 100 μl. Blanks were prepared with tris/saline/EDTA buffer (10 mM/140 mM/1 mM), pH 7.4, and negative controls with non-transgenic mice plasma. The incubations were carried out at 40°C for 2 hours. The apo B containing lipoproteins were precipitated using a mixture of 1.6% dextran sulfate/1 M MgCl2 solution (1:1) and the radioactivity was measured in the remaining supernatant in a scintillation solution Ultima Gold (Eastman Kodak Co., NY) in a LS6000 Beckman Beta Counter. The % CE transferred from [14C]-CE-HDL to VLDL+LDL was calculated as: (dpm in the blank tube - dpm in the plasma sample/dpm in the blank tube) × 100.
Reverse transcriptase-polymerase chain reaction (RT-PCR) for liver CETP mRNA
Total liver RNA was extracted from ~200 mg of tissue using Trizol reagent (Invitrogen, Grand Island, NY). The integrity of the RNA was checked in tris-borate 1.2% agarose gels stained with ethidium bromide. The amount and purity of the RNA were determined by OD readings at 260 and 280 nm (Gene Quant, Amersham-Pharmacia Biotech, Uppsala, Sweden). Genomic DNA contamination was excluded by running a PCR on the RNA samples. cDNA was obtained from 1 μg of total RNA by reverse transcription using 150 ng of random primers, 10 mM of dNTPs and 200 U of Moloney murine leukemia virus reverse transcriptase (Superscript II; Invitrogen, Grand Island, NY) in a final volume of 20 μl. The tubes were incubated for 60 min at 42°C followed by 15 min at 70°C to inactivate the enzyme. For PCR, a pair of primers was designed to amplify the region spanning part of exon 7 and exons 8 to 14 of the human CETP cDNA, which generated a 668 bp fragment. PCR mixtures consisted of 1 μl of cDNA, 15 pmol of primers, 2.5 U Taq polymerase and 200 μM of dNTPs in a final volume of 25 μl. A initial denaturation at 94°C for 4 min was followed by 28 cycles of 1 min at 94°C, 1.5 min at 58°C and 2 min at 72°C in a Gene Amp PCR System 9700 (Perkin-Elmer, Norwalk, CT). In parallel tubes, the same cDNA was amplified with primers (10 pmol) for rat β-actin cDNA, which generated a 533 bp fragment as an internal standard for the samples. The PCR conditions for β-actin were 2 min at 94°C followed by 23 cycles of 30 s at 94°C, 30 s at 57°C and 45 s at 72°C. The PCR products were separated by electrophoresis on 1.8% agarose gels and the DNA was visualized by ethidium bromide staining. The band intensities were determined by digital scanning and quantitation using Scion Image analysis software (Scion Corp., Frederick, USA).
Kinetic studies with labeled HDL
HDL was labeled with 3H-cholesteryl oleoyl ether (Amersham Biosciences, Buckinghamshire, England) as described by Oliveira and Quintão [42]. More than 95% of 3H-HDL was recovered in the cholesteryl ester band after extraction and thin layer chromatography. Labeling efficiency was about 30%. Labeled HDL was filtered in 0.22 μm Millipore membrane and used freshly. Awaken mice received an intraperitoneal (ip) injection of 3H-HDL (1 × 106 dpm). Blood samples (50 μl) were obtained by the tail tip at 0.5, 1, 2, 3, 4 and 6 hours after ip injection. Cholesterol radioactivity was determined in the supernatant of apo B lipoprotein precipitated plasma samples in a liquid scintillation solution in a beta counter (Beckman LS 6000 TA). Area under the radioactivity versus time curve was determined as previously reported [43]. Final plasma samples obtained by retro orbital bleeding of anesthetized mice were submitted to FPLC, as described above, to determine radioactivity distribution in the lipoprotein fractions.
