
Abstract
Background
Segmental duplications (SD) have been found in genomes of various organisms, often accumulated at the ends of chromosomes. It has been assumed that the sequence homology in-between the SDs allow for ectopic interactions that may contribute to the emergence of new genes or gene variants through recombinatorial events.
Methods
In silico analysis of the 3D7 Plasmodium falciparum genome, conducted to investigate the subtelomeric compartments, led to the identification of subtelomeric SDs. Sequence variation and copy number polymorphisms of the SDs were studied by DNA sequencing, real-time quantitative PCR (qPCR) and fluorescent in situ hybridization (FISH). The levels of transcription and the developmental expression of copy number variant genes were investigated by qPCR.
Results
A block of six genes of >10 kilobases in size, including var, rif, pfmc-2tm and three hypothetical genes (n-, o- and q-gene), was found duplicated in the subtelomeric regions of chromosomes 1, 2, 3, 6, 7, 10 and 11 (SD1). The number of SD1 per genome was found to vary from 4 to 8 copies in between different parasites. The intragenic regions of SD1 were found to be highly conserved across ten distinct fresh and long-term cultivated P. falciparum. Sequence variation was detected in a ≈ 23 amino-acid long hypervariable region of a surface-exposed loop of PFMC-2TM. A hypothetical gene within SD1, the n-gene, encoding a PEXEL/VTS-containing two-transmembrane protein was found expressed in ring stage parasites. The n-gene transcription levels were found to correlate to the number of n-gene copies. Fragments of SD1 harbouring two or three of the SD1-genes (o-gene, pfmc-2tm, q-gene) were also found in the 3D7 genome. In addition a related second SD, SD2, of ≈ 55% sequence identity to SD1 was found duplicated in a fresh clinical isolate but was only present in a single copy in 3D7 and in other P. falciparum lines or clones.
Conclusion
Plasmodium falciparum carries multiple sequence conserved SDs in the otherwise highly variable subtelomeres of its chromosomes. The uniqueness of the SDs amongst plasmodium species, and the conserved nature of the genes within, is intriguing and suggests an important role of the SD to P. falciparum.
Similar content being viewed by others

Background
Plasmodium falciparum, the causative agent of severe human malaria, carries a haploid nuclear genome of approximately 23 Mb distributed onto 14 chromosomes [1]. The subtelomeric regions of the chromosomes are adjacent to telomeric repeats and appear to be species-specific, highly polymorphic and to consist of a patchworks of repetitive blocks [2]. Many families of variable genes are located in these regions. In addition to the well-studied var [3–5], rif [6, 7] and stevor [8] genes, other multi-gene families such as etramp/sep [9, 10], surf [11], pfmc-2tm [12], phist [13] and fikk [14, 15] have recently been identified, most of which share the trait of sub-telomeric localization. These genes encode polypeptides many of which have been predicted to be involved in immune-evasion [16].
Syntenic comparisons of the Plasmodium chabaudi, Plasmodium berghei, and Plasmodium yoelii genomes with that of P. falciparum have revealed a striking conservation within the central cores of the chromosomes, whilst the subtelomeric regions display vast genetic variation [17, 18]. For example, about 78% of the orphan genes of the P. falciparum genome with no detectable orthologs in the rodent parasites are located subtelomerically [18], indicating that most of the genetic variability is derived in this compartment. Therefore, genomic and genetic analyses of the subtelomeric content might provide information of the evolutionary forces driving speciation and factors contributing to biological variation among malaria parasites.
The plasticity and dynamic nature of the subtelomeres allow genes located in these regions to evolve more rapidly than their centromeric counterparts. Functional roles of telomeres and subtelomeres in antigenic variation include reversible gene silencing mediated by telomere-protein complexes [19] and engagement in ectopic exchange with other chromosomal ends [20, 21]. The subtelomeres are polymorphic but they are also highly ordered within the end structures [22] where each subtelomere harbours five subtelomeric blocks (SB1 – 5) [17]. Recent studies have shown that the SB-3 (pRepHind or rep20) plays a role in mediating the formation and/or stabilization of telomere clusters [23, 24]. Such physical tethering of chromosomes promotes recombination between virulence genes located at the end of heterologous chromosomes [24, 25].
Genetic events such as transpositions, deletions, translocations and segmental duplications enable rapid adaptation to new environments. In genomes of primates and humans, a segmental duplication has been defined as a duplication of a DNA segment equal to or longer than 1 kb with a high level of sequence identity (> 90%) between copies transposed to new locations [26, 27]. Due to the sequence identity between duplicated sequences and the resulting potential of genetic recombination, segmental duplications have contributed to the emergence of new genes or gene variants and thereby to the total genetic variation of genomes [26–28]. Well-described examples of such genetic changes is the generation of the repertoire of olfactory receptors in humans [29, 30] and disease resistance genes in plants [31]. Further, it was recently shown that duplications and deletions in the human genome often are population specific [32].
