
Abstract
Insulin resistance condition is associated to the development of several syndromes, such as obesity, type 2 diabetes mellitus and metabolic syndrome. Although the factors linking insulin resistance to these syndromes are not precisely defined yet, evidence suggests that the elevated plasma free fatty acid (FFA) level plays an important role in the development of skeletal muscle insulin resistance. Accordantly, in vivo and in vitro exposure of skeletal muscle and myocytes to physiological concentrations of saturated fatty acids is associated with insulin resistance condition. Several mechanisms have been postulated to account for fatty acids-induced muscle insulin resistance, including Randle cycle, oxidative stress, inflammation and mitochondrial dysfunction. Here we reviewed experimental evidence supporting the involvement of each of these propositions in the development of skeletal muscle insulin resistance induced by saturated fatty acids and propose an integrative model placing mitochondrial dysfunction as an important and common factor to the other mechanisms.
Similar content being viewed by others
Introduction
Insulin resistance is broadly defined as the reduction in insulin ability to stimulate glucose uptake from body peripheral tissues. At physiological conditions, insulin activates glucose uptake by stimulating the canonical IRS-PI3K-Akt pathway and by phosphorylating and inactivating Akt substrate 160 (AS160), a protein that, when activated, prevents glucose transporter (GLUT) 4 translocation to the membrane. Thus, by inhibiting AS160, insulin promotes the GLUT4 translocation from inner vesicules, promoting fusion to the plasma membrane and consequently glucose uptake [1].
Although insulin resistance is a key component of several chronic syndromes associated with obesity such as type 2 diabetes mellitus and metabolic syndrome, the involved factors and their underlying mechanisms linking excessive adiposity to insulin resistance were not completely elucidated yet [2–5]. Evidence suggests that fatty acids, whose circulating levels are markedly increased in obesity and associated-diseases, might play a role in the development of skeletal muscle insulin resistance [6, 7]. In this sense, prolonged exposure of skeletal muscle and myocytes to high levels of fatty acids leads to severe insulin resistance [8, 9]. Among the different types of fatty acids, saturated long-chain fatty acids such as palmitic and stearic acids were demonstrated to be potent inducers of insulin resistance [5, 10]. Several mechanisms have been suggested by us [2, 5, 11, 12] and others [6, 8, 13–16] to explain how saturated fatty acids impair insulin actions such as the Randle cycle, accumulation of intracellular lipid derivatives (diacylglycerol and ceramides), oxidative stress, modulation of gene transcription, inflammation and mitochondrial dysfunction. In the present review, we discuss evidence supporting the involvement of these mechanisms in the regulation of insulin sensitivity by saturated fatty acids and propose the mitochondrial dysfunction found in conditions of elevated fatty acid levels has a central role in the pathogenesis of insulin resistance.
Mechanisms underlying the insulin resistance induced by saturated fatty acids
Competition between fatty acids and glucose: the randle cycle
The first mechanistic explanation for the inverse relationship between fatty acids availability and glucose utilization was proposed by Randle et al.[13]. In this study, it was shown that an elevation in fatty acids supply to diaphragm and isolated heart is associated with an increase in fatty acid oxidation and an impairment in glycolytic flux and glucose utilization, such effect being mediated by alosteric inhibition of glycolytic enzymes. More specifically, the proposed hypothesis was that increased fatty acid oxidation raises the production of acetyl-CoA resulting in inhibition of pyruvate dehydrogenase activity and elevation of citrate levels at the tricarboxylic acid cycle. Citrate together with an increased ATP/ADP ratio reduce the activity of phosphofructokinase and consequently glucose flux through the glycolytic pathway, resulting in glucose 6-phosphate accumulation, hexokinase II inhibition, increase in intracellular glucose content and, consequently, reduction in glucose uptake [17, 18].
In accordance with Randle's hypothesis, elevation in circulating fatty acids levels by either intralipid/heparin or lipid infusion in rats, humans and type 2 diabetes mellitus patients is associated with impairments in glucose uptake, utilization and oxidation in insulin-sensitive tissues (heart, skeletal muscle and adipose tissue) [19–21]. Acutely, fatty acids lead to Randle cycle effect, increasing intracellular content of citrate and glucose-6-phosphate and decreasing glycolytic pathway flux [2, 11]. It has been also demonstrated that palmitate accutely increases glucose uptake in L6 myotubes by activating insulin signaling pathways (Akt and ERK1/2) [22].
However, in contrast to Randle's hypothesis, in which intracellular glucose accumulation must precede the inhibition of glucose uptake, further studies demonstrated that the insulin resistance induced by fatty acids is primarily associated with impaired glucose uptake rather than changes in hexose metabolism [8, 18]. In studies involving lipid infusion associated with other techniques including glucose and insulin clamp and nuclear magnetic resonance a rapid reduction in glycolysis (previous to 2 hours) followed by impaired glucose disposal and glycogen synthesis (between 4-6 hours) was observed [7, 14]. Roden et al.[14] demonstrated that the reduction in muscle glycogen synthesis is preceded by a decrease in intramuscular glucose 6-phosphate, suggesting that the increase in plasma fatty acid concentration initially induces insulin resistance by inhibiting glucose transport or its phosphorylation. Other studies also demonstrated that lipid infusion decreases intracellular glucose and glucose 6-phosphate content, due to inhibition of glucose uptake by skeletal muscle [8, 23]. These studies demonstrated that Randle cycle does not completely explain the effects of FFA on glucose metabolism indicating that other mechanisms are also involved in the FFA-induced insulin resistance.
Inhibition of skeletal muscle insulin signaling by saturated fatty acids
In addition to its important effects on glucose metabolism directly, saturated fatty acids were demonstrated to affect insulin intracellular signaling pathways in skeletal muscle and myocytes [5, 14, 24, 25]. Studies have demonstrated a marked reduction in IRS-1 tyrosine phosphorylation [9], IRS-1 and -2-associated PI3-kinase activity [9, 26], and Akt phosphorylation and activity [26] in skeletal muscle after lipid infusion in euglycemic-hyperinsulinemic clamp. Along with a direct effect of saturated fatty acids on skeletal muscle insulin signaling, palmitic acid was shown to decrease insulin receptor expression and activity [27] and phosphorylation of IRS-1 and -2 at tyrosine residues [2, 28], Akt [5, 28–31] and GSK-3, as reviewed by Schmitz-Peiffer et al.[32] in isolated soleus muscle, primary culture of rat myocytes, pmi28 myotubes, C2C12 and L6 myocytes. Similarly to skeletal muscle, palmitic acid inhibits Akt phosphorylation and activity in rat perfused heart and in HL-1 cells, an immortalized cardiomyocyte like cell lineage [33].
Several mechanisms have been postulated to account for the inhibition of insulin signaling by saturated fatty acids, including the activation of various kinases such as PKCs, IKK β, JNK, and p38 MAP kinase. These kinases have been postulated to catalyze the phosphorylation of serine residues in IRS-1 inhibiting its activity and directing it for degradation by the proteasome [34, 35]. Such effects culminate with a reduction in the phosphorylation of tyrosine residues of IRS-1 by insulin, blocking its downstream signal transduction [36–38]. Other serine/threonine kinase activated in high-fat diet-induced or palmitate-induced insulin resistance is mammalian target of rapamycin (mTOR) [39, 40], but the mechanisms involved are unknown yet.
Lipotoxic intramyocellular lipid accumulation induced by fatty acids
When the amount of circulating lipids chronically exceeds white adipose tissue ability for uptake and storage, like obesity, fatty acids accumulate in other tissues with limited capacity for lipid storage such as liver and skeletal muscles. Such abnormal ectopic lipid accumulation (lipotoxicity) is strongly associated with insulin resistance [41, 42].