Results
Mice expressing apo CIII and/or CETP and non-transgenic littermates (CIII, CIII/CETP, CETP, non-Tg) were treated with ciprofibrate during 3 weeks. After this period, fibrate treatment did not change body weight but relative liver weight increased significantly (48-69%, p < 0.05) in all four genotypic groups of mice, an expected effect for PPARα agonists [18]. Liver and muscle fat and glycogen content and gonadal adipose tissue weight were not affected by ciprofibrate treatment (data not shown).
Plasma lipid, glucose and insulin concentrations before and after ciprofibrate treatment are shown in Table 1. Cipro treatment reduced TG (30-43%) and FFA (19-47%) levels, did not change significantly the total cholesterol and insulin levels, and increased (11-43%) glycemia. The 10-40% elevation in glucose concentrations was confirmed in a shorter 2-week period of treatment (data not shown).
Cholesterol (chol) distribution in plasma lipoproteins is shown in Table 2. The response to ciprofibrate treatment was markedly dependent on the genotypes. Treated CIII mice presented elevation in VLDL-chol (88%) and reduction in HDL-chol (45%), resulting in a 3-fold increase in the (V+LDL)/HDL ratio. CIII/CETP mice responded to ciprofibrate with increases in VLDL (178%) and reductions in LDL (61%) and HDL (39%). This also led to a 2-fold increase in the (V+LDL)/HDL ratio. In ciprofibrate treated CETP mice, there was a reduction in LDL-chol fraction (29%) while in non-transgenic mice the LDL-chol increased (122%). In both, CETP and non-Tg, there were no changes in the (V+LDL)/HDL ratio.
As expected, ciprofibrate treatment increased plasma post heparin LPL activity (1.3-2.1 fold) in all groups except in CIII/CETP group (Table 3). Hepatic lipase was not altered in apo CIII expressing mice (CIII and CIII/CETP) but decreased significantly in normotriglyceridemic mice (CETP and non-Tg) (Table 3).
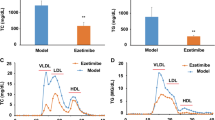
The effects of ciprofibrate on the CETP plasma levels and liver mRNA abundance are shown in figure 1. CIII/CETP and CETP mice treated with ciprofibrate showed 30 and 50% increase in plasma CETP activity, respectively, and 100 and 50% increase in relative liver mRNA levels, respectively.

Ciprofibrate increases CETP plasma levels and liver mRNA. Plasma CETP levels was measured as cholesteryl ester (CE) transfer rates from exogenous labeled HDL to V+LDL acceptors (A). Panel B shows the relative hepatic CETP mRNA levels. Dark and open bars correspond to ciprofibrate treated and control CETP expressing mice, respectively. Mean ± SE, n = 7-10. Student t test: *P < 0.05; **p < 0.005.
We hypothesized that higher plasma CETP and lower HL activities in fibrate treated CETP mice could change the cholesterol flow through the reverse cholesterol transport, increasing indirect removal through LDL and decreasing direct removal through HDL liver uptake. Thus, 3H-cholesteryl ether (CEt) labelled HDL was injected into ciprofibrate treated and control CETP mice and followed plasma radioactivity up to 6 hours. Results are shown in figure 2. The area under the radioactivity versus time curve was 23% larger for the treated than for control CETP mice (704 ± 39 vs. 572 ± 55 × 103 dpm.h, respectively, p < 0.05), indicating that greater amount of HDL remained in the plasma of treated mice during this 6 h-period. Radioactivity distribution in plasma lipoprotein fractions at 6 h after the 3H-HDL injection was determined by FPLC (Table 4). Ciprofibrate treated CETP mice presented significantly higher levels of labeled HDL (16%) and a marked reduction (56%) of 3H-CEt remaining in LDL fraction (p < 0.005) when compared to control CETP mice. This means that 3H-CEt transferred from HDL to LDL was more efficiently removed from the plasma of fibrate treated than from control CETP mice and explains the LDL-cholesterol mass reduction observed in treated CETP mice (Table 2). To confirm this interpretation, we measured liver uptake of 3H-CEt 6 hours after labeled HDL injection. Indeed, whole liver 3H-CEt content was significantly higher in ciprofibrate treated than in control CETP mice (36614 ± 4925 vs. 23946 ± 5745 dpm/g, p < 0.05).