Previous comparative genomic hybridization (CGH) approaches have demonstrated copy number polymorphisms located within internal regions of the P. falciparum chromosomes. The suitability of the same approach to detect duplications/deletions in the subtelomeres is limited due to the high degree of sequence variability genes in these compartments exhibit [33–35]. Nevertheless, a DNA segment on the right end of chromosome 1, spanning the genes PFA0685c, PFA0690w and PFA0695c, was previously suggested to be duplicated in a fresh clinical isolate [35]. Further analysis of this segment revealed covered genes being paralogous to genes within a >10 kb segmental duplication in the 3D7 subtelomeres (eight copies). In this study, the gene content, sequence polymorphism and copy number variation of the SDs have been investigated in distinct clones and lines of the parasite. In addition, transcription levels were monitored in parasites harbouring different numbers of SDs.
Materials and methods
Genome information
Sequence information, chromosomal locations and transcriptional directions of genes in the 3D7 genome were obtained from the Plasmodium Genome Resource [36]. Sequences from the Hb3 and Dd2 sequencing projects were retrieved from the Microbial Sequencing Center, Broad Institute [37]. Sequences from the Ghanaian isolate and the It/FCR3 strain were downloaded from The Plasmodium genome project, Welcome Trust Sanger Institute [38]. The coverage of Hb3, Dd2 and the Ghanaian isolate were 8.07×, 7.13× and 8× respectively, whereas the coverage of the It strain was estimated to be 3.84× by averaging the sizes of Hb3, Dd2 and Ghanaian isolate genomes and comparing the number of reads sequenced for the four strains.
Sequence reads were aligned to the n-, o-, pfmc-2tm and q-genes using BLASTN without low complexity filtering. The identity cutoff was set to 95% with a minimum accepted length of an overlap of ≥ 36 bp. The number of bps aligning to the genes was compared to the length of each gene, which yielded an estimated coverage for each gene in each of the parasite lines. This gene specific coverage was subsequently compared to the total coverage for the strains and a copy number estimate was calculated.
Graphical presentation of the subtelomeres
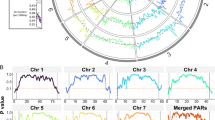
A graphical output of all genes in the subtelomeric block 4–5 for all 14 chromosomes was generated (Figure 1). The boundaries of the subtelomeric ends were defined based on the whole genome synteny mapping of P. falciparum with rodent malaria parasites (P. berghei, P. chabaudi and P. yoelii) [18]. Subtelomeric gene-families are categorized into 18 groups (Additional File 1) and are displayed in different colors. Grouping of the subtelomeric genes was based on information from literature, the OrthoMCL Database [39] and/or protein features (possession of PEXEL/VTS domain and transmembrane regions) acquired from the Plasmodium database [36] where protein domains were predicted using HMM against the Pfam database, version 17.

High-resolution display of gene families in the subtelomeric compartment of P. falciparum 3D7. Subtelomeric genes are plotted according to their chromosomal positions and color labeled. For additional information see Additional File 1. The 8 segmental duplications SD1 are located in the subtelomeres of multiple chromosomes, and are here depicted in pink shaded ellipses. A second segmental duplication on chromosome 1 named SD2 is marked with a pale blue shaded ellipse.
Nucleic acid extraction
Nucleic acids (gDNA and RNA) were extracted using either the Easy-DNA™ (Invitrogen) or the RNeasy® (Qiagen) kits according to the recommendations of the suppliers. Total RNA was isolated from 3D7AH1, FCR3 and 7G8 at 8 to 28 hours post invasion with four-hour intervals for two consecutive parasite cycles. To ensure DNA-free RNA, the isolated RNA was treated with TURBO DNA-free™ DNAse (Ambion).
PCR amplification and sequencing
Standard polymerase chain reaction (PCR) was used for the amplification of n-, o-, pfmc-2tm-, and q- genes of the SDs. Primers were designed based on the published 3D7 sequences: n-gene: forward 5'-TTT TTT TCA AGT AAG AGA TGC-3', reverse 5'-CCA CAA CCA CAC AAG AAG-3'; o-gene: forward 5'-CAA TAA ATA TAG CAA GTC G-3', reverse 5'-TAA ATC ATG TTC TGT GTG-3'; pfmc-2tm: forward 5'-ATC ATA CCA TAA TGG AGG-3', reverse 5'-ACC TAT TTT CAT GTC AGG-3' and q-gene: forward 5'-TGA AAA TAC CAA AGT ACC-3', reverse 5'-ATT GTA ATC CTT TAG CTC-3'. Amplification products were cloned into Topo vectors (TOPO TA cloning kit, Invitrogen) before transformation into TOP10 competent E.coli. DNA from at least four bacterial clones was sequenced for each target using M13 forward and reverse primers. ClustalW multiple alignments were performed thereafter, using BioEdit software version 7.0.5 (Tom hall, Ibis Therapeutics, Carlsbad, CA).