Fatty acids accumulate intracellularly in myocytes mainly as long-chain fatty acyl-CoA, monoacylglcyerol, diacylglycerol, phosphatidic acid, triacylglycerol and ceramides [32, 43–46]. Among these fatty acid derivatives, high intramyocellular levels of diacylglycerol, triacylglycerol, and ceramides are directly associated with insulin resistance. Corroborating with this hypothesis, high fat feeding is associated with an increase in intramyocellular content of diacylglycerol and triacylglycerol and insulin resistance, such effects being abolished by inhibition of muscle lipid accumulation due to genetic deletion of lipoprotein lipase, fatty acid transporters (CD36 and FATP1), and diacylglycerol acyl transferase-1 (DGAT-1) [47–49]. Diacylglycerol accumulation is associated with the activation of subgroup of novel kinases, members of the large protein kinase C (PKC) family. Among the novel kinases, diacylglycerol directly activates PKCθ that catalyzes the phosphorylation of serine-307 residue at IRS-1, reducing its tyrosine phosphorylation and activation by insulin.
Consistent with this, Schmitz-Peiffer et al.[50] reported increased concentration of DAG in rodent's muscle and activation of PKCs induced by high-fat diet. Similarly, infusion of lipid and heparin caused insulin resistance in muscles that was associated with accumulation of intracellular DAG and specific activation of PKCθ [8]. Insulin resistance in this model was due to lipid-induced defects in the insulin signaling pathway that was caused by a reduction in tyrosine phosphorylation of IRS1, increasing its phosphorylation in serine-307 residue [9]. However, there are still no evidence to explain how the activation of novel PKCs might relate to serine phosphorylation of IRS1, and which kinases might have a role in the pathway, as reviewed by Samuel et al.[51].
In addition to diacylglycerol accumulation, high-fat diet or palmitate treatment increases production of ceramide and sphingosines in skeletal muscle cells, which is associated with glucose intolerance and insulin resistance [52, 53]. Evidence suggests that ceramide and phosphatidic acid mediate the deleterious effects of palmitic acid on insulin signaling in cultured myotubes and insulin mediated Akt and GSK phosphorylation in C2C12 myotubes [52, 54]. Ceramides also affects insulin signaling by two distinct mechanisms involving the activation of Akt dephosphorylation at threonine 308 and inhibition of its translocation to the plasma membrane [55], such effects being dependent on ceramides activation of protein phosphatase 2A (PP2A) and PKCζ, respectively [56, 57]. In addition, glycosylceramide, a glycosyl derivative of ceramides was shown to inhibit insulin receptor activity inducing insulin resistance [56, 57]. Recently, it has been demonstrated that increased lysophosphatidylcholine content, a phosphatidic acid, in L6 myotubes treated with palmitate also leads to JNK activation and IRS-1 Ser307 phosphorylation, contributing to the development of muscle insulin resistance [58].
Activation of inflammatory signaling pathways by saturated fatty acids
Saturated fatty acids activate inflammatory signaling pathways directly through interaction with members of Toll-like receptor (TLR) family and indirectly through the secretion of cytokines including TNF-α, IL-1β and IL-6 [59–61]. TLRs are an evolutionarily ancient pattern-recognition class of receptors that facilitate the detection of microbes. Saturated fatty acids activate TLR-4 in skeletal muscle promoting c-Jun NH(2)-terminal kinase (JNK) and Iκb kinase (IKK) complex activation, which results in degradation of the inhibitor of κB (IκBα) and nuclear factor-κB (NFκB) activation. Activation of JNK and IKKβ by saturated fatty acids is associated with a marked inhibition of insulin action due to the phosphorylation of serine residues on the insulin IRS-1 and inhibition of its stimulatory phosphorylation of tyrosine residues by the insulin receptor [62, 63]. Corroborating with an important contribution of TLR-4 to muscle insulin resistance, mice containing a loss of function by mutation of this receptor are partially protected from fat-induced inflammation and insulin resistance [64]. In addition, diabetic and obese mice have increased skeletal muscle IKK and JNK activities, whose pharmacological and genetic inhibition leads to an improvement in insulin sensitivity and glucose tolerance [65–67].
Coletta and Mandarino [68] demonstrated that changes in genes and proteins from inflammatory pathway contribute to the mitochondrial dysfunction observed in insulin resistant muscle and it can lead to decreased fat oxidation, ectopic fat accumulation, insulin signaling abnormalities and finally insulin resistance. These authors indicate that this mechanism is compatible with and complementary to other hypotheses regarding the vicious cycle connecting inflammation, mitochondrial changes, lipid accumulation and insulin signaling defects. The novel aspect of this mechanism is that it connects inflammatory processes with changes in insulin sensitivity by means of altered mechanosignal transduction due to fibrotic changes.
Fatty acids were also demonstrated to reduce mitochondrial function through induction of pro-inflammatory cytokines. Saturated fatty acids, palmitic and stearic acids, stimulate the secretion of TNF-α, IL-1β and IL-6 in human leukocytes [60, 69]. Wen et al.[61] showed increased IL-1β production and release by palmitic acid in macrophages through activation of the NLRP3-ASC inflammasome. The pro-inflammatory cytokines have been associated to impaired mitochondrial function, establishing a link between fatty acids and mitochondrial dysfunction. Indeed, some studies reported reduced mitochondrial function in cells exposed to TNF-α, IL-1β or IL-6 [70, 71].
Alteration in gene expression by saturated fatty acids
Evidence has been obtained that fatty acids modulate expression of genes involved in glucose and lipid metabolism. Schmid et al.[72] demonstrated that C57BL/6 mice submitted to high-fat diet present reduced expression of enolase, a glycolytic enzyme, and ATP synthase in skeletal muscle. In addition, other enzymes of the glycolytic pathway have been shown to be modulated by fatty acids, such as pyruvate dehydrogenase kinase isozyme 1 (PDK-1), whose expression is increased in pancreatic islets incubated with saturated fatty acids [73], and lactate dehydrogenase A (LDHA), which was downregulated in white adipose tissue from high-fat-fed animals [74]. Moreover, increased intramyocellular lipid content has been associated with down-regulation of PGC-1α and of other genes encoding protein mitochondrial respiratory complexes I, II, III, and IV [75], resulting in impaired mitochondrial biogenesis and function [76]. Some studies identified transcription factors that recognize conserved motifs at the promoters of mitochondrial oxidative phosphorylation genes, such as nuclear respiratory factor (NRF)-1 and GA-binding protein (GABP) (also known as NRF-2) [77]. Studies showed that the peroxisome proliferator activator receptors (PPARs) control mitochondrial gene subsets, modulating fatty acid oxidation (FAO) and uncoupling [77, 78]. Later, studies showed that PGC-1α is a transcriptional coactivator of NRF-1, GABP, and PPARs, demonstrating the ability of PGC-1α to integrate physiological signals and to increase mitochondrial biogenesis and oxidative function [79, 80]. Thus, a reduction of PGC-1α content in conditions of high fatty acid levels might be associated with impairment of mitochondrial function [5, 76]. In addition, insulin-resistant subjects have reduced expression of mitochondrial genes, such as cytochrome c oxidase and complexes I and III subunits of the electron transport chain [81]. Activities of carnitine palmitoyltransferase-1 (CPT-1) and other key mitochondrial enzymes, such as citrate synthase and β-hydroxyacyl-CoA dehydrogenase, have also been found decreased in skeletal muscle from obese and type 2 diabetic individuals [26, 82, 83]. These changes in gene expression and enzyme activities induced by fatty acids contribute to the reduced mitochondrial oxidative capacity consequently leading to mitochondrial dysfunction.