Ciprofibrate treatment delays plasma removal of 3H-cholesteryl ether (3HCet) derived from HDL. Plasma kinetic of 3HCet labeled HDL injected intraperitoneally into ciprofibrate treated and control CETP transgenic mice was followed through 6 hours. Each point represents the average of radioactivity in plasma from 6 mice per group. The area under the radioactivity versus time curve was 23% larger for the treated than for control CETP mice (704 ± 39 vs. 572 ± 55 × 103 dpm.h, respectively, p < 0.05).
Discussion
Fibrates have been extensively and successfully used as hypotriglyceridemic drugs. However, differential efficiency and lipid responses have been found, which are probably related to the type of fibrates and to genetic variation in treated patients. A paradoxal increase in total or LDL-cholesterol in fibrate treated subjects is not very uncommon [44–46].
In this work we showed that some important changes in lipoprotein profile responses to ciprofibrate treatment are largely dependent on the baseline genotype-phenotype, specifically on the high levels of apoCIII and CETP. Fibrate treatment reduced TG levels in both hypertriglyceridemic (CIII and CIII/CETP) and normotriglyceridemic (CETP and non-Tg) mice. Thus, this effect is independent of the lipemic phenotype and of the presence of the proteins apo CIII and CETP. However, cholesterol distribution in plasma lipoproteins was affected in a genotype dependent manner. Fibrates induced VLDL-chol increases and HDL-chol reductions only in apo CIII hypertriglyceridemic mice (CIII and CIII/CETP). Although ciprofibrate increased plasma CETP activity, the reduction in HDL-chol was independent of CETP, since it occurred also in the absence of this protein (CIII only mice). HDL is probably reduced because in rodents fibrates decrease apo AI synthesis [21]. The preservation of HDL-cholesterol levels in treated normotriglyceridemic CETP and non-Tg mice is probably associated to the decreased activity of HL observed in these treated mice. Staels et al. [47] also reported that fenofibrate induced reduction in HL. On the other hand, ciprofibrate induced reductions in LDL-chol only in the presence of CETP, either in normo- and in hypertriglyceridemic mice (CIII/CETP and CETP), whereas in mice that did not express CETP, the fibrate treatment increased LDL cholesterol levels (CIII and non-Tg).
Conflicting results have been reported regarding the effect of fibrate treatments on the plasma CETP activity. In the present study, we demonstrate that ciprofibrate induced elevation of CETP plasma protein and liver mRNA levels. The mechanism underlying the CETP gene activation is likely transcriptional, either directly through a putative PPAR response element in the CETP promoter [48], or through PPARα induction of LXR which, in its turn, enhances CETP gene transcription [49].
The opposite effect of fibrate in increasing CETP and reducing HL levels in CETP transgenic mice led us to suppose that the indirect reverse cholesterol transport would be facilitated in these treated mice. Indeed, we observed that higher amounts of 3H-HDL and lower 3H-LDL remained in the plasma of ciprofibrate treated CETP mice 6 hours after the 3H-HDL injection. Thus, ciprofibrate increased CETP mediated transfer of 3H-CEt from HDL to LDL and accelerated plasma removal of 3H-CEt-LDL. Since LDL receptor expression seems not to be affected by fibrates, it is conceivable to think that ciprofibrate, or the consequent elevated CETP activity, could improve LDL particle affinity for its receptor, and hence speed up LDL plasma removal process. Accordingly, Sakai et al [50] reported that CETP deficient patients presented a retarded plasma clearance of LDL particles because of a lower affinity for their receptor. Alternatively, fibrate stimulated LPL and repressed HL activities could have generated more IDL. Since IDL is removed from plasma much faster than LDL this would explain the lower levels of LDL. In addition, CETP, alone or in conjunction with HL, must play a critical role in either IDL or LDL removal process, since ciprofibrate did not lower the plasma LDL levels in non-transgenic mice, which also presented higher LPL and lower HL activities.