Real-time quantitative PCR
Copy numbers relative to the 3D7AH1 parasite of the n-gene, PFA0675w, PFA0685c, PFA0690w and PFA0700c were determined for FCR3, 7G8, UAM25, HB3, Dd2, TM180 and TM284. Primers specific for the n-gene (5'-AGG GCA ATT GAT TTT AGC AGG TAT-3' and 5'-CAA AAC TAC TGA ATG CTA TAA ATG AAG GA-3'), PFA0675w (5'-TAT AAG ACC AAC TCT TTT CAT TTG TCT TTA C-3' and 5'-AAA ATC CTG TTG TAT GTA CGA TTA GCA T-3'), PFA0685c (5'-AAT ATA TAA CAA GTC GAG CAC TAA CGG A-3' and 5'-TCC TCT TAT TTG TGG ATT TTT ATT TCC-3'), PFA0690w (5'-ACC AAG AGC CTT GTG AAA CGA-3' and 5'-TTT CTT CCT TCT TCA GTT TTT TTG TG-3'), PFA0700c (5'-AGG AGA TTA CTA GCC GAA CCA CAC-3' and 5'-TTT ATG GGT TTT CAA TAT ATG TGA TTT GT-3') and the endogenous control gene PF10_0084 (5'-ACA ACG AAG CAA CAG GAG GTA GAT-3' and 5'-AGT CCA TCA ATA TAG CTC TTG GAA CAT A-3') were all designed using Primer Express 2.0 (Applied Biosystems) towards perfectly conserved stretches of the genes. Approximately 1 ng of DNA was used as template in quadruplicate amplification reactions in MicroAmp 96 well plates in 20 μl containing SYBR Green master mix and 300 nm of each primer. Amplifications were carried out in an ABI sequence detector 7500 (Applied Biosystems) for 40 cycles (95°C for 15 seconds and 60°C for 1 min). PCR-efficiencies of all primer-pairs were evaluated on dilution series of 3D7AH1 genomic DNA and found to be sufficiently close to obviate the need for any correction factor. Results were analysed using the ΔΔCt method (User bulletin 2, Applied Biosystems) based on the tested assumption that the target genes are amplified with the same efficiency as the endogenous control.
Total RNA was reversibly transcribed with SuperScript III Rnase H reverse transcriptase (Invitrogen), random hexamers and oligo(dT)12–18 (300 ng/μl and 25 ng/μl respectively, both from Invitrogen) for two hours at 50°C. For each cDNA synthesis reaction, a control reaction without reverse transcriptase was performed with identical amounts of template. For qPCR-based determination of n-gene transcription the same primers were used as listed above except for the endogenous control, where seryl-tRNA synthetase was employed. The primers were: 5'-TAT CAT CTC AAC AGG TAT CTA CAT CTC CTA-3' and 5'-TTT GAG AGT TAC ATG TGG TAT CAT CTT TT-3'. The amplification reactions were conducted as described above, with the only difference that 2 ng of template was used. Transcription levels were achieved by dividing the with the for each strain and time point. The standard deviation of the quotient was calculated according to the User Bulletin 2, Applied Biosystems. Results were visualized as log2 transformed values and plotted using SigmaPlot 9.0 (Systat Software Inc.).
Fluorescent in-situ hybridization
FISH targeting the n-gene was conducted according to previously described methodology [35]. The fluorescein labeled (Fluorescein-High Prime, Roche Applied Science) n-gene probe was generated from 3D7AH1 gDNA using the primers 5'-TTT TTT TCA AGT AAG AGA TGC-3' and 5'-CCA CAA CCA CAC AAG AAG-3'.
Results
Segmental duplications in the subtelomeres
Comparative analysis of the P. falciparum genome with rodent plasmodium species has disclosed synteny breaks at the boundaries of the subtelomeric compartments [18]. Here, we have analysed the subtelomeric gene content of the 3D7 genome by grouping the genes into families as shown in Figure 1. Eight homologous regions were found, all sharing the same genomic organization being located on seven chromosomes (Chromosomes 1, 2, 3, 6, 7, 10 and 11). This duplicated DNA segment (named SD1) was found to contain six genes: rif, pfmc-2tm, a var pseudogene and three hypothetical genes (n-, o- and q-gene) (Figure 2A). The breakpoints of these segmental duplicons vary slightly, with the 5' break point being either within or downstream with respect to the rif gene and the 3' break point being either upstream or downstream of the var pseudogene. The most extended duplicated loci (approximately 32 kb in size) are both located on chromosome 6, but on opposite chromosomal ends. Although the rif genes are not identical in-between the SD1, homologous rif copies can be found within all SD1 (Figure 2B). Most of the genes within SD1 encode PEXEL-containing export proteins, with the exception of the q-gene and the var pseudogenes (Additional File 2). SD1-fragments harbouring only two or three of the SD1- genes (o-gene, pfmc-2tm, q-gene) were also found in the 3D7 genome (Additional File 1).