Increase in reactive oxygen species by saturated fatty acids
Type 2 diabetes mellitus, obesity and the metabolic syndrome are strongly correlated with increased skeletal muscle content of reactive oxygen species (ROS) [84–86]. All conditions cited above contribute to an oxidative environment, modulating insulin sensitivity either by increasing insulin signaling or impairing glucose tolerance. The mechanisms by which this occurs are often multifactorial and complex, involving several cell signaling pathways [87].
Production of ROS can occur in response to diverse stimuli including: (1) intracellular factors, such as nutrient metabolism, endoplasmic reticulum stress, and detoxification of various xenobiotics; (2) extracellular factors like signaling through plasma membrane receptors, such as hormones and growth factors and by pro-inflammatory cytokines; and (3) physical-environmental factors (e.g. ultraviolet irradiation) [88–91]. When moderately produced, ROS are involved in important physiological processes that lead to desired cellular responses. However, high ROS production is negatively associated with different biological signaling pathways [87]. ROS can react with multiple cellular components, such as proteins, lipids and nucleic acids, generating reversible or irreversible oxidative modifications. Pathophysiological processes mediated by ROS are more likely to induce irreversible modifications in cellular components, a reasonable definition of the term oxidative stress [88].
Control of vascular tone, cell adhesion, immune responses, and growth factors and hormone action are examples of ROS participation in normal physiology [92, 93]. Conversely, a negative role of ROS has been implicated in ageing-related diseases, malignant transformation, atherosclerosis, neurodegenerative diseases, obesity, and diabetes [88, 94, 95]. Insulin signaling can be also impaired by oxidative stress, but the mechanisms involved are not fully understood. Studies have been demonstrated that ROS lead to impaired insulin response by inducing IRS serine/threonine phosphorylation, decreasing GLUT4 gene transcription, and decreasing mitochondrial activity [37, 96].
Chronic elevation in plasma lipids levels and excessive intramyocellular fatty acid disposal are characterized by increased ROS and reactive nitrogen species (RNS) production [15, 97, 98]. Diet-induced obese mice have an increased expression of inducible nitric oxide synthase (iNOS) and RNS generation in skeletal muscle such effects being associated with impaired insulin sensitivity [15, 99], since mice with iNOS disruption in muscle are protected from insulin resistance induced by obesity [100, 101]. Diabetic patients present elevated ROS production in endothelial cells through NADPH oxidase activation, a mechanism mediated by PKC [102]. NADPH-oxidase complex is also found in skeletal muscles, raising the possibility that a similar mechanism occurs under elevated FA availability. Our group recently demonstrated that palmitate induces superoxide production in cultured skeletal muscle cells via NADPH oxidase activation, at least in part [98]. Some evidence also links xanthine oxidase (XO) as a source of increased ROS generation in diabetes and obesity. XO protein and activity is found increased in muscle arterioles and livers from animal models of type 1 diabetes and diet-induced obesity [88, 103, 104].
Animals treated either with high-fat diet or oxidant drugs such as buthionine sulfoximine (BSO), an inhibitor of gluthatione synthase, have increased skeletal muscle ROS production, oxidative stress and are insulin-resistant [105, 106]. On the other hand, animals food restricted or treated with antioxidant drugs such as N-acetyl-cysteine (NAC), lipoic acid, vitamin E, and taurine, have reduced oxidative stress and improved insulin sensitivity [106–108]. In addition, mice overexpressing SOD2 have decreased ROS levels, improved hepatic insulin sensitivity, normalization of blood glucose and insulin levels, and reduced activation of cellular stress signalling pathways [109]. These data suggest that mitochondrial ROS is important for the development of insulin resistance.
The involvement of oxidative stress in insulin resistance was also observed in studies performed in myocytes. In L6 muscle cells, H2O2 reduced insulin-stimulated glucose uptake and glutathione content, effects that were prevented by preincubation with the antioxidant lipoic acid [110]. Rat soleus muscle exposed to nitric oxide (NO) donors have decreased insulin-stimulated glucose uptake and glycogen synthesis, effects that were associated with reduced insulin-stimulated phosphorylation of IR, IRS-1, and Akt [15].
Since mitochondria is the main site of ROS production in skeletal muscle, mitochondrion DNA, protein and lipids components are exposed to high levels of these metabolites suffering structural modification and damage which at long term can result in impaired function of this organelle [111, 112].
Mitochondrial dysfunction or reduced mitochondrial biogenesis and density can lead to a decrease in mitochondrial fatty acids oxidation, which results in increased levels of fatty acil-CoA and DAG that can activate stress-related Ser/Thr kinase activity and inhibit glucose transport, as reviewed by Lowell and Shulman [113].
In the case of stress-activated kinases, oxidative stress also contributes to impair insulin signaling by increased uncoupling protein-2 (UCP-2) activity. When these proteins are activated, it results in a heat generation that does not contribute to ATP production [114, 115]. UCP-2 negatively regulates glucose-stimulated insulin secretion by reducing the ATP production, which is key to provide energy for almost all cellular processes [113]. In addition, UCP-2-/- mice demonstrate enhanced insulin secretory capacity after a high-fat diet due to improved β-cell functions in a type 2 diabetes animal model [116].
Mitochondrial uncoupling is a powerful tool to control ROS formation and consequently to preserve mitochondrial function. It can be hypothesized that fatty-acid induced uncoupling decrease mitochondrial ROS production and thus it can prevent mitochondrial lipotoxicity. In fact, UCP-3 is upregulated using high-fat diets [117, 118], fasting [119], etomoxir treatment (which inhibits the mitochondrial fatty acid oxidation) [120, 121] and lipid infusion [122], all conditions being associated with excessive lipid accumulation in skeletal muscle. On the other hand, when fat oxidative capacity is improved, like with endurance training [123, 124], weight loss [125], or lowering circulatory fatty acids [126, 127] there is a decrease in UCP-3. Interestingly, UCPs are activated by fatty acids and/or its peroxidation products, reducing mitochondrial ROS production [128, 129]. So, it can be suggested that UCP-3 is involved in the protection against mitochondrial lipotoxicity by decreasing ROS production when activated by FA, as reviewed by Schrauwen et al.[130].
In summary, oxidative stress seems to play an important role in mitochondrial dysfunction, which can further exacerbate stress signals and reduce ATP production. The pathways leading to insulin resistance may be synergistic and mitochondrial dysfunction can create a feedback loop, adding to the overall oxidative stress environment [87].
Impairment of skeletal muscle mitochondrial function by saturated fatty acids
Several studies have shown that mitochondrial content, mitochondrial function, and oxidative capacity are decreased in insulin-resistant obese and type 2 diabetic individuals [131, 132] suggesting that mitochondrial dysfunction might play an important role in the pathophysiology of insulin resistance. Corroborating with this hypothesis, impaired mitochondrial function and reduced fatty acid oxidative capacity were found in isolated primary myocytes, isolated rectus abdominal muscle strips and muscle homogenates from insulin-resistant obese and type 2 diabetic patients [26, 83, 133]. Moreover, mitochondrial density is reduced in insulin-resistant skeletal muscle from children, offspring of people with type 2 diabetes, suggesting that impaired mitochondrial oxidative capacity can be an inherited defect and an early marker for the development of insulin resistance [96]. Some studies have also reported alterations (mutations, polymorphisms, and epigenetics) in the mitochondrial DNA in conditions of insulin resistance, such as obesity, type 2 diabetes, and metabolic syndrome [134–137].
Evidence points for an important role of fatty acids in the genesis of the mitochondrial dysfunction associated with obesity and type 2 diabetes mellitus. In this sense, lipid infusion or administration of high-fat diet to health human and rodents were associated with impaired mitochondrial function characterized by a reduction in ATP synthesis, oxygen consumption and oxidative phosphorylation [16, 75, 138, 139]. These findings were corroborated by in vitro studies in which treatment of cultured skeletal muscle cells with palmitic acid increased ROS production, impaired fatty acid oxidation and decreased PGC-1 expression [103, 104, 140, 141]. Studies from our group demonstrated that saturated fatty acids directly induces mitochondrial dysfunction in C2C12 skeletal muscle cells, as evidenced by reduced ATP synthesis and mitochondrial polarization [5].