Higher CETP activity induced by ciprofibrate did not result in reduction of HDL-cholesterol because plasma removal of HDL was also impaired in fibrate treated mice. Besides reducing HL activity, fibrate treatment may have also diminished SRBI expression, as described by Mardones et al. [51]. Nonetheless, whole liver cholesterol uptake was increased in ciprofibrate treated CETP transgenic mice, suggesting that the indirect (through LDL) reverse cholesterol transport was more effective in CETP treated mice, as depicted in the diagram in figure 3.

Diagram showing increased indirect reverse cholesterol transport steps as a response to ciprofibrate treatment. Changes in the steps 3, 4 and 5 were verified in the present work, and other steps were previously described for different fibrates and animal species, as follow: Step 1: Chinetti G, et al. Nat Med. 2001;7:53-58; Step 2: Staels et al., 1992 and Bouly et al [ref [23, 47]]; Step 4: Berger & Moller, Annu Rev Med 2002;53:409-35; Step 5: Staels et al., 1992 [ref [47]]; Step 6: Mardones et al., 2003 [ref [51]]; Step 7: Staels et al., 1998 [ref [19]]. ABCA1: ATP-binding cassette transporter-A1; LCAT: lecithin cholesterol acyl transferase; LDLr: LDL receptor; LRP: LDL receptor related protein; SRB1:Scavenger receptor class B type I.
Conclusion
Together these data suggest that ciprofibrate treatment of human CETP expressing model is highly beneficial by improving the flow of cholesterol through the indirect reverse cholesterol transport system and preserving HDL particles in the plasma compartment to play their pleiotropic antiatherogenic actions. In humans, these effects could be even more positive considering that fibrates also increase apo AI synthesis.
Abbreviations
- Apo :
-
apolipoprotein
- CE :
-
cholesteryl ester
- CEt :
-
cholesteryl ether
- CHD :
-
coronary heart disease
- chol :
-
cholesterol
- FFA :
-
free fatty acids
- FPLC :
-
fast protein liquid chromatography
- HL :
-
hepatic lipase
- LPL :
-
lipoprotein lipase
- PPAR :
-
peroxissome proliferator activated receptors
- TG :
-
triglycerides
References
McConathy WJ, Gesquiere JC, Bass H, Tartar A, Fruchart JC, Wang CS: Inhibition of lipoprotein lipase activity by synthetic peptides of apolipoprotein C-III. J Lipid Res. 1992, 33: 995-1003.
Sehayek E, Eisenberg S: Mechanisms of inhibition by apolipoprotein C of apolipoprotein E-dependent cellular metabolism of human triglyceride-rich lipoproteins through the low density lipoprotein receptor pathway. J Biol Chem. 1991, 266: 18259-18267.
Ebara T, Ramakrishnan R, Steiner G, Shachter NS: Chylomicronemia due to apolipoprotein CIII overexpression in apolipoprotein E-null mice. Apolipoprotein CIII-induced hypertriglyceridemia is not mediated by effects on apolipoprotein E. J Clin Invest. 1997, 99: 2672-2681. 10.1172/JCI119456
Ito Y, Azrolan N, O'Connell A, Walsh A, Breslow JL: Hypertriglyceridemia as a result of human apo CIII gene expression in transgenic mice. Science. 1990, 249: 790-793. 10.1126/science.2167514
Sacks FM, Alaupovic P, Moye LA, Cole TG, Sussex B, Stampfer MJ, Pfeffer MA, Braunwald E: VLDL, apolipoproteins B, CIII, and E, and risk of recurrent coronary events in the Cholesterol and Recurrent Events (CARE) trial. Circulation. 2000, 102: 1886-1892.