Gene content and organization of the segmental duplicon SD1. (A) Example of a typical SD1 containing six complete genes. Genes encoding PEXEL-containing proteins are depicted in black. A red arrow indicates the position of the hypervariable loop in pfmc2tm. (B) The SD1 exists in eight copies in the 3D7 genome with a slight variation in respect to the rif gene. Homologous rif copies, with > 95% sequence homology, in between the SD1s are shown with the same color. Crosses indicate breakpoints of each SD1.
A previous CGH project from this laboratory revealed a subtelomeric gene segment (PFA0685c, PFA0690w and PFA0695c), located on the right end of chromosome 1 in the 3D7 strain, to be duplicated in a fresh clinical isolate (UAM25) [35] (Figure 3A). Further analysis indicates that this locus shares three of the same paralogous genes as SD1s described above, with the same gene order and orientation but with less sequence homology (55% identity). This SD was named SD2. Compared to the eight SD1, SD2 was found to carry the n-gene as a pseudogene and the q-gene (PFA0675w) was found to harbour RESA-like repeats and a DNAJ domain (PFAM database: PF0026; amino acid 1097–1160), which the q-gene of SD1 does not possess. PSI-BLAST analyses of the genes in the SD2 (converged at iteration 3) showed that the q-gene has orthologous genes in P. vivax and in rodent malaria parasites (P. yoelii, P. chabaudi and P. berghei). However, no orthologous genes could be identified for the other SD2 gene-members.

Copy numbers of the SD2 in different P. falciparum strains. (A) Ratio based differences (UAM25 over 3D7AH1) of microarray oligonucleotides mapped according to the gene locations on chromosome 1 in 3D7. A black arrow indicates the SD2 found duplicated in UAM25. (B) Genetic organization of the SD2 on the right arm of chromosome 1 in 3D7. Genes found duplicated in UAM25 relative to 3D7, according to CGH data and qPCR, are shown in red. (C) Copy numbers of the SD2 genes in different strains relative to 3D7 parasite confirmed by qPCR.
Sequence variation within the segmental duplicons
To elucidate whether the sequence conservation of the SD1 remains across different P. falciparum parasites, we sequenced the n-, o-, pfmc-2tm and q-gene of five parasites originating from different geographical areas: FCR3 (The Gambia), TM180 (Thailand), 7G8 (Brazil), UAS31 and UAS39 (both from Uganda). In addition, sequence information for HB3 (Honduras) and Dd2 (Indochina) [37] and It (Brazil) [38] was retrieved for the analysis. ClustalW multiple alignments revealed that genes within the SD1s are of a high sequence identity (99%), with the exception of a ≈ 23 amino acid hypervariable loop within pfmc-2tm which is predicted to be surface-exposed [12, 40]. Polymorphisms other than those of pfmc-2tm in the eight SD1s of 3D7 were mainly situated within repetitive sequence stretches of the intra- and intergenic regions. Comparisons of sequences to single nucleotide polymorphism (SNPs) data published recently [41] (Additional File 3) revealed four novel non-synonymous SNPs in the n-gene, and four non-synonymous and two synonymous SNPs in the q-gene.
Copy number polymorphism of the segmental duplicons
Using the n-gene as a representative member of SD1, the SD1 copy number in different P. falciparum strains relative to the 3D7 parasite was estimated using qPCR. The genomes of HB3 and the clinical isolate (UAM25) were found to contain the same number of SD1 copies as 3D7 (n = 8), whereas Dd2 was found to carry ≤ 4 (Figure 4A). Comparable numbers of pfmc-2tm was previously reported for HB3 relative to 3D7 [40], signifying a copy number association between the n-gene and pfmc-2tm.

Copy number polymorphisms of the n-gene in different P. falciparum strains and isolates. (A) Copy numbers of the n-gene in different parasite lines relative to 3D7 detected by qPCR. (B) Visualization of copy numbers and localization of n-gene (green) in 3D7, FCR3 and 7G8. Distribution of fluorescent signals at the rim of the parasite nuclei (blue) confirms the position of the SD at the chromosomal ends.
The results were further confirmed by fluorescent in situ hybridizations (FISH). In addition to a clear pattern of variable copy numbers (Figure 4A) most of the signals were distributed at the rim of the parasite-nuclei where chromosomal ends are known to tether [23], confirming the subtelomeric localization of the SD1s (Figure 4B).
The amplification of SD2 was also verified by qPCR targeting the pseudo n-gene (PFA0690w), as well as the adjacent genes, PFA0675w (pseudo q-gene), PFA0685c (pseudo o-gene) and PFA0700c (Figure 3B). In contrast to SD1, the SD2 in UAM25 did not include PFA0675w (paralogous to the q-gene).
Transcriptional analysis
The intraerythrocytic developmental expression of the genes in the SDs was previously studied using microarrays ([42] : E-MEXP-128) [43, 44]. Only the n-gene was found significantly transcribed, with maximum expression in the ring stages. In addition, the pseudo n-gene (PFA0690w) of SD2 was found to be expressed, despite of its supposedly truncated ORF, with maximum transcript abundance at 36h post-invasion [44].