Mitochondrial dysfunction plays a central role in the fatty acid-induced insulin resistance
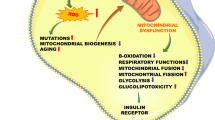
As discussed above, several mechanisms have been proposed to explain the insulin resistance induced by saturated fatty acids. All these mechanisms operate in coordinated, integrated manner linking fatty acids availability to skeletal muscle insulin resistance. To account for this multifactorial characteristic of saturated fatty acid actions, we propose herein an integrative model centered on mitochondrial dysfunction as an important factor in the genesis of insulin resistance induced by fatty acids (Figure 1).

Role of mitochondria in the insulin resistance induced by free fatty acids (FFA). In the normal condition (A), mitochondrial function is normal and FFA are rapidly metabolized with low reactive oxygen species (ROS) production and without accumulation of lipid metabolites; normal insulin response is preserved in this condition. In pathological condition (B), the excess of plasma FFA levels induces high FFA uptake into the cell, modulating negatively the expression of genes related to mitochondrial biogenesis and oxidative capacity, and into the mitochondrion, increasing the electron flux through to electron transport chain and, consequently, ROS and RNS production. As a result, mitochondrial biogenesis and function are impaired, decreasing mitochondrial mass and oxidative capacity, leading to abnormal intracellular accumulation of lipid metabolites and ROS and RNS, which activate some protein kinases involved in the phosphorylation of IRS-1 on threonine and serine residues. When phosphorylated in threonine and serine residues, IRS-1 is not phosphorylated on tyrosine residues, preventing activation of downstream signalling pathways by insulin. In addition, RNS increases IRS-1 nitrosilation, resulting in high degradation of these protein, which can contribute to impaired insulin response
In physiological conditions, fatty acids are normally and rapidly oxidized with low ROS production, little intracellular lipid accumulation and preservation of insulin sensitivity (Figure 1A). In pathological conditions, chronic elevation in circulating fatty acid levels reduces the expression of genes involved in mitochondrial biogenesis oxidative capacity and increase production of ROS, impairing mitochondrial biogenesis and function (Figure 1B). As a consequence, oxidative capacity is impaired and mitochondrial mass is reduced, increasing still further ROS production, leading to accumulation of fatty acid-derived metabolites such as diacylglycerol and ceramides.
ROS and lipid metabolites have been positively associated with insulin resistance and activation of several kinases, such as NFκB, p38 MAP kinase, JNK, and some novel and atypical PKC isoforms, as PKC-ζ and -ε, in skeletal muscle [27, 36, 50, 141–144]. These kinases impair the insulin signaling pathway by inducing serine/threonine phosphorylation in IRS-1. Under this condition, insulin-stimulated tyrosine phosphorylation of IRS-1 is inhibited, impairing activation of downstream signaling pathways and decreasing glucose uptake and metabolism in response to the hormone [5, 6, 145, 146] (Figure 2).

Mechanisms underlying insulin resistance by impaired mitochondrial function.
Concluding remarks
We discussed herein the mechanisms involved in insulin resistance in skeletal muscle cells induced by high availability of FFA. Several mechanisms have been proposed, such as Randle cycle, inhibition of insulin signaling pathway, regulation of gene expression and enzymatic activities, increase in ROS and RNS production, and impairment in mitochondrial function. Although numerous studies have been performed in order to investigate each one of these mechanisms, the conclusive proposition has not been defined yet. Evidence suggests that these mechanisms are not exclusive and there are several data in the literature pointing out that more than one mechanism is involved in the insulin resistance induced by FFA. We proposed herein a unifying hypothesis that places the importance of mitochondria in the establishment of FFA-induced insulin resistance.
References
Sakamoto K, Holman GD: Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am J Physiol Endocrinol Metab. 2008, 295: 29-37.
Hirabara SM, Silveira LR, Abdulkader F, Carvalho CR, Procopio J, Curi R: Time-dependent effects of fatty acids on skeletal muscle metabolism. Cell Physiol. 2007, 210: 7-15.
Silveira LR, Fiamoncini J, Hirabara SM, Procópio J, Cambiaghi TD, Pinheiro CH, Lopes LR, Curi R: Updating the effects of fatty acids on skeletal muscle. J Cell Physiol. 2008, 217: 1-12.
Ragheb R, Shanab GM, Medhat AM, Seoudi DM, Adeli K, Fantus IG: Free fatty acid-induced muscle insulin resistance and glucose uptake dysfunction: evidence for PKC activation and oxidative stress-activated signaling pathways. Biochem Biophys Res Commun. 2009, 389: 211-216.
Hirabara SM, Curi R, Maechler P: Saturated fatty acid-induced insulin resistance is associated with mitochondrial dysfunction in skeletal muscle cells. J Cell Physiol. 2010, 222: 187-194.
Shulman GI: Cellular mechanisms of insulin resistance. J Clin Invest. 2000, 106: 171-176.
Roden M: How free fatty acids inhibit glucose utilization in human skeletal muscle. News Physiol Sci. 2004, 19: 92-96.
Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci N, Lee D, Goodyear LJ, Kraegen EW, White MF, Shulman GI: Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes. 1999, 48: 1270-1274.
Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, Atcheson B, White MF, Kraegen EW, Shulman GI: Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002, 277: 50230-50236.
Yuzefovych L, Wilson G, Rachek L: Different effects of oleate vs. palmitate on mitochondrial function, apoptosis, and insulin signaling in L6 skeletal muscle cells: role of oxidative stress. Am J Physiol Endocrinol Metab. 2010, 299: 1096-1105.
Hirabara SM, Carvalho CRO, Mendonça JR, Piltcher Haber E, Fernandes LC, Curi R: Palmitate acutely raises glycogen synthesis in rat soleus muscle by a mechanism that requires its metabolization (Randle cycle). FEBS Lett. 2003, 541: 109-114.
Hirabara SM, Silveira LR, Alberici LC, Leandro CV, Lambertucci RH, Polimeno GC, Cury Boaventura MF, Procopio J, Vercesi AE, Curi R: Acute effect of fatty acids on metabolism and mitochondrial coupling in skeletal muscle. Biochim Biophys Acta. 2006, 1757: 57-66.
Randle PJ, Garland PB, Hales CN, Newsholme EA: The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963, 1: 785-789.
Roden M, Price TB, Perseghin G, Petersen KF, Rothman DL, Cline GW, Shulman GI: Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest. 1996, 97: 2859-2865.
Carvalho-Filho MA, Ueno M, Hirabara SM, Seabra AB, Carvalheira JB, de Oliveira MG, Velloso LA, Curi R, Saad MJ: S-nitrosation of the insulin receptor, insulin receptor substrate 1, and protein kinase B/Akt: a novel mechanism of insulin resistance. Diabetes. 2005, 54: 959-967.
Brehm A, Krssak M, Schmid AI, Nowotny P, Waldhäusl W, Roden M: Increased lipid availability impairs insulin-stimulated ATP synthesis in human skeletal muscle. Diabetes. 2006, 55: 136-140.
Randle PJ: Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab Rev. 1998, 14: 263-283.
Dresner A, Laurent D, Marcucci M, Griffin ME, Dufour S, Cline GW, Slezak LA, Andersen DK, Hundal RS, Rothman DL, Petersen KF, Shulman GI: Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest. 1999, 103: 253-259.