Alaupovic P, Mack WJ, Knight-Gibson C, Hodis HN: The role of triglyceride-rich lipoprotein families in the progression of atherosclerotic lesions as determined by sequential coronary angiography from a controlled clinical trial. Arterioscler Thromb Vasc Biol. 1997, 17: 715-722.
Lee SJ, Campos H, Moye LA, Sacks FM: LDL containing apolipoprotein CIII is an independent risk factor for coronary events in diabetic patients. Arterioscler Thromb Vasc Biol. 2003, 23: 853-858. 10.1161/01.ATV.0000066131.01313.EB
Tall AR, Jiang X, Luo Y, Silver D: 1999 George Lyman Duff memorial lecture: lipid transfer proteins, HDL metabolism, and atherogenesis. Arterioscler Thromb Vasc Biol. 2000, 20: 1185-1188.
Barter PJ, Brewer HB, Chapman MJ, Hennekens CH, Rader DJ, Tall AR: Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol. 2003, 23: 160-167. 10.1161/01.ATV.0000054658.91146.64
Neeli H, Rader DJ: Cholesteryl ester transfer protein (CETP) inhibitors: is there life after torcetrapib?. Cardiol Clin. 2008, 26: 537-546. 10.1016/j.ccl.2008.06.005
Thompson A, Di Angelantonio E, Sarwar N, Erqou S, Saleheen D, Dullaart RP, Keavney B, Ye Z, Danesh J: Association of cholesteryl ester transfer protein genotypes with CETP mass and activity, lipid levels, and coronary risk. JAMA. 2008, 23: 2777-2788. 10.1001/jama.299.23.2777.
Zhong S, Sharp DS, Grove JS, Bruce C, Yano K, Curb JD, Tall AR: Increased coronary heart disease in Japanese-American men with mutation in the cholesteryl ester transfer protein gene despite increased HDL levels. J Clin Invest. 1996, 97: 2917-2923. 10.1172/JCI118751
Harder C, Lau P, Meng A, Whitman SC, McPherson R: Cholesteryl ester transfer protein (CETP) expression protects against diet induced atherosclerosis in SR-BI deficient mice. Arterioscler Thromb Vasc Biol. 2007, 27: 858-864. 10.1161/01.ATV.0000259357.42089.dc
Westerterp M, Hoogt van der CC, de Haan W, Offerman EH, Dallinga-Thie GM, Jukema JW, Havekes LM, Rensen PC: Cholesteryl ester transfer protein decreases high-density lipoprotein and severely aggravates atherosclerosis in APOE*3-Leiden mice. Arterioscler Thromb Vasc Biol. 2006, 26: 2552-2559. 10.1161/01.ATV.0000243925.65265.3c
Marotti KR, Castle CK, Boyle TP, Lin AH, Murray RW, Melchior GW: Severe atherosclerosis in transgenic mice expressing simian cholesteryl ester transfer protein. Nature. 1993, 364: 73-75. 10.1038/364073a0
Cazita PM, Berti JA, Aoki C, Gidlund M, Harada LM, Nunes VS, Quintao EC, Oliveira HC: Cholesteryl ester transfer protein expression attenuates atherosclerosis in ovariectomized mice. J Lipid Res. 2003, 44: 33-40. 10.1194/jlr.M100440-JLR200
Berti JA, Salerno AG, Bighetti EJ, Casquero AC, Boschero AC, Oliveira HC: Effects of diabetes and CETP expression on diet-induced atherosclerosis in LDL receptor-deficient mice. APMIS. 2005, 113: 37-44. 10.1111/j.1600-0463.2005.apm1130106.x
Auwerx J, Schoonjans K, Fruchart JC, Staels B: Transcriptional control of triglyceride metabolism: fibrates and fatty acids change the expression of the LPL and apo C-III genes by activating the nuclear receptor PPAR. Atherosclerosis. 1996, 124 (Suppl): S29-37. 10.1016/0021-9150(96)05854-6
Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart JC: Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation. 1998, 98: 2088-2093.