In order to investigate the impact of gene dosage on transcription levels, n-gene transcription was investigated for three parasites with varying numbers of SD1s. 3D7AH1, FCR3 and 7G8 parasites were harvested at 4-hour intervals from eight to 28 hours post-invasion and relative mRNA levels were studied by qPCR. The maximum level of transcription of the n-gene was found in ring-stage parasites, which coincides with previous transcription data [43, 44]. A clear transcriptional difference was observed when comparing 3D7AH1 and 7G8, which carry eight and five copies in the genome, respectively, but similar level of transcription was found for 3D7AH1 and FCR3, although the latter carries fewer copies of the n-gene (Figure 5).

Temporal and relative transcript abundance of the n-gene in 3D7, FCR3 and 7G8. The transcript levels of the n-gene, in relation to the endogenous control gene seryl-tRNA synthetase, were measured during 8–28 hours post invasion. Data was log2 transformed and plotted at four-hour intervals for each particular parasite.
Discussion
Genomic variability of P. falciparum has been shown to be of importance to the parasite and to underly the ability of local adaptation, antigenic variation and drug resistance. A substantial degree of genetic variation in between different P. falciparum strains is confined to the subtelomeric compartment [33, 34], where families of variable genes and a number of repetitive regions are located. This paper reports the presence of a >10 kb segment duplicated and translocated onto multiple chromosomal ends. These paralogous regions span at least six genes and include members from the previously identified multigene families rif, pfmc-2tm and var as well as three families of hypothetical genes (n-, o-, q-genes) present as multiple homologous copies.
Pfmc-2tm, together with rif and stevor, belong to a large topological super family based on the presence of the erythrocyte-trafficking motif PEXEL/VTS [45–47] and two predicted transmembrane regions. While rif and stevor encode >150 RIFIN-related and >35 STEVOR-related proteins respectively, pfmc-2tm possesses a lower number of paralogous members (13 in the 3D7 genome) [12, 40]. Sequences of pfmc-2tm genes are relatively conserved with differences located in a ≈ 23 amino acid region situated between the two predicted transmembrane regions. This hypervariable stretch is presumably forming a loop exposed on the infected red cell surface. Among the 13 pfmc-2tm genes in the 3D7 genome, 9 are located within, and the remaining adjacent to the SD. A possible explanation for this observation is that the pfmc-2tm gene family is expanded through segmental duplication. Subsequent modification of the duplicated genes, most likely by single nucleotide mutations within the variable loop region, may provide the diversity of this gene family. In contrast to pfmc-2tm, var gene diversity is probably generated mainly through ectopic recombination. This is facilitated by the neighbouring rep20 sequences, which mediate clustering of the telomeres and hence bring subtelomeric var genes into close proximity [21, 24, 25]. Although this study also suggests that members of the rif family might be expanded through segmental duplicative events, this still only represents a small fraction of this large gene family of more than 150 members [48].
Evolution of multigene families involves different rates of gene duplication, maintenance and loss, often accompanied by formation of pseudogenes [49]. Pseudogenes provide a record of how genomic DNA has been changed without such evolutionary pressure and can be used as a model for determining the underlying rates of nucleotide substitutions, insertions and deletions in the genome. The subtelomeric regions, due to their dynamic nature, are breeding grounds for generation of pseudogenes. In the 3D7 genome, 57 out of 73 pseudogenes belong to the three big multigene families (rif, stevor and var), of which the majority (44/57) are subtelomerically located (Figure 1, shown as). Except for SD1 on chromosome 7, all other SDs end with a var pseudogene. The sequence identity of these var pseudogenes is high, not only within the 3D7 but also across other genomes of P. falciparum parasites.
In addition to FISH and qPCR we also adopted an in silico strategy similar to the one used by Bailey et al. for locating copy number variations in the human genome [50] (see Material and Methods). Sequences from four strains (Hb3, Dd2, It and Ghanaian isolate) were downloaded and aligned to the n-, o-, pfmc-2tm and q-gene of the SDs. Different copy numbers of n-, o-, pfmc-2tm and q-gene were identified in all parasites (Additional File 4), but the copy numbers estimated with this method were lower compared to those obtained by qPCR. The most likely reason for this discrepancy is an incomplete assembly of these recently sequenced parasite genomes.
The n-gene belongs to the same PEXEL-containing two-transmembrane superfamily as rif, stevor and pfmc-2tm (Additional File 2). Previous microarray data from isogenic clones of 3D7 revealed that the n-gene is the only gene in the SD1 that is transcribed during the intraerythrocytic cycle. The transcription levels of the n-gene correlated in part with the copy number abundance (in 3D7 vs. 7G8 but not in 3D7 vs. FCR3). The gene-copy number may be one of the reasons for a relatively low level of n-gene transcription in Dd2 and high levels in HB3 as shown by Llinas et al [51]. Taken together it seems that the abundance of the n-gene affects the levels of mRNA.