Jenkins AB, Storlien LH, Chisholm DJ, Kraegen EW: Effects of nonesterified fatty acid availability on tissue-specific glucose utilization in rats in vivo. J Clin Invest. 1988, 82: 293-299.
Johnson AB, Argyraki M, Thow JC, Cooper BG, Fulcher G, Taylor R: Effect of increased free fatty acid supply on glucose metabolism and skeletal muscle glycogen synthase activity in normal man. Clin Sci. 1992, 82: 219-226.
Boden G, Chen X: Effects of fat on glucose uptake and utilization in patients with non-insulin-dependent diabetes. J Clin Invest. 1995, 96: 1261-1268.
Pu J, Peng G, Li L, Na H, Liu Y, Liu P: Palmitic acid acutely stimulates glucose uptake via activation of Akt and ERK1/2 in skeletal muscle cells. J Lipid Res. 2011, 52: 1319-1327.
Rothman DL, Shulman RG, Shulman GI: 31P nuclear magnetic resonance measurements of muscle glucose-6-phosphate. Evidence for reduced insulin-dependent muscle glucose transport or phosphorylation activity in non-insulin-dependent diabetes mellitus. J Clin Invest. 1992, 89: 1069-1075.
Hawley JA, Burke LM, Angus DJ, Fallon KE, Martin DT, Febbraio MA: Effect of altering substrate availability on metabolism and performance during intense exercise. Br J Nutr. 2000, 84: 829-838.
Savage DB, Petersen KF, Shulman GI: Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol Rev. 2007, 87: 507-520.
Kim JY, Hickner RC, Cortright RL, Dohm GL, Houmard JA: Lipid oxidation is reduced in obese human skeletal muscle. Am J Physiol Endocrinol Metab. 2000, 279: 1039-1044.
Dey D, Mukherjee M, Basu D, Datta M, Roy SS, Bandyopadhyay A, Bhattacharya S: Inhibition of insulin receptor gene expression and insulin signaling by fatty acid: interplay of PKC isoforms therein. Cell Physiol Biochem. 2005, 16: 217-228.
Storz P, Döppler H, Wernig A, Pfizenmaier K, Müller G: Cross-talk mechanisms in the development of insulin resistance of skeletal muscle cells palmitate rather than tumour necrosis factor inhibits insulin-dependent protein kinase B (PKB)/Akt stimulation and glucose uptake. Eur J Biochem. 1999, 266: 17-25.
Thompson AL, Lim-Fraser MY, Kraegen EW, Cooney GJ: Effects of individual fatty acids on glucose uptake and glycogen synthesis in soleus muscle in vitro. Am J Physiol Endocrinol Metab. 2000, 279: 577-584.
Cazzolli R, Carpenter L, Biden TJ, Schmitz-Peiffer C: A role for protein phosphatase 2A-like activity, but not atypical protein kinase Czeta, in the inhibition of protein kinase B/Akt and glycogen synthesis by palmitate. Diabetes. 2001, 50: 2210-2218.
Hajduch E, Balendran A, Batty IH, Litherland GJ, Blair AS, Downes CP, Hundal HS: Ceramide impairs the insulin-dependent membrane recruitment of protein kinase B leading to a loss in downstream signalling in L6 skeletal muscle cells. Diabetologia. 2001, 44: 173-183.
Schmitz-Peiffer C: Signalling aspects of insulin resistance in skeletal muscle: mechanisms induced by lipid oversupply. Cell Signal. 2000, 12: 583-594.
Soltys CL, Buchholz L, Gandhi M, Clanachan AS, Walsh K, Dyck JR: Phosphorylation of cardiac protein kinase B is regulated by palmitate. Am J Physiol Heart Circ Physiol. 2002, 283: 1056-1064.
Paz K, Hemi R, LeRoith D, Karasik A, Elhanany E, Kanety H, Zick Y: A molecular basis for insulin resistance. Elevated serine/threonine phosphorylation of IRS-1 and IRS-2 inhibits their binding to the juxtamembrane region of the insulin receptor and impairs their ability to undergo insulin-induced tyrosine phosphorylation. J Biol Chem. 1997, 272: 29911-29918.
Zhande R, Mitchell JJ, Wu J, Sun XJ: Molecular mechanism of insulin-induced degradation of insulin receptor substrate 1. Mol Cell Biol. 2002, 22: 1016-1026.
Tirosh A, Potashnik R, Bashan N, Rudich A: Oxidative stress disrupts insulin-induced cellular redistribution of insulin receptor substrate-1 and phosphatidylinositol 3-kinase in 3T3-L1 adipocytes. A putative cellular mechanism for impaired protein kinase B activation and GLUT4 translocation. J Biol Chem. 1999, 274: 10595-10602.
Bloch-Damti A, Bashan N: Proposed mechanisms for the induction of insulin resistance by oxidative stress. Antioxid Redox Signal. 2005, 7: 1553-1567.
Evans JL, Maddux BA, Goldfine ID: The molecular basis for oxidative stress-induced insulin resistance. Antioxid Redox Signal. 2005, 7: 1040-1052.
Khamzina L, Veilleux A, Bergeron S, Marette A: Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology. 2005, 146: 1473-1481.
Rivas DA, Yaspelkis BB, Hawley JA, Lessard SJ: Lipid-induced mTOR activation in rat skeletal muscle reversed by exercise and 5'-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside. J Endocrinol. 2009, 202: 441-451.
Consitt LA, Bell JA, Houmard JA: Intramuscular lipid metabolism, insulin action, and obesity. IUBMB Life. 2009, 61: 47-55.
Samuel VT: Fructose induced lipogenesis: from sugar to fat to insulin resistance. Trends Endocrinol Metab. 2011, 22: 60-65.
Cooney GJ, Thompson AL, Furler SM, Ye J, Kraegen EW: Muscle long-chain acyl CoA esters and insulin resistance. Ann N Y Acad Sci. 2002, 967: 196-207.
Itani SI, Ruderman NB, Schmieder F, Boden G: Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002, 51: 2005-2011.
Summers SA: Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. 2006, 45: 42-72.
Nagle CA, Klett EL, Coleman RA: Hepatic triacylglycerol accumulation and insulin resistance. J Lipid Res. 2009, 50: S74-S79.
Choi JS, Koh IU, Jung MH, Song J: Effects of three different conjugated linoleic acid preparations on insulin signalling, fat oxidation and mitochondrial function in rats fed a high-fat diet. Br J Nutr. 2007, 98: 264-275.
Wang H, Knaub LA, Jensen DR, Young Jung D, Hong EG, Ko HJ, Coates AM, Goldberg IJ, de la Houssaye BA, Janssen RC, McCurdy CE, Rahman SM, Soo Choi C, Shulman GI, Kim JK, Friedman JE, Eckel RH: Skeletal muscle-specific deletion of lipoprotein lipase enhances insulin signaling in skeletal muscle but causes insulin resistance in liver and other tissues. Diabetes. 2009, 58: 116-124.
Liu L, Yu S, Khan RS, Ables GP, Bharadwaj KG, Hu Y, Huggins LA, Eriksson JW, Buckett LK, Turnbull AV, Ginsberg HN, Blaner WS, Huang LS, Goldberg IJ: DGAT1 deficiency decreases PPAR expression and does not lead to lipotoxicity in cardiac and skeletal muscle. J Lipid Res. 2011, 52: 732-744.
Schmitz-Peiffer C, Browne CL, Oakes ND, Watkinson A, Chisholm DJ, Kraegen EW, Biden TJ: Alterations in the expression and cellular localization of protein kinase C isozymes epsilon and theta are associated with insulin resistance in skeletal muscle of the highfat-fed rat. Diabetes. 1997, 46: 169-178.
Samuel VT, Petersen KF, Shulman GI: Lipid-induced insulin resistance: unravelling the mechanism. Lancet. 2010, 375: 2267-2277.