Gnasso A, Lehner B, Haberbosch W, Leiss O, von Bergmann K, Augustin J: Effect of gemfibrozil on lipids, apoproteins, and postheparin lipolytic activities in normolipidemic subjects. Metabolism. 1986, 35: 387-393. 10.1016/0026-0495(86)90125-3
Berthou L, Duverger N, Emmanuel F, Langouet S, Auwerx J, Guillouzo A, Fruchart JC, Rubin E, Denefle P, Staels B, Branellec D: Opposite regulation of human versus mouse apolipoprotein A-I by fibrates in human apolipoprotein A-I transgenic mice. J Clin Invest. 1996, 97: 2408-2416. 10.1172/JCI118687
Vu-Dac N, Chopin-Delannoy S, Gervois P, Bonnelye E, Martin G, Fruchart JC, Laudet V, Staels B: The nuclear receptors peroxisome proliferator-activated receptor alpha and Rev-erbalpha mediate the species-specific regulation of apolipoprotein A-I expression by fibrates. J Biol Chem. 1998, 273: 25713-25720. 10.1074/jbc.273.40.25713
Bouly M, Masson D, Gross B, Jiang XC, Fievet C, Castro G, Tall AR, Fruchart JC, Staels B, Lagrost L, Luc G: Induction of the phospholipid transfer protein gene accounts for the high density lipoprotein enlargement in mice treated with fenofibrate. J Biol Chem. 2001, 276: 25841-25847. 10.1074/jbc.M101160200
Watts GF, Ji J, Chan DC, Ooi EM, Johnson AG, Rye KA, Barrett PH: Relationships between changes in plasma lipid transfer proteins and apolipoprotein B-100 kinetics during fenofibrate treatment in the metabolic syndrome. Clin Sci (Lond). 2006, 111: 193-199. Erratum in: Clin Sci (Lond) 2007, 112:625, 10.1042/CS20060072
Durrington PN, Mackness MI, Bhatnagar D, Julier K, Prais H, Arrol S, Morgan J, Wood GN: Effects of two different fibric acid derivatives on lipoproteins, cholesteryl ester transfer, fibrinogen, plasminogen activator inhibitor and paraoxonase activity in type IIb hyperlipoproteinaemia. Atherosclerosis. 1998, 138: 217-225. 10.1016/S0021-9150(98)00003-3
Guerin M, Bruckert E, Dolphin PJ, Turpin G, Chapman MJ: Fenofibrate reduces plasma cholesteryl ester transfer from HDL to VLDL and normalizes the atherogenic, dense LDL profile in combined hyperlipidemia. Arterioscler Thromb Vasc Biol. 1996, 16: 763-772.
Jonkers IJ, Smelt AH, Hattori H, Scheek LM, van Gent T, de Man FH, Laarse van der A, van Tol A: Decreased PLTP mass but elevated PLTP activity linked to insulin resistance in HTG: effects of bezafibrate therapy. J Lipid Res. 2003, 44: 1462-1469. 10.1194/jlr.M300008-JLR200
McPherson R, Agnani G, Lau P, Fruchart JC, Edgar AD, Marcel YL: Role of Lp A-I and Lp A-I/A-II in cholesteryl ester transfer protein-mediated neutral lipid transfer. Studies in normal subjects and in hypertriglyceridemic patients before and after fenofibrate therapy. Arterioscler Thromb Vasc Biol. 1996, 16: 1340-1346.