The presence of four to eight copies of SD1 in all parasites studied and up to two copies of a second segmental duplication (SD2) in a freshly isolated parasite suggests that segmental duplications do occur in P. falciparum and that they are of biological importance in vivo. It may be that the SDs are part of a transposon-like system in P. falciparum but this remains to be investigated.
Conclusion
Plasmodium falciparum carries multiple SD in the subtelomeres of its chromosomes. The unique presence of the SDs in P. falciparum compared to other Plasmodium species and the conserved nature of the genes within, suggests a functional role of the SDs to P. falciparum.
Abbreviations
- FISH:
-
fluorescent in situ hybridization
- PEXEL/VTS:
-
Plasmodium export element or the vacuolar transport signal
- qPCR:
-
real-time quantitative PCR
- SD:
-
segmental duplication.
References
Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S: Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002, 419: 498-511. 10.1038/nature01097.
Mefford HC, Trask BJ: The complex structure and dynamic evolution of human subtelomeres. Nat Rev Genet. 2002, 3: 91-102. 10.1038/nrg727.
Su XZ, Heatwole VM, Wertheimer SP, Guinet F, Herrfeldt JA, Peterson DS, Ravetch JA, Wellems TE: The large diverse gene family var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum-infected erythrocytes. Cell. 1995, 82: 89-100. 10.1016/0092-8674(95)90055-1.
Baruch DI, Pasloske BL, Singh HB, Bi X, Ma XC, Feldman M, Taraschi TF, Howard RJ: Cloning the P. falciparum gene encoding PfEMP1, a malarial variant antigen and adherence receptor on the surface of parasitized human erythrocytes. Cell. 1995, 82: 77-87. 10.1016/0092-8674(95)90054-3.
Smith JD, Chitnis CE, Craig AG, Roberts DJ, Hudson-Taylor DE, Peterson DS, Pinches R, Newbold CI, Miller LH: Switches in expression of Plasmodium falciparum var genes correlate with changes in antigenic and cytoadherent phenotypes of infected erythrocytes. Cell. 1995, 82: 101-110. 10.1016/0092-8674(95)90056-X.
Kyes SA, Rowe JA, Kriek N, Newbold CI: Rifins: a second family of clonally variant proteins expressed on the surface of red cells infected with Plasmodium falciparum. Proc Natl Acad Sci USA. 1999, 96: 9333-9338. 10.1073/pnas.96.16.9333.
Fernandez V, Hommel M, Chen Q, Hagblom P, Wahlgren M: Small, clonally variant antigens expressed on the surface of the Plasmodium falciparum-infected erythrocyte are encoded by the rif gene family and are the target of human immune responses. J Exp Med. 1999, 190: 1393-1404. 10.1084/jem.190.10.1393.
Cheng Q, Cloonan N, Fischer K, Thompson J, Waine G, Lanzer M, Saul A: stevor and rif are Plasmodium falciparum multicopy gene families which potentially encode variant antigens. Mol Biochem Parasitol. 1998, 97: 161-176. 10.1016/S0166-6851(98)00144-3.
Birago C, Albanesi V, Silvestrini F, Picci L, Pizzi E, Alano P, Pace T, Ponzi M: A gene-family encoding small exported proteins is conserved across Plasmodium genus. Mol Biochem Parasitol. 2003, 126: 209-218. 10.1016/S0166-6851(02)00275-X.
Spielmann T, Fergusen DJ, Beck HP: etramps, a new Plasmodium falciparum gene family coding for developmentally regulated and highly charged membrane proteins located at the parasite-host cell interface. Mol Biol Cell. 2003, 14: 1529-1544. 10.1091/mbc.E02-04-0240.
Winter G, Kawai S, Haeggstrom M, Kaneko O, von Euler A, Kawazu S, Palm D, Fernandez V, Wahlgren M: SURFIN is a polymorphic antigen expressed on Plasmodium falciparum merozoites and infected erythrocytes. J Exp Med. 2005, 201: 1853-1863. 10.1084/jem.20041392.
Sam-Yellowe TY, Florens L, Johnson JR, Wang T, Drazba JA, Le Roch KG, Zhou Y, Batalov S, Carucci DJ, Winzeler EA, Yates JR: A Plasmodium gene family encoding Maurer's cleft membrane proteins: structural properties and expression profiling. Genome Res. 2004, 14: 1052-1059. 10.1101/gr.2126104.
Sargeant TJ, Marti M, Caler E, Carlton JM, Simpson K, Speed TP, Cowman AF: Lineage-specific expansion of proteins exported to erythrocytes in malaria parasites. Genome Biol. 2006, 7: R12-10.1186/gb-2006-7-2-r12.
Schneider AG, Mercereau-Puijalon O: A new Apicomplexa-specific protein kinase family: multiple members in Plasmodium falciparum, all with an export signature. BMC Genomics. 2005, 6: 30-10.1186/1471-2164-6-30.