Chavez JA, Holland WL, Bär J, Sandhoff K, Summers SA: Acid ceramidase overexpression prevents the inhibitory effects of saturated fatty acids on insulin signaling. J Biol Chem. 2005, 280: 20148-20153.
Hu W, Bielawski J, Samad F, Merrill AH, Cowart LA: Palmitate increases sphingosine-1-phosphate in C2C12 myotubes via upregulation of sphingosine kinase message and activity. J Lipid Res. 2009, 50: 1852-1862.
Chavez JA, Summers SA: Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3-L1 adipocytes and C2C12 myotubes. Arch Biochem Biophys. 2003, 419: 101-109.
Summers SA: Sphingolipids and insulin resistance: the five Ws. Curr Opin Lipidol. 2010, 21: 128-135.
Holland WL, Summers SA: Sphingolipids, insulin resistance, and metabolic disease: new insights from in vivo manipulation of sphingolipid metabolism. Endocr Rev. 2008, 29: 381-402.
Lipina C, Hundal HS: Sphingolipids: agents provocateurs in the pathogenesis of insulin resistance. Diabetologia. 2011, 54: 1596-1607.
Han MS, Lim YM, Quan W, Kim JR, Chung KW, Kang M, Kim S, Park SY, Han JS, Park SY, Cheon HG, Dal Rhee S, Park TS, Lee MS: Lysophosphatidylcholine as an effector of fatty acid-induced insulin resistance. J Lipid Res. 2011, 52: 1234-1246.
Hotamisligil GS: Inflammation and metabolic disorders. Nature. 2006, 444: 860-867.
Håversen L, Danielsson KN, Fogelstrand L, Wiklund O: Induction of proinflammatory cytokines by long-chain saturated fatty acids in human macrophages. Atherosclerosis. 2009, 202: 382-393.
Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP: Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011, 12: 408-415.
Hotamisligil GS, Shargill NS, Spiegelman BM: Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993, 259: 87-91.
Hotamisligil GS: Inflammation and endoplasmic reticulum stress in obesity and diabetes. Int J Obes (Lond). 2008, 32: 52-54.
Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, Prada PO, Hirabara SM, Schenka AA, Araújo EP, Vassallo J, Curi R, Velloso LA, Saad MJ: Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007, 56: 1986-1998.
Hirosumi J, Tuncman G, Chang L, Görgün CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS: A central role for JNK in obesity and insulin resistance. Nature. 2002, 420: 333-336.
Kaneto H, Nakatani Y, Kawamori D, Miyatsuka T, Matsuoka TA, Matsuhisa M, Yamasaki Y: Role of oxidative stress, endoplasmic reticulum stress, and c-Jun N-terminal kinase in pancreatic beta-cell dysfunction and insulin resistance. Int J Biochem Cell Biol. 2006, 38: 782-793.
Solinas G, Naugler W, Galimi F, Lee MS, Karin M: Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc Natl Acad Sci USA. 2006, 103: 16454-16459.
Coletta DK, Mandarino LJ: Mitochondrial dysfunction and insulin resistance from the outside in: extracellular matrix, the cytoskeleton, and mitochondria. Am J Physiol Endocrinol Metab. 2011, 301: 749-755.
Bunn RC, Cockrell GE, Ou Y, Thrailkill KM, Lumpkin CK, Fowlkes JL: Palmitate and insulin synergistically induce IL-6 expression in human monocytes. Cardiovasc Diabetol. 2010, 9: 73-
Ji C, Chen X, Gao C, Jiao L, Wang J, Xu G, Fu H, Guo X, Zhao Y: IL-6 induces lipolysis and mitochondrial dysfunction, but does not affect insulin-mediated glucose transport in 3T3-L1 adipocytes. J Bioenerg Biomembr. 2011, 43: 367-375.
Yasuhara R, Miyamoto Y, Akaike T, Akuta T, Nakamura M, Takami M, Morimura N, Yasu K, Kamijo R: Interleukin-1beta induces death in chondrocyte-like ATDC5 cells through mitochondrial dysfunction and energy depletion in a reactive nitrogen and oxygen species-dependent manner. Biochem J. 2005, 389: 315-323.
Schmid GM, Converset V, Walter N, Sennitt MV, Leung KY, Byers H, Ward M, Hochstrasser DF, Cawthorne MA, Sanchez JC: Effect of high-fat diet on the expression of proteins in muscle, adipose tissues, and liver of C57BL/6 mice. Proteomics. 2004, 4: 2270-2282.
Xu J, Han J, Epstein PN, Liu YQ: Regulation of PDK mRNA by high fatty acid and glucose in pancreatic islets. Biochem Biophys Res Commun. 2006, 344: 827-833.
López IP, Milagro FI, Martí A, Moreno-Aliaga MJ, Martínez JA, De Miguel C: Gene expression changes in rat white adipose tissue after a high-fat diet determined by differential display. Biochem Biophys Res Commun. 2004, 318: 234-239.
Sparks LM, Xie H, Koza RA, Mynatt R, Hulver MW, Bray GA, Smith SR: A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes. 2005, 54: 1926-1933.
Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI: Impaired mitochondrial activity in the insulin-resistant offspring of patientswith type 2 diabetes. N Engl J Med. 2004, 350: 664-671.
Scarpulla RC: Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008, 88: 611-638.
Evans RM, Barish GD, Wang YX: PPARs and the complex journey to obesity. Nat Med. 2004, 10: 355-361.
Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM: A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998, 92: 829-839.
Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM: Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999, 98: 115-124.
Heilbronn LK, Gan SK, Turner N, Campbell LV, Chisholm DJ: Markers of mitochondrial biogenesis and metabolism are lower in overweight and obese insulin-resistant subjects. J Clin Endocrinol Metab. 2007, 92: 1467-1473.
Simoneau JA, Kelley DE: Altered glycolytic and oxidative capacities of skeletal muscle contribute to insulin resistance in NIDDM. J Appl Physiol. 1997, 83: 166-171.
Hulver MW, Berggren JR, Cortright RN, Dudek RW, Thompson RP, Pories WJ, MacDonald KG, Cline GW, Shulman GI, Dohm GL, Houmard JA: Skeletal muscle lipid metabolism with obesity. Am J Physiol Endocrinol Metab. 2003, 284: 741-747.
Abdul-Ghani MA, Muller FL, Liu Y, Chavez AO, Balas B, Zuo P, Chang Z, Tripathy D, Jani R, Molina-Carrion M, Monroy A, Folli F, Van Remmen H, DeFronzo RA: Deleterious action of FA metabolites on ATP synthesis: possible link between lipotoxicity, mitochondrial dysfunction, and insulin resistance. Am J Physiol Endocrinol Metab. 2008, 295: 678-685.
Bonnard C, Durand A, Peyrol S, Chanseaume E, Chauvin MA, Morio B, Vidal H, Rieusset J: Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. 2008, 118: 789-800.
Kumashiro N, Tamura Y, Uchida T, Ogihara T, Fujitani Y, Hirose T, Mochizuki H, Kawamori R, Watada H: Impact of oxidative stress and peroxisome proliferator-activated receptor gamma coactivator-1alpha in hepatic insulin resistance. Diabetes. 2008, 57: 2083-2091.
Rains JL, Jain SK: Oxidative stress, insulin signaling, and diabetes. Free Radic Biol Med. 2011, 50: 567-575.
Bashan N, Kovsan J, Kachko I, Ovadia H, Rudich A: Positive and negative regulation of insulin signaling by reactive oxygen and nitrogen species. Physiol Rev. 2009, 89: 27-71.
Gomez-Mendikute A, Cajaraville MP: Comparative effects of cadmium, copper, paraquat and benzo[a]pyrene on the actin cytoskeleton and production of reactive oxygen species (ROS) in mussel haemocytes. Toxicol Vitr. 2003, 17: 539-546.