Mann CJ, Yen FT, Grant AM, Bihain BE: Mechanism of plasma cholesteryl ester transfer in hypertriglyceridemia. J Clin Invest. 1991, 88: 2059-2066. 10.1172/JCI115535
Ponsin G, Girardot G, Berthezene F: Mechanism of the gemfibrozil-induced decrease in the transfer of cholesterol esters from high density lipoproteins to very low and low density lipoproteins. Biochem Med Metab Biol. 1994, 52: 58-64. 10.1006/bmmb.1994.1034
Franceschini G, Lovati MR, Manzoni C, Michelagnoli S, Pazzucconi F, Gianfranceschi G, Vecchio G, Sirtori CR: Effect of gemfibrozil treatment in hypercholesterolemia on low density lipoprotein (LDL) subclass distribution and LDL-cell interaction. Atherosclerosis. 1995, 114: 61-71. 10.1016/0021-9150(94)05465-U
Kahri J, Sane T, van Tol A, Taskinen MR: Effect of gemfibrozil on the regulation of HDL subfractions in hypertriglyceridaemic patients. J Intern Med. 1995, 238: 429-436. 10.1111/j.1365-2796.1995.tb01220.x
Beyer TP, Chen Y, Porter RK, Lu D, Schmidt RJ, Mantlo NB, Konrad RJ, Cao G: Peroxisome proliferator-activated receptor alpha agonists regulate cholesterol ester transfer protein. Lipids. 2008, 43: 611-618. 10.1007/s11745-008-3187-0
Hoogt van der CC, de Haan W, Westerterp M, Hoekstra M, Dallinga-Thie GM, Romijn JA, Princen HM, Jukema JW, Havekes LM, Rensen PC: Fenofibrate increases HDL-cholesterol by reducing cholesteryl ester transfer protein expression. J Lipid Res. 2007, 48: 1763-1771. 10.1194/jlr.M700108-JLR200
Jiang XC, Agellon LB, Walsh A, Breslow JL, Tall A: Dietary cholesterol increases transcription of the human cholesteryl ester transfer protein gene in transgenic mice. Dependence on natural flanking sequences. J Clin Invest. 1992, 90: 1290-1295. 10.1172/JCI115993
Walsh A, Azrolan N, Wang K, Marcigliano A, O'Connell A, Breslow JL: Intestinal expression of the human apoA-I gene in transgenic mice is controlled by a DNA region 3' to the gene in the promoter of the adjacent convergently transcribed apoC-III gene. J Lipid Res. 1993, 34: 617-623.
Berti JA, Amaral ME, Boschero AC, Nunes VS, Harada LM, Castilho LN, Oliveira HC: Thyroid hormone increases plasma cholesteryl ester transfer protein activity and plasma high-density lipoprotein removal rate in transgenic mice. Metabolism. 2001, 50: 530-536. 10.1053/meta.2001.22514
Jiao S, Cole TG, Kitchens RT, Pfleger B, Schonfeld G: Genetic heterogeneity of lipoproteins in inbred strains of mice: analysis by gel-permeation chromatography. Metabolism. 1990, 39: 155-160. 10.1016/0026-0495(90)90069-O
Scott AM, Atwater I, Rojas E: A method for the simultaneous measurement of insulin release and B cell membrane potential in single mouse islets of Langerhans. Diabetologia. 1981, 21: 470-475. 10.1007/BF00257788
Ehnholm C, Kuusi T: Preparation, characterization, and measurement of hepatic lipase. Methods Enzymol. 1986, 129: 716-738. full_text
McPherson R, Mann CJ, Tall AR, Hogue M, Martin L, Milne RW, Marcel YL: Plasma concentrations of cholesteryl ester transfer protein in hyperlipoproteinemia. Relation to cholesteryl ester transfer protein activity and other lipoprotein variables. Arterioscler Thromb. 1991, 11: 797-804.