Nunes MC, Goldring JP, Doerig C, Scherf A: A novel protein kinase family in Plasmodium falciparum is differentially transcribed and secreted to various cellular compartments of the host cell. Mol Microbiol. 2007, 63: 391-403. 10.1111/j.1365-2958.2006.05521.x.
Miller LH, Good MF, Milon G: Malaria pathogenesis. Science. 1994, 264: 1878-1883. 10.1126/science.8009217.
Carlton JM, Angiuoli SV, Suh BB, Kooij TW, Pertea M, Silva JC, Ermolaeva MD, Allen JE, Selengut JD, Koo HL: Genome sequence and comparative analysis of the model rodent malaria parasite Plasmodium yoelii yoelii. Nature. 2002, 419: 512-519. 10.1038/nature01099.
Hall N, Karras M, Raine JD, Carlton JM, Kooij TW, Berriman M, Florens L, Janssen CS, Pain A, Christophides GK: A comprehensive survey of the Plasmodium life cycle by genomic, transcriptomic, and proteomic analyses. Science. 2005, 307: 82-86. 10.1126/science.1103717.
Figueiredo L, Scherf A: Plasmodium telomeres and telomerase: the usual actors in an unusual scenario. Chromosome Res. 2005, 13: 517-524. 10.1007/s10577-005-0996-3.
Barry AE, Leliwa-Sytek A, Tavul L, Imrie H, Migot-Nabias F, Brown SM, McVean GA, Day KP: Population genomics of the immune evasion (var) genes of Plasmodium falciparum. PLoS Pathog. 2007, 3: e34-10.1371/journal.ppat.0030034.
Taylor HM, Kyes SA, Newbold CI: Var gene diversity in Plasmodium falciparum is generated by frequent recombination events. Mol Biochem Parasitol. 2000, 110: 391-397. 10.1016/S0166-6851(00)00286-3.
Figueiredo LM, Pirrit LA, Scherf A: Genomic organisation and chromatin structure of Plasmodium falciparum chromosome ends. Mol Biochem Parasitol. 2000, 106: 169-174. 10.1016/S0166-6851(99)00199-1.
Figueiredo LM, Freitas-Junior LH, Bottius E, Olivo-Marin JC, Scherf A: A central role for Plasmodium falciparum subtelomeric regions in spatial positioning and telomere length regulation. Embo J. 2002, 21: 815-824. 10.1093/emboj/21.4.815.
O'Donnell RA, Freitas-Junior LH, Preiser PR, Williamson DH, Duraisingh M, McElwain TF, Scherf A, Cowman AF, Crabb BS: A genetic screen for improved plasmid segregation reveals a role for Rep20 in the interaction of Plasmodium falciparum chromosomes. Embo J. 2002, 21: 1231-1239. 10.1093/emboj/21.5.1231.
Freitas-Junior LH, Bottius E, Pirrit LA, Deitsch KW, Scheidig C, Guinet F, Nehrbass U, Wellems TE, Scherf A: Frequent ectopic recombination of virulence factor genes in telomeric chromosome clusters of P. falciparum. Nature. 2000, 407: 1018-1022. 10.1038/35039531.
Bailey JA, Yavor AM, Massa HF, Trask BJ, Eichler EE: Segmental duplications: organization and impact within the current human genome project assembly. Genome Res. 2001, 11: 1005-1017. 10.1101/gr.GR-1871R.
Samonte RV, Eichler EE: Segmental duplications and the evolution of the primate genome. Nat Rev Genet. 2002, 3: 65-72. 10.1038/nrg705.
Linardopoulou EV, Williams EM, Fan Y, Friedman C, Young JM, Trask BJ: Human subtelomeres are hot spots of interchromosomal recombination and segmental duplication. Nature. 2005, 437: 94-100. 10.1038/nature04029.
Trask BJ, Massa H, Brand-Arpon V, Chan K, Friedman C, Nguyen OT, Eichler E, van den Engh G, Rouquier S, Shizuya H, Giorgi D: Large multi-chromosomal duplications encompass many members of the olfactory receptor gene family in the human genome. Hum Mol Genet. 1998, 7: 2007-2020. 10.1093/hmg/7.13.2007.
Yue Y, Haaf T: 7E olfactory receptor gene clusters and evolutionary chromosome rearrangements. Cytogenet Genome Res. 2006, 112: 6-10. 10.1159/000087507.
Baumgarten A, Cannon S, Spangler R, May G: Genome-level evolution of resistance genes in Arabidopsis thaliana. Genetics. 2003, 165: 309-319.
Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W: Global variation in copy number in the human genome. Nature. 2006, 444: 444-454. 10.1038/nature05329.
Carret CK, Horrocks P, Konfortov B, Winzeler E, Qureshi M, Newbold C, Ivens A: Microarray-based comparative genomic analyses of the human malaria parasite Plasmodium falciparum using Affymetrix arrays. Mol Biochem Parasitol. 2005, 144: 177-186. 10.1016/j.molbiopara.2005.08.010.