Haynes CM, Titus EA, Cooper AA: Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Molecular cell. 2004, 15: 767-776.
He YY, Hader DP: UV-B-induced formation of reactive oxygen species and oxidative damage of the cyanobacterium Anabaena sp.: protective effects of ascorbic acid and N-acetyl-L-cysteine. J Photochem Photobiol B Biol. 2002, 66: 115-124.
Gutterman DD, Miura H, Liu Y: Redox modulation of vascular tone: focus of potassium channel mechanisms of dilation. Arterioscler Thromb Vasc Biol. 2005, 25: 671-678.
Taddei ML, Parri M, Mello T, Catalano A, Levine AD, Raugei G, Ramponi G, Chiarugi P: Integrin-mediated cell adhesion and spreading engage different sources of reactive oxygen species. Antioxid redox Signaling. 2007, 9: 469-481.
Lee HC, Wei YH: Oxidative stress, mitochondrial DNA mutation, and apoptosis in aging. Exp Biol Med. 2007, 232: 592-606.
Madamanchi NR, Vendrov A, Runge MS: Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol. 2005, 25: 29-38.
Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, Shatzkes N, Neschen S, White MF, Bilz S, Sono S, Pypaert M, Shulman GI: Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest. 2005, 115: 3587-3593.
Duval C, Cámara Y, Hondares E, Sibille B, Villarroya F: Overexpression of mitochondrial uncoupling protein-3 does not decrease production of the reactive oxygen species, elevated by palmitate in skeletal muscle cells. FEBS Lett. 2007, 581: 955-961.
Lambertucci RH, Hirabara SM, Silveira L, dos R, Levada-Pires AC, Curi R, Pithon-Curi TC: Palmitate increases superoxide production through mitochondrial electron transport chain and NADPH oxidase activity in skeletal muscle cells. J Cell Physiol. 2008, 216: 796-804.
Pilon G, Charbonneau A, White PJ, Dallaire P, Perreault M, Kapur S, Marette A: Endotoxin mediated-iNOS induction causes insulin resistance via ONOO- induced tyrosine nitration of IRS-1 in skeletal muscle. PLoS One. 2010, 5: 15912-
Perreault M, Marette A: Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat Med. 2001, 7: 1138-1143.
Dallaire P, Bellmann K, Laplante M, Gélinas S, Centeno-Baez C, Penfornis P, Peyot ML, Latour MG, Lamontagne J, Trujillo ME, Scherer PE, Prentki M, Deshaies Y, Marette A: Obese mice lacking inducible nitric oxide synthase are sensitized to the metabolic actions of peroxisome proliferator-activated receptor-gamma agonism. Diabetes. 2008, 57: 1999-2011.
Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H: High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000, 49: 1939-1945.
Desco MC, Asensi M, Marquez R, Martinez-Valls J, Vento M, Pallardo FV, Sastre J, Vina J: Xanthine oxidase is involved in free radical production in type 1 diabetes: protection by allopurinol. Diabetes. 2002, 51: 1118-1124.
Erdei N, Toth A, Pasztor ET, Papp Z, Edes I, Koller A, Bagi Z: High-fat diet-induced reduction in nitric oxide-dependent arteriolar dilation in rats: role of xanthine oxidase-derived superoxide anion. Am J Physiol Heart Circulatory Physiol. 2006, 291: H2107-H2115.
Ogihara T, Asano T, Katagiri H, Sakoda H, Anai M, Shojima N, Ono H, Fujishiro M, Kushiyama A, Fukushima Y, Kikuchi M, Noguchi N, Aburatani H, Gotoh Y, Komuro I, Fujita T: Oxidative stress induces insulin resistance by activating the nuclear factor-kappa B pathway and disrupting normal subcellular distribution of phosphatidylinositol 3-kinase. Diabetologia. 2004, 47: 794-805.
Blouet C, Mariotti F, Mikogami T, Tome D, Huneau JF: Meal cysteine improves postprandial glucose control in rats fed a high-sucrose meal. J Nutr Biochem. 2007, 18: 519-524.
Maddux BA, Goldfine ID: Membrane glycoprotein PC-1 inhibition of insulin receptor function occurs via direct interaction with the receptor alpha-subunit. Diabetes. 2000, 49: 13-19.
Haber CA, Lam TK, Yu Z, Gupta N, Goh T, Bogdanovic E, Giacca A, Fantus IG: N-acetylcysteine and taurine prevent hyperglycemia-induced insulin resistance in vivo: possible role of oxidative stress. Am J Physiol Endocrinol Metab. 2003, 285: 744-753.
Zhai L, Ballinger SW, Messina JL: Role of reactive oxygen species in injury-induced insulin resistance. Mol Endocrinol. 2011, 25: 492-502.
Maddux BA, See W, Lawrence JC, Goldfine AL, Goldfine ID, Evans JL: Protection against oxidative stress-induced insulin resistance in rat L6 muscle cells by mircomolar concentrations of alpha-lipoic acid. Diabetes. 2001, 50: 404-410.
Schrauwen P, Hesselink MK: Oxidative capacity, lipotoxicity, and mitochondrial damage in type 2 diabetes. Diabetes. 2004, 53: 1412-1417.
Indo HP, Davidson M, Yen HC, Suenaga S, Tomita K, Nishii T, Higuchi M, Koga Y, Ozawa T, Majima HJ: Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion. 2007, 7: 106-118.
Lowell BB, Shulman GI: Mitochondrial dysfunction and type 2 diabetes. Science. 2005, 307: 384-387.
Rousset S, Alves-Guerra MC, Mozo J, Miroux B, Cassard-Doulcier AM, Bouillaud F, Ricquier D: The biology of mitochondrial uncoupling proteins. Diabetes. 2004, 53: 130-135.
Fisler JS, Warden CH: Uncoupling proteins, dietary fat and the metabolic syndrome. Nutr Metab (Lond). 2006, 3: 38-
Joseph JW, Koshkin V, Zhang CY, Wang J, Lowell BB, Chan CB, Wheeler MB: Uncoupling protein 2 knockout mice have enhanced insulin secretory capacity after a high-fat diet. Diabetes. 2002, 51: 3211-3219.
Hesselink MK, Greenhaff PL, Constantin-Teodosiu D, Hultman E, Saris WH, Nieuwlaat R, Schaart G, Kornips E, Schrauwen P: Increased uncoupling protein 3 content does not affect mitochondrial function in human skeletal muscle in vivo. J Clin Invest. 2003, 111: 479-486.
Weigle DS, Selfridge LE, Schwartz MW, Seeley RJ, Cummings DE, Havel PJ, Kuijper JL, BeltrandelRio H: Elevated free fatty acids induce uncoupling protein 3 expression in muscle: a potential explanation for the effect of fasting. Diabetes. 1998, 47: 298-302.
Millet L, Vidal H, Andreelli F, Larrouy D, Riou JP, Ricquier D, Laville M, Langin D: Increased uncoupling protein-2 and -3 mRNA expression during fasting in obese and lean humans. J Clin Invest. 1997, 100: 2665-2670.
Schrauwen P, Hinderling V, Hesselink MK, Schaart G, Kornips E, Saris WH, Westerterp-Plantenga M, Langhans W: Etomoxir-induced increase in UCP3 supports a role of uncoupling protein 3 as a mitochondrial fatty acid anion exporter. FASEB J. 2002, 16: 1688-1690.
Samec S, Seydoux J, Dulloo AG: Role of UCP homologues in skeletal muscles and brown adipose tissue: mediators of thermogenesis or regulators of lipids as fuel substrate?. FASEB J. 1998, 12: 715-724.