Oliveira HC, Quintao EC: 'In vitro' cholesteryl ester bidirectional flow between high-density lipoproteins and triglyceride-rich emulsions: effects of particle concentration and composition, cholesteryl ester transfer activity and oleic acid. J Biochem Biophys Methods. 1996, 32: 45-57. 10.1016/0165-022X(94)00060-Q
Tai MM: A mathematical model for the determination of total area under glucose tolerance and other metabolic curves. Diabetes Care. 1994, 17: 152-154. 10.2337/diacare.17.2.152
Eisenberg S, Gavish D, Oschry Y, Fainaru M, Deckelbaum RJ: Abnormalities in very low, low and high density lipoproteins in hypertriglyceridemia. Reversal toward normal with bezafibrate treatment. J Clin Invest. 1984, 74: 470-482. 10.1172/JCI111444
Pasternak RC, Brown LE, Stone PH, Silverman DI, Gibson CM, Sacks FM: Effect of combination therapy with lipid-reducing drugs in patients with coronary heart disease and "normal" cholesterol levels. A randomized, placebo-controlled trial. Harvard Atherosclerosis Reversibility Project (HARP) Study Group. Ann Intern Med. 1996, 125: 529-540.
Turpin G, Bruckert E: Efficacy and safety of ciprofibrate in hyperlipoproteinaemias. Atherosclerosis. 1996, 124 (Suppl): S83-87. 10.1016/0021-9150(96)05861-3
Staels B, Peinado-Onsurbe J, Auwerx J: Down-regulation of hepatic lipase gene expression and activity by fenofibrate. Biochim Biophys Acta. 1992, 1123: 227-230.
Cheema SK, Rashid-Kolvear F: Streptozotocin-induced increase in cholesterol ester transfer protein (CETP) and its reversal by insulin in transgenic mice expressing human CETP. Can J Physiol Pharmacol. 2003, 81: 997-1004. 10.1139/y03-093
Luo Y, Tall AR: Sterol upregulation of human CETP expression in vitro and in transgenic mice by an LXR element. J Clin Invest. 2000, 105: 513-520. 10.1172/JCI8573
Sakai N, Yamashita S, Hirano K, Ishigami M, Arai T, Kobayashi K, Funahashi T, Matsuzawa Y: Decreased affinity of low density lipoprotein (LDL) particles for LDL receptors in patients with cholesteryl ester transfer protein deficiency. Eur J Clin Invest. 1995, 25: 332-339. 10.1111/j.1365-2362.1995.tb01710.x
Mardones P, Pilon A, Bouly M, Duran D, Nishimoto T, Arai H, Kozarsky KF, Altayo M, Miquel JF, Luc G, Clavey V, Staels B, Rigotti A: Fibrates down-regulate hepatic scavenger receptor class B type I protein expression in mice. Biol Chem. 2003, 278: 7884-7890. 10.1074/jbc.M211627200.
Acknowledgements
This work was supported by grants from Fundação de Amparo a Pesquisa do Estado de S. Paulo (Fapesp) and Conselho Nacional para o Desenvolvimento Científico e Tecnológico (INCT-CNPq, National Institute for Science and Technology in Diabetes and Obesity). The authors wish to thank Maria Esméria C. Amaral and Valéria Nunes Sutti for helping with the insulin and lipoprotein lipase assays and Léscio D. Teixeira for animal care.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
EJBB and PRP equally participated in all experiments, analysis and data interpretation and helped to draft the manuscript. ACC and JAB carried out mice breeding and screening and helped with plasma biochemical analysis and CETP activity assay. HCFO conceived the study and its design, obtained research grants for its development, supervised all technical activities, coordinated data interpretation and wrote the final version of the manuscript. All authors read and approved the final manuscript.
Eliete JB Bighetti, Patrícia R Patrício contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Bighetti, E.J., Patrício, P.R., Casquero, A.C. et al. Ciprofibrate increases cholesteryl ester transfer protein gene expression and the indirect reverse cholesterol transport to the liver. Lipids Health Dis 8, 50 (2009). https://doi.org/10.1186/1476-511X-8-50
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-511X-8-50