Kidgell C, Volkman SK, Daily J, Borevitz JO, Plouffe D, Zhou Y, Johnson JR, Le Roch K, Sarr O, Ndir O: A systematic map of genetic variation in Plasmodium falciparum. PLoS Pathog. 2006, 2: e57-10.1371/journal.ppat.0020057.
Ribacke U, Mok BW, Wirta V, Normark J, Lundeberg J, Kironde F, Egwang TG, Nilsson P, Wahlgren M: Genome wide gene amplifications and deletions in Plasmodium falciparum. Mol Biochem Parasitol. 2007, 155: 33-44. 10.1016/j.molbiopara.2007.05.005.
Plasmodium Genome Resource. [http://www.plasmodb.org]
Plasmodium falciparum Sequencing Project Broad Institute of Harvard and MIT. [http://www.broad.mit.edu]
Plasmodium genome project, Wellcome Trust Sanger Institute. [http://www.sanger.ac.uk]
OrthoMCL Database. [http://orthomcl.cbil.upenn.edu]
Lavazec C, Sanyal S, Templeton TJ: Hypervariability within the Rifin, Stevor and Pfmc-2TM superfamilies in Plasmodium falciparum. Nucleic Acids Res. 2006, 34: 6696-6707. 10.1093/nar/gkl942.
Mu J, Awadalla P, Duan J, McGee KM, Keebler J, Seydel K, McVean GA, Su XZ: Genome-wide variation and identification of vaccine targets in the Plasmodium falciparum genome. Nat Genet. 2007, 39: 126-130. 10.1038/ng1924.
ArrayExpress at European Bioinformatics Institute (EBI). [http://www.ebi.ac.uk/arrayexpress]
Mok BW, Ribacke U, Winter G, Yip BH, Tan CS, Fernandez V, Chen Q, Nilsson P, Wahlgren M: Comparative transcriptomal analysis of isogenic Plasmodium falciparum clones of distinct antigenic and adhesive phenotypes. Mol Biochem Parasitol. 2007, 151: 184-192. 10.1016/j.molbiopara.2006.11.006.
Bozdech Z, Llinas M, Pulliam BL, Wong ED, Zhu J, DeRisi JL: The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003, 1: E5-10.1371/journal.pbio.0000005.
Hiller NL, Bhattacharjee S, van Ooij C, Liolios K, Harrison T, Lopez-Estrano C, Haldar K: A host-targeting signal in virulence proteins reveals a secretome in malarial infection. Science. 2004, 306: 1934-1937. 10.1126/science.1102737.
Lopez-Estrano C, Bhattacharjee S, Harrison T, Haldar K: Cooperative domains define a unique host cell-targeting signal in Plasmodium falciparum-infected erythrocytes. Proc Natl Acad Sci USA. 2003, 100: 12402-12407. 10.1073/pnas.2133080100.
Marti M, Good RT, Rug M, Knuepfer E, Cowman AF: Targeting malaria virulence and remodeling proteins to the host erythrocyte. Science. 2004, 306: 1930-1933. 10.1126/science.1102452.
Joannin N, Abhiman S, Sonnhammer EL, Wahlgren M: Sub-grouping and sub-functionalization of the RIFIN multi-copy protein family. BMC Genomics. 2008, 9: 19-10.1186/1471-2164-9-19.
Rooney AP, Ward TJ: Evolution of a large ribosomal RNA multigene family in filamentous fungi: birth and death of a concerted evolution paradigm. Proc Natl Acad Sci USA. 2005, 102: 5084-5089. 10.1073/pnas.0409689102.
Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE: Recent segmental duplications in the human genome. Science. 2002, 297: 1003-1007. 10.1126/science.1072047.
Llinas M, Bozdech Z, Wong ED, Adai AT, DeRisi JL: Comparative whole genome transcriptome analysis of three Plasmodium falciparum strains. Nucleic Acids Res. 2006, 34: 1166-1173. 10.1093/nar/gkj517.
Acknowledgements
The work was funded by grants from the Swedish Research Council, the Swedish Institute for Infectious Disease Control (SMI) and the European Union (BioMalPar).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
BWM carried out the design of the study, sequencing, data analysis and wrote the manuscript. UR designed and carried out the qPCR and FISH and helped to finalize the manuscript. ES was involved in the in silico work on copy number predictions. MW participated in the study design and helped to finalize the manuscript. All authors have read and approved the final manuscript.
Electronic supplementary material
12936_2007_521_MOESM2_ESM.pdf
Additional file 2: Topology of proteins encoded by the genes in the SDs. Red bars show transmembrane regions predicted by TMHMM; PEXEL motifs are indicated by green bars. (PDF 365 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Mok, B.W., Ribacke, U., Sherwood, E. et al. A highly conserved segmental duplication in the subtelomeres of Plasmodium falciparum chromosomes varies in copy number. Malar J 7, 46 (2008). https://doi.org/10.1186/1475-2875-7-46
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-7-46