Hoeks J, Hesselink MK, Sluiter W, Schaart G, Willems J, Morrisson A, Clapham JC, Saris WH, Schrauwen P: The effect of high-fat feeding on intramuscular lipid and lipid peroxidation levels in UCP3-ablated mice. FEBS Lett. 2006, 580: 1371-1375.
Russell AP, Gastaldi G, Bobbioni-Harsch E, Arboit P, Gobelet C, Dériaz O, Golay A, Witztum JL, Giacobino JP: Lipid peroxidation in skeletal muscle of obese as compared to endurance-trained humans: a case of good vs. bad lipids?. FEBS Lett. 2003, 551: 104-106.
Schrauwen P, Troost FJ, Xia J, Ravussin E, Saris WH: Skeletal muscle UCP2 and UCP3 expression in trained and untrained male subjects. Int J Obes Relat Metab Disord. 1999, 23: 966-972.
Mingrone G, Rosa G, Greco AV, Manco M, Vega N, Hesselink MK, Castagneto M, Schrauwen P, Vidal H: Decreased uncoupling protein expression and intramyocytic triglyceride depletion in formerly obese subjects. Obes Res. 2003, 11: 632-640.
Hoeks J, van Baak MA, Hesselink MK, Hul GB, Vidal H, Saris WH, Schrauwen P: Effect of beta1- and beta2-adrenergic stimulation on energy expenditure, substrate oxidation, and UCP3 expression in humans. Am J Physiol Endocrinol Metab. 2003, 285: E775-E782.
Samec S, Seydoux J, Dulloo AG: Interorgan signaling between adipose tissue metabolism and skeletal muscle uncoupling protein homologs: is there a role for circulating free fatty acids?. Diabetes. 1998, 47: 1693-1698.
Echtay KS, Esteves TC, Pakay JL, Jekabsons MB, Lambert AJ, Portero-Otín M, Pamplona R, Vidal-Puig AJ, Wang S, Roebuck SJ, Brand MD: A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J. 2003, 22: 4103-4110.
Nabben M, Hoeks J, Briedé JJ, Glatz JF, Moonen-Kornips E, Hesselink MK, Schrauwen P: The effect of UCP3 overexpression on mitochondrial ROS production in skeletal muscle of young versus aged mice. FEBS Lett. 2008, 582: 4147-4152.
Schrauwen P, Schrauwen-Hinderling V, Hoeks J, Hesselink MK: Mitochondrial dysfunction and lipotoxicity. Biochim Biophys Acta. 2010, 1801: 266-271.
Holloway GP, Thrush AB, Heigenhauser GJ, Tandon NN, Dyck DJ, Bonen A, Spriet LL: Skeletal muscle mitochondrial FAT/CD36 content and palmitate oxidation are not decreased in obese women. Am J Physiol Endocrinol Metab. 2007, 292: 1782-1789.
Schrauwen-Hinderling VB, Kooi ME, Hesselink MK, Jeneson JA, Backes WH, van Echteld CJ, van Engelshoven JM, Mensink M, Schrauwen P: Impaired in vivo mitochondrial function but similar intramyocellular lipid content in patients with type 2 diabetes mellitus and BMI-matched control subjects. Diabetologia. 2007, 50: 113-120.
Ukropcova B, McNeil M, Sereda O, de Jonge L, Xie H, Bray GA, Smith SR: Dynamic changes in fat oxidation in human primary myocytes mirror metabolic characteristics of the donor. J Clin Invest. 2005, 115: 1934-1941.
Lim S, Park KS, Kim MS, Cho BY, Lee HK: Relationship between various surrogate indices of insulin resistance and mitochondrial DNA content in the peripheral blood of 18 healthy volunteers. Mitochondrion. 2001, 1: 71-77.
Guo LJ, Oshida Y, Fuku N, Takeyasu T, Fujita Y, Kurata M, Sato Y, Ito M, Tanaka M: Mitochondrial genome polymorphisms associated with type-2 diabetes or obesity. Mitochondrion. 2005, 5: 15-33.
Juo SHH, Lu MY, Bai RK, Liao YC, Trieu RB, Yu ML, Wong LJC: A common mitochondrial polymorphism 10398A > G is associated metabolic syndrome in a Chinese population. Mitochondrion. 2010, 10: 294-299.
Wallace DC, Fan W: Energetics, epigenetics, mitochondrial genetics. Mitochondrion. 2010, 10: 12-31.
Chanseaume E, Malpuech-Brugère C, Patrac V, Bielicki G, Rousset P, Couturier K, Salles J, Renou JP, Boirie Y, Morio B: Diets high in sugar, fat, and energy induce muscle typespecific adaptations in mitochondrial functions in rats. J Nutr. 2006, 136: 2194-2200.
Szendroedi J, Schmid AI, Meyerspeer M, Cervin C, Kacerovsky M, Smekal G, Graser-Lang S, Groop L, Roden M: Impaired mitochondrial function and insulin resistance of skeletal muscle in mitochondrial diabetes. Diabetes Care. 2009, 32: 677-679.
Coll T, Jové M, Rodríguez-Calvo R, Eyre E, Palomer X, Sánchez RM, Merlos M, Laguna JC, Vázquez-Carrera M: Palmitate-mediated downregulation of peroxisome proliferator-activated receptorgamma coactivator 1alpha in skeletal muscle cells involves MEK1/2 and nuclear factor-kappaB activation. Diabetes. 2006, 55: 2779-2787.
Pimenta AS, Gaidhu MP, Habib S, So M, Fediuc S, Mirpourian M, Musheev M, Curi R, Ceddia RB: Prolonged exposure to palmitate impairs fatty acid oxidation despite activation of AMP-activated protein kinase in skeletal muscle cells. J Cell Physiol. 2008, 217: 478-485.
Jove M, Laguna JC, Vázquez-Carrera M: Agonist-induced activation releases peroxisome proliferator-activated receptor beta/delta from its inhibition by palmitate-induced nuclear factor-kappaB in skeletal muscle cells. Biochim Biophys Acta. 2005, 1734: 52-61.
Bloch-Damti A, Potashnik R, Gual P, Le Marchand-Brustel Y, Tanti JF, Rudich A, Bashan N: Differential effects of IRS1 phosphorylated on Ser307 or Ser632 in the induction of insulin resistance by oxidative stress. Diabetologia. 2006, 49: 2463-2473.
Watt MJ, Steinberg GR, Chen ZP, Kemp BE, Febbraio MA: Fatty acids stimulate AMP-activated protein kinase and enhance fatty acid oxidation in L6 myotubes. J Physiol. 2006, 574: 139-147.
Krebs M, Roden M: Molecular mechanisms of lipid-induced insulin resistance in muscle, liver and vasculature. Diabetes Obes Metab. 2005, 7: 621-632.
Talukdar I, Szeszel-Fedorowicz W, Salati LM: Arachidonic acid inhibits the insulin induction of glucose-6-phosphate dehydrogenase via p38 MAP kinase. J Biol Chem. 2005, 280: 40660-40667.
Acknowledgements
This study is supported by grants from FAPESP, CAPES, CNPq/National Institute of Sciences and Technology in Obesity and Diabetes, and Center of Lipid Research And Education (CLEAR).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
ARM, RTN, and RHL participated of the acquisition, analysis, and interpretation of the data from literature. RG, MAV, and WTF organized the structure of the manuscript and had substantial contribution to the conception and design. MFCB and LRS were involved in the drafting of the manuscript and figures. SMH and RC critically revised the text and figures for the final version of the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Martins, A.R., Nachbar, R.T., Gorjao, R. et al. Mechanisms underlying skeletal muscle insulin resistance induced by fatty acids: importance of the mitochondrial function. Lipids Health Dis 11, 30 (2012). https://doi.org/10.1186/1476-511X-11-30
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-511X-11